
Abstract
Chitosan fibers were prepared by coagulation from 4% chitosan solutions in 2% acetic acid solution. The synthesis method for polypyrrole (PPy) involving in situ oxidative polymerization of gaseous pyrrole on the surface of chitosan fibers was developed. Structure of the composites was studied by scanning electron microscopy. Electrical conductivity and mechanical characteristics of the obtained composite systems were determined. It was shown that formation of PPy layers on chitosan fibers leads to appearance of electrical conductivity. Conductivity of these layers, as well as their mechanical characteristics, depends on polymerization conditions. The MTT test (a colorimetric assay for assessing cell metabolic activity) showed good biocompatibility of chitosan–PPy composite films; these films contain higher amounts of viable cells than the films cast from solutions of pure chitosan.
In recent years, research endeavors in the field of functional biomaterials have been focused on the development of improved scaffolds (devices or constructions possessing specific and complex physical and biological functions that interact with cells and (when implanted) with the body environment through biochemical and physical signals). 1 These scaffolds are used in regenerative medicine and tissue engineering. As tissue analogs, artificial scaffolds are able to support directed cellular response; the ultimate goal of their preparation is regeneration of lost or damaged tissues and organs.2,3 A scaffold imitating target extracellular matrix (ECM) with suitable cells (mostly stem or somatic cells) seeded on the surface is implanted into a human or animal organism; then, cell differentiation and proliferation proceed under the action of biologically active media and “growth factors” introduced into the matrix.
In view of the foregoing considerations, scaffolds are fabricated to perform several or all of the following functions: (a) facilitate matrix biocompatibility; (b) promote strong interactions between cells and a biomaterial, cell adhesion, and ECM deposition; (c) contribute to determination of cell fate; (d) allow for sufficient transport of gases, nutrients, and regulatory factors that facilitate cell survival, proliferation, and differentiation; (e) undergo biodegradation at a controllable rate that approximates the rate of tissue regeneration under the influence of active biological media; (f) provoke a minimal degree of inflammation or cytotoxicity in vivo and low antigenicity; and (g) have a certain level of strength and sufficient elastic characteristics necessary for manipulations with scaffolds in liquid media. Mechanical parameters of the materials should provide ability for scaffolds to withstand the physiological loads applied during functioning in vivo.3,4 In addition, the architecture of a tissue engineering scaffold should allow for full infiltration of target cells in order to promote ingrowth of both the tissue and the necessary vascular support networks. 2
Thus, scaffolds are designed in different shapes, sizes, structures, and architectures by various technologies and methods and can be produced from different materials, depending on their intended use.3–5 Biological scaffolds are derived from human or animal tissues, and synthetic scaffolds are prepared from organic (mostly polymeric) or inorganic materials.3,4 Polymeric materials are preferred, because the important characteristics of scaffolds, such as their strength, rate of degradation, porosity, microstructure, shape and size, are relatively easily controlled and can be reproduced in polymer-based scaffolds. 4 Chitosan (poly-(β-1/4)-2-amino-2-deoxy-D-glucopyranose), the N-deacetylated derivative of natural polysaccharide chitin, is the main component of the shells of crustaceans, such as crab, shrimp, and crawfish. 6 Chitosan is one of the most promising polymers for tissue engineering due to its numerous favorable medical properties, including biocompatibility, bioresorption capability, absence of cytotoxicity, and low environmental impact of processing technologies. 3 Chitosan is soluble in dilute aqueous solutions of acids (formic, acetic and propionic acids) and possesses good fiber- and film-forming properties so that it is easily processed into two-dimensional matrices, 7 fibers,8–13 nanofibers, 14 three-dimensional porous materials,15–18 and hydrogels.19,20 Thus, a wide selection of chitosan matrices makes it possible to simulate surface properties and mechanical characteristics of any natural tissue. The majority of research works devoted to fabricating chitosan-based biomaterials have been aimed at preparation of three-dimensional porous scaffolds. However, one-dimensional matrices can be of particular interest, because these matrices are promising for regenerating nerve 21 or muscle tissue, ligaments, and tendons; 22 they can be used in preparation of hemostatic materials.23,24
Although the chitosan-based materials have advantageous medicinal characteristics, they do not generally possess electrical conductivity. Because electric signals are involved in various biological events (such as cell communications and behavior, tissue regeneration), scaffolds should have a certain level of electrical conductivity to enhance their biocompatibility, modulate cell/tissue responses, facilitate survival, promote growth, and enable differentiation of stem cells and their stimulation.25,26
To improve functional characteristics of chitosan scaffolds and to facilitate better cell communication and tissue responses, chitosan-based composite materials that include conducting polymers are prepared. Among other conductive polymers, polypyrrole (PPy) best meets the biomedical requirements. A large number of publications are devoted to analysis of its properties and possible applications. 27 PPy exhibits redox properties and paramagnetism; its electronic and ionic conductivities are controllable and sensitive to external influences; they change reversibly in the range of 10−6–101 S/cm. This polymer is biocompatible both in vivo and in vitro,27–29 and affects the mammalian immune system only minimally. 30 PPy is chemically stable in air and water, and demonstrates relatively high electrical conductivity in physiological conditions. This polymer is considered a promising basis for preparation of “smart” bioactive materials. 27
PPy is synthesized by chemical or electrochemical oxidative polymerization of pyrrole. In the case of chemical oxidation, a broad range of oxidants and various solvents (including water) are used. The polymer is obtained in the form of nanoparticles with different structures, which are aggregated to give various morphological forms (from densely packed to loose structures) with large surface areas and controllable porosity parameters. To adapt the material to various biomedical applications, PPy can be modified with a number of doping agents including bioactive molecules. 27 PPy can also be “grown” during metabolic processes within living microorganisms whose polysaccharide cell walls are structurally similar to chitosan;31–34 in this way, charge transfer through cell walls can be improved. 35 Properties of this polymer can be changed reversibly by applying electric potential. 27
PPy is often used as a component of composite materials, in which the other component (or components) ensures the required mechanical properties of the material (strength and flexibility), which are absent in PPy. 36 However, preparation of these composites is difficult due to poor processability of PPy (lack of solubility and meltability) and the fact that conventional methods used in polymer synthesis cannot be applied in the case of PPy. For this reason, PPy-containing composites are usually obtained by the one-stage in situ process, which combines pyrrole polymerization and the formation of the composite. The synthesis is performed in situ, that is, in the presence of the other component that plays the role of a support. In the course of polymerization, PPy is adsorbed on the support in the form of a nanolayer, which coats the surface and penetrates into pores. The preparation method for each composite is developed individually, because the properties of PPy and characteristics of the resulting polymer layer (thickness, homogeneity, and structure) are very sensitive to the nature of a support and synthesis conditions. 27
The aim of this work was to develop conducting chitosan–PPy composite fibers that possess the mechanical properties necessary for cell cultivation, show good biocompatibility and bioactivity (that provides biological response) and can be applied as advanced matrices in tissue engineering.
Materials and methods
Preparation of fibers and films
Chitosan (Biolog Heppe GmbH, Germany) with molecular mass of 1.64 × 105 and deacetylation degree of 92% was used in fiber preparation. Pyrrole solution (98%) (Aldrich) and anhydrous FeCl3 salt (Aldrich) were used in the synthesis of PPy.
Chitosan fibers were spun by coagulation from 4% chitosan solution in 2% acetic acid solution.8,37
To prepare fibers, the mixture of chitosan and water (pH = 4–5) was stirred at room temperature for 30 min until partial dissolution of chitosan and its swelling occurred. Then aqueous solution of acetic acid was added to the mixture at continuous mechanical stirring. The resulting mixture was stirred in the glass flask at room temperature for 90 min, then filtered and deaerated for 24 h at a pressure of 0.1 atm.
The fibers were spun by coagulation9,37 using the laboratory equipment developed at the Institute of Macromolecular Compounds, Russian Academy of Sciences (RAS). The scheme of this process is presented in Figure 1. The mixture of 10% solution of NaOH and C2H5OH in the 1:1 ratio was used as a precipitant. Monofilaments were prepared using the die hole with a diameter of 0.6 mm; to spun polyfilaments, the 100-hole die with a hole diameter of 0.1 mm was employed. The solution was fed through the dies at rates of 0.2 mL/min and 0.3 mL/min, respectively; precipitation time was 150 s. The degree of orientation drawing (λ, as a percentage) of the monofilament in the coagulation bath was varied from 0% to 100%; for polyfilaments, this value was 50%. The fibers were washed with distilled water and dried at 50°C for 10 min.
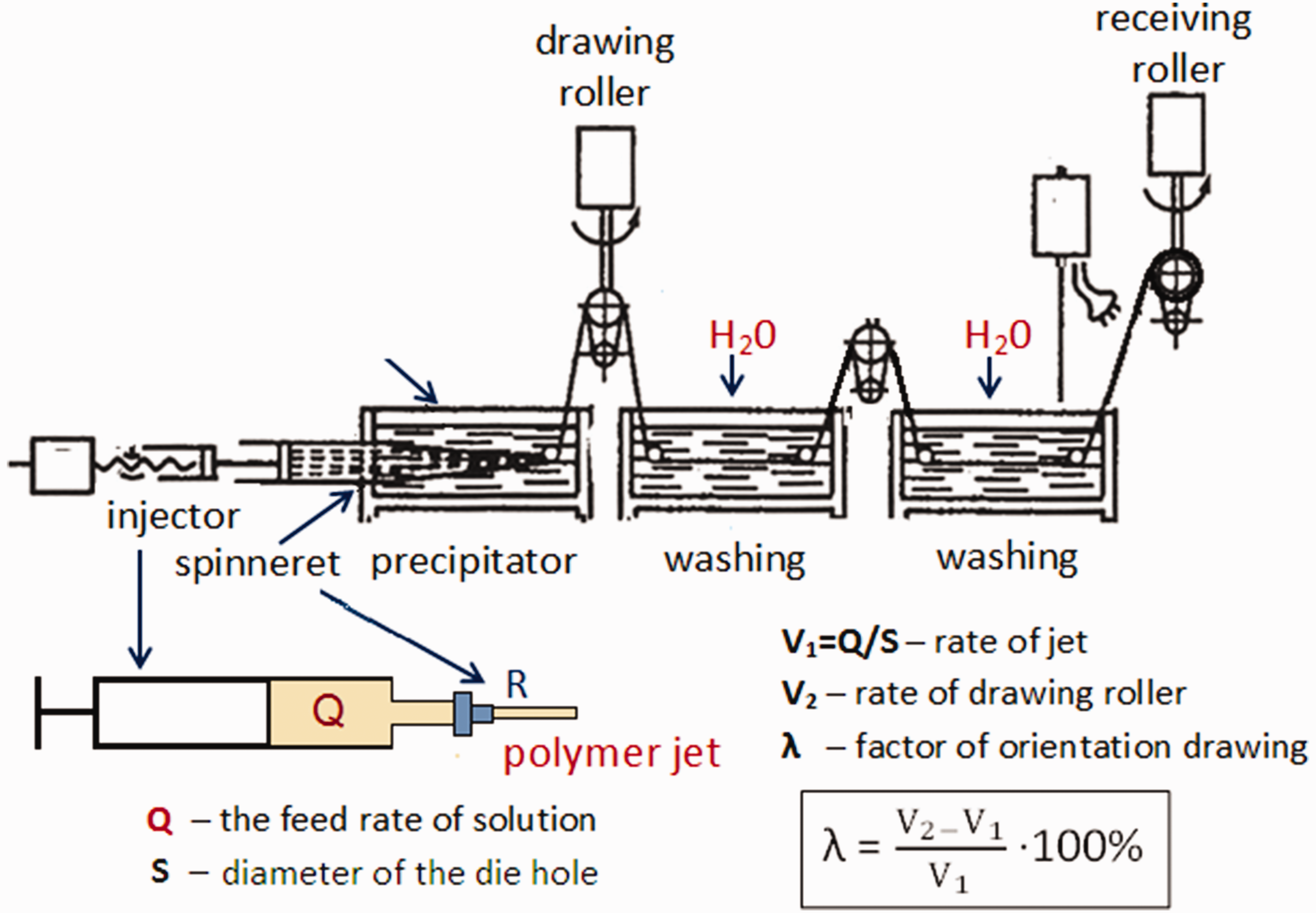
Scheme of chitosan fiber spinning.
Chitosan films were prepared under similar conditions. The samples were obtained by passing the chitosan solution through a slit draw die onto a glass substrate followed by drying at room temperature for 24 h. The film thickness was 40 ± 15 µm. Then the initial films were left in a precipitator for 10 min, washed with distilled water, and air dried.
Rheological measurements of chitosan solutions
Rheological measurements of polymer solutions were carried out with the aid of a Physica MCR 301 rheometer (Anton Paar GmbH, Austria) at 20°C according to the “cylinder in cylinder” method in the shear flow regime, at shear rates varying from 10−4 to 100 s−1. Next, 5 mL of solution was placed into the rheological cuvette, and the dependence of viscosity (η) on shear rate (
Swelling measurements
The swelling ratios of chitosan fibers in water and methanol were measured by hydrostatic weighing using an ER-182A electronic analytical balance ( ER-182A, Japan). The samples were pre-dried at a temperature of 80°C38 until constant weight was reached. The weight of the swollen samples was determined directly in the media where they swelled to an accuracy of ±0.0001 g; the samples were placed onto the balance in the test liquid. When a sample swells, the liquid gradually fills the free volume of the polymer, and the free volume of the fibers decreases; therefore, the buoyant force also decreases, and the weight of the fiber in a non-inert medium grows. Thus, if we take the partial density of water in a polymer equal to the density of bulk liquid, the swelling (H, as a percentage) will be equal to
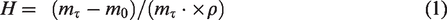
PPy polymerization
The chitosan–PPy composite fibers were prepared by oxidative polymerization of pyrrole; FeCl3 was used as an oxidant. 39
The PPy layer on the surface of chitosan fibers was obtained by saturation of chitosan fibers with an oxidant (1 M solution of ferric chloride) followed by in situ polymerization of pyrrole monomer vapors on the chitosan fiber surface. The duration of the oxidation was varied from 30 min to 24 h, and the duration of pyrrole polymerization was varied from 30 min to 72 h.
Studies of sample structure
The structural studies were performed using a Supra-55 VP scanning electron microscope (Carl Zeiss, Germany) and a Micromed-2 optical microscope.
Mechanical testing of fibers
Mechanical properties of chitosan and composite fibers were tested using an Instron 5943 machine at room temperature; the loading speed was 10 mm/min; the basic length of fibers was 100 mm. The cross-section (S, in square meters) of the monofilaments was estimated by the formula

Study of electrical resistivity
To determine the electrical resistivity values of composite fibers, current–voltage characteristics were measured at room temperature using the special automated device based on the double-probe system.40,41
The sample was placed into the screened chamber. The contacts between the sample and electrodes were made of the carbon paste (Conductive Carbon Paint manufactured by SPI Supplies, USA (dispersity: 1 µm, specific electrical resistance: 10−1 Ω cm)). The power supply gave constant voltage on one of the electrodes. The electric current passed through a sample to the second electrode and further to a picoammeter, showing the current value in the chain.
The electrical resistivity ρV (in ohm centimeters) of the samples was estimated by the equation
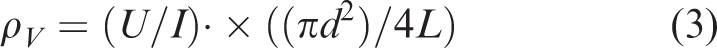
Taking into account the small diameter of the fiber and the fact that it is not possible to correctly determine the PPy layer thickness, the calculations were performed for the fiber as a whole, and not exclusively for the PPy layer.
Attachment, viability, proliferation, and morphology of fibroblasts
Biological compatibility is the fundamental parameter that determines the safety of medical implants. Currently, the majority of the techniques used for evaluation of cytotoxicity of degradable implants involve the indirect methods described in the ISO 10993 series of standards.42,43 According to these standards, the MTT assay of human fibroblasts is suggested as a cytotoxicity test for evaluating biocompatibility of biodegradable materials.
Human dermal fibroblasts were obtained from the cell culture collection of the Institute of Cytology RAS (Saint Petersburg, Russia) and used up to the 12th passage. The cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 1% penicillin, 1% streptomycin, 1% fungizone, 2 mM
Studies of viability and proliferation of the cells in the samples were performed using the MTT test. Film matrices were placed in 24-well plates, and culture medium was added. Sterilized silicone rings were placed on top to keep the scaffolds submersed, and 25 × 103 fibroblasts were seeded on top of the scaffolds. The cells were re-suspended in culture medium and then incubated in humidified atmosphere containing 5% of CO2. After 4 days of incubation, the culture medium was removed, and each well was treated with 10 µL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) (Thermo Fisher Scientific, USA), 5 mg/mL in culture medium, and incubated for 4 h at 37°C in humidified atmosphere of 5% CO2. The yellow MTT is reduced to blue-purple formazan in the presence of the mitochondrial dehydrogenase. This enzyme is contained in intact living cells, hence the blue-purple color produced should be proportional to the number of viable cells present. The MTT solution was then replaced with 100 µL per well of dimethylsulfoxide (Paneco Ltd, Russian Federation) to dissolve the formazan salts, followed by 10 min of slow agitation, yielding a blue-purple solution. The absorbance of this solution was measured at 570 nm using a SPECTROstar® Nano microplate reader for absorbance measurements (BMG LABTECH, Germany).
Studies of cellular morphology and cell attachment were performed using a Primo Vert inverted light microscope (Zeiss, Germany); the images were taken with a digital camera.
All measurements were performed in triplicates, and the t-test was used to determine significant differences (p < 0.05) between chitosan–PPy composites and chitosan matrix.
Results and discussion
Rheological properties of chitosan solutions
To optimize the fiber spinning conditions, it is necessary to obtain information about rheological properties of polymer solutions. Chitosan is a rigid chain polymer. It has been shown8,37 that oriented structure of the chitosan fibers is formed during flowing of the solution through the die hole. In turn, orientation of the macromolecules depends on the shear stress occurring in the die hole. The shear rate during spinning of the polymer solution through a die strongly affects the strength of the resulting fibers.
Figure 2 represents the dependences of the viscosity on the shear rate for the 3.5, 4, and 4.5 wt.% chitosan solutions in 2% acetic acid solution.
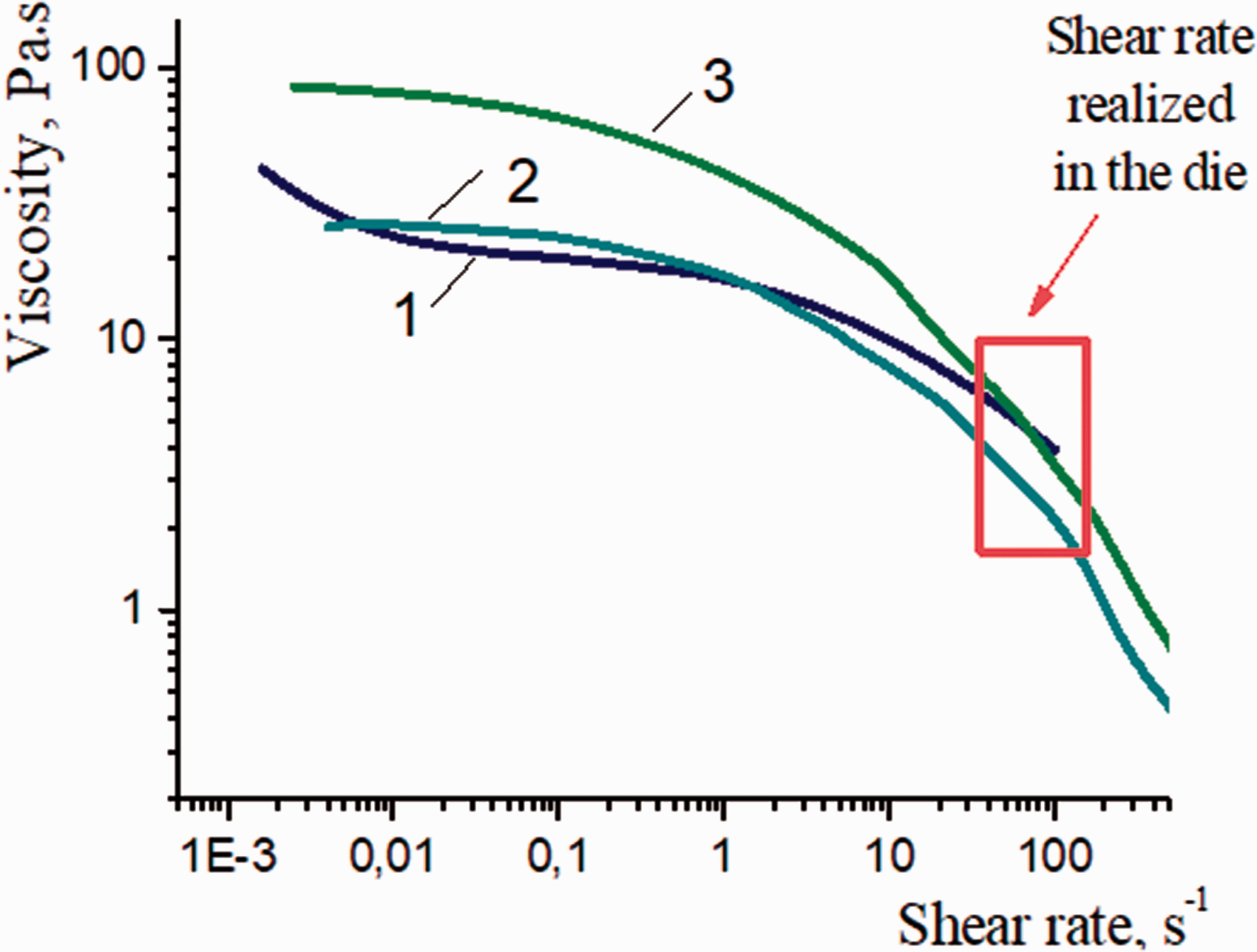
Rheological properties of chitosan solutions: 1 = 3.5 wt.%; 2 = 4 wt.%; 3 = 4.5 wt.%.
All dependences of viscosity on shear rate for chitosan solutions demonstrate nonlinear behavior. The slopes of the curves increase substantially with an increase in the shear rate; the significant decrease in viscosity is observed at the shear rate of 10 s−1 or higher. The decrease in viscosity upon an increase in the shear rate is related to the destruction of the initial structure of the polymer solution and appearance of a new oriented structure; that is, transition from an isotropic state to an anisotropic state occurs. The formation of the anisotropic structure of the polymer solution under the action of the shear field is typical of the majority of rigid chain polymers.8,44
The 3.5 and 4 wt.% chitosan solutions have almost similar viscosity values in the shear rate, with a range of 1–10 s−1. The dependences for the 4 and 4.5 wt.% chitosan solutions have a similar behavior and show the most noticeable decrease in the shear rate. These solutions are able to form more ordered structures during the spinning process. At the same time, the viscosity of the 4.5 wt.% chitosan solution is too high, which may cause difficulties in fiber processing. Thus, the optimal concentration of chitosan in the solution for fiber spinning is 4%.
The studies of rheological properties also allowed us to calculate the optimum feed rate of the chitosan solution through the die hole. The optimum feed rate Q (in millimeters per minute) of the polymer solution through the die hole of the radius R can be calculated according to the following equation

Mechanical properties of chitosan fibers
In our previous work, 37 it has been shown that chitosan fibers with the best mechanical properties could be prepared from 4 wt.% chitosan solution.
Mechanical properties of chitosan fibers obtained from the 4 wt.% chitosan solution are illustrated in Figure 3.
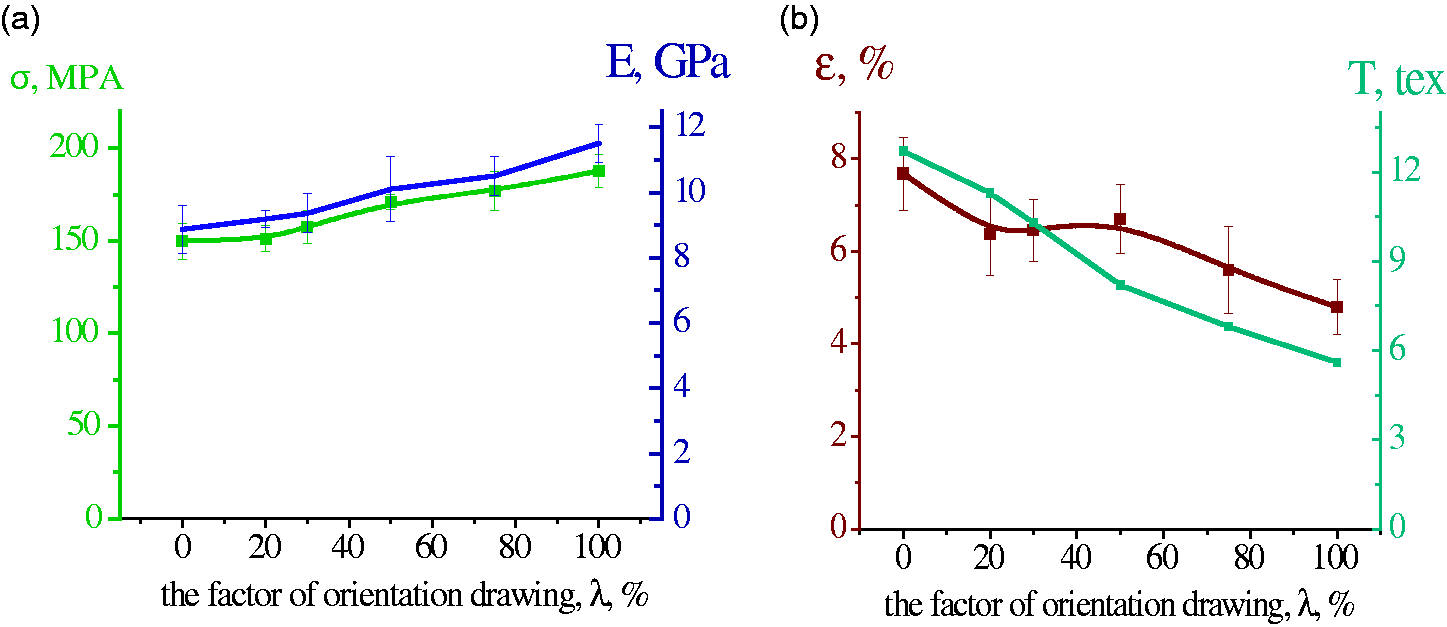
Dependences on the degree of orientation drawing (λ) for chitosan fibers obtained from the 4 wt.% chitosan solution: (a) tensile strength (σ), and Young’s modulus (E) and (b) elongation at break (ε) and fineness (T).
It is seen that tensile strength (σ) and Young’s modulus (E) of the spun fibers increase considerably with an increase in the degree of orientation drawing (λ), whereas the elongation at break (ε) and fineness (T) decrease. The maximum degree of orientation drawing of the monofilament is λ = 100%. The fibers with the degree of orientation drawing λ = 50% have the optimal mechanical properties: tensile strength is 171.0 ± 4.1 MPa, Young’s modulus is 10.1 ± 1.0 GPa, and elongation at break is 6.7 ± 0.8%. These fibers do not break under dynamic mechanical loads, and thus can be used for further processing.
PPy polymerization
PPy layers were formed on the surface of mono- and polyfilament chitosan fibers in the course of oxidative polymerization. Ferric chloride solution (1 M) was used as an oxidizing agent. This reaction can be described by the following equation
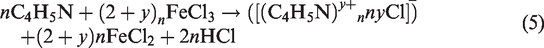
It has been established46,47 that the synthesis of pyrrole in the absence of any material in the reaction medium yields spherical particles with diameters ranging from 100 to 200 nm.
Solutions of the oxidant were prepared in water and methanol. These solvents were used because chitosan fibers have different swelling degrees in these two media. The initial chitosan fibers have a high swelling degree in water (∼120%) and practically do not swell in methanol (Figure 4). Degrees of swelling determine the amount of oxidant adsorbed by fibers, and, consequently, the structure of the PPy layer formed on the surface of fibers and the duration of its formation.
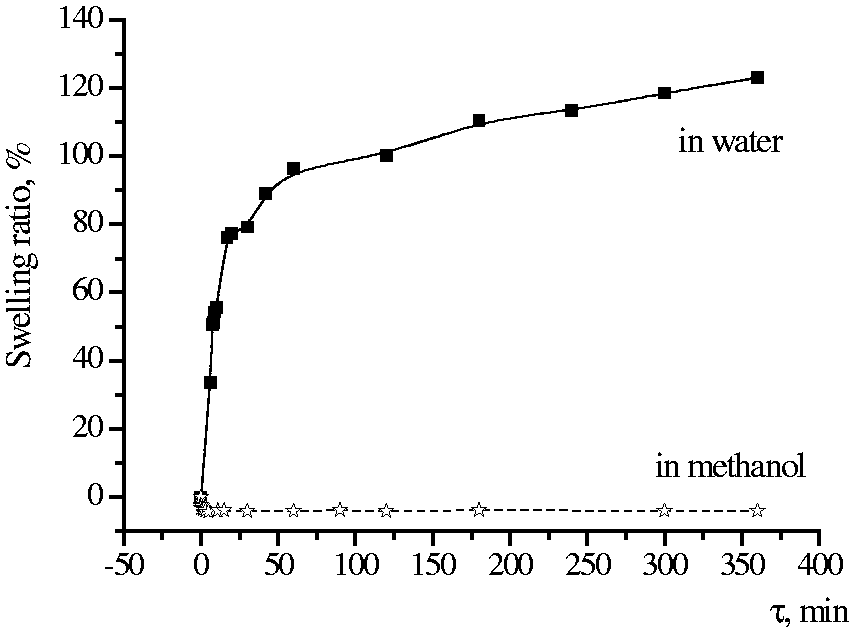
Swelling kinetics of chitosan fibers.
Polymerization of pyrrole was carried out in gas phase. In this case, polymerization occurs only on the fiber surface, and does not proceed in the bulk of the polymerization medium.
Modification of chitosan fibers with PPy was carried out in the following manner (Figure 5). The initial fibers were left in the oxidant solution and then dried in air at room temperature. The time of exposure to the oxidant varied from 30 min to 24 h.

Processing of chitosan fibers and preparation of composites.
It was shown that the amount of oxidant adsorbed on the fiber was higher when using FeCl3 solution in methanol compared with that in the case of the aqueous oxidant solution (maximum 30% and 18%, respectively).
The oxidant-treated fibers were placed in monomer vapors. The polymerization time varied from 30 min to 72 h. The resulting composite fibers were washed with methanol to remove the residual unreacted oxidant.
The electron microscopy investigations of the surface of composite fibers (Figure 6) showed that the duration of the PPy layer formation on the chitosan fibers’ surface and its structure depend on the type of the solvent used. In the case of the aqueous solution, the PPy layer on the surface of fibers is not formed if the time of exposure to the oxidant is less than 24 h (irrespective of polymerization time). The formation of the PPy layer occurs when the oxidant treatment time exceeds 24 h, and the polymerization time varies from 24 to 48 h. However, the structure of the PPy layer is neither homogeneous nor uniform. When the polymerization time reaches 72 h, the “excess” PPy is formed. Because PPy is a rigid chain polymer, the defects in the form of cracks and “growths” appear.

Scanning electron microscopy images of pure chitosan fibers and composite samples (chitosan fibers coated with polypyrrole layer.
When the methanol solution of FeCl3 is used, the composite fiber surface is even and homogeneous shortly after beginning the oxidant treatment and polymerization. This result can be explained by the fact that chitosan fibers do not swell in methanol, which leads to the formation of a denser and more uniform initial fiber surface. As a result, the uniform and homogeneous PPy layer is obtained in the course of polymerization. Similarly, in the above case (aqueous solutions), the increase in polymerization time up to 72 h leads to formation of defects.
Mechanical properties of the chitosan–PPy composite fibers
Mechanical characteristics, such as tensile strength, elongation at break, and Young’s modulus, were measured for all samples (Table 1).
Mechanical properties of the composite chitosan–polypyrrole fibers
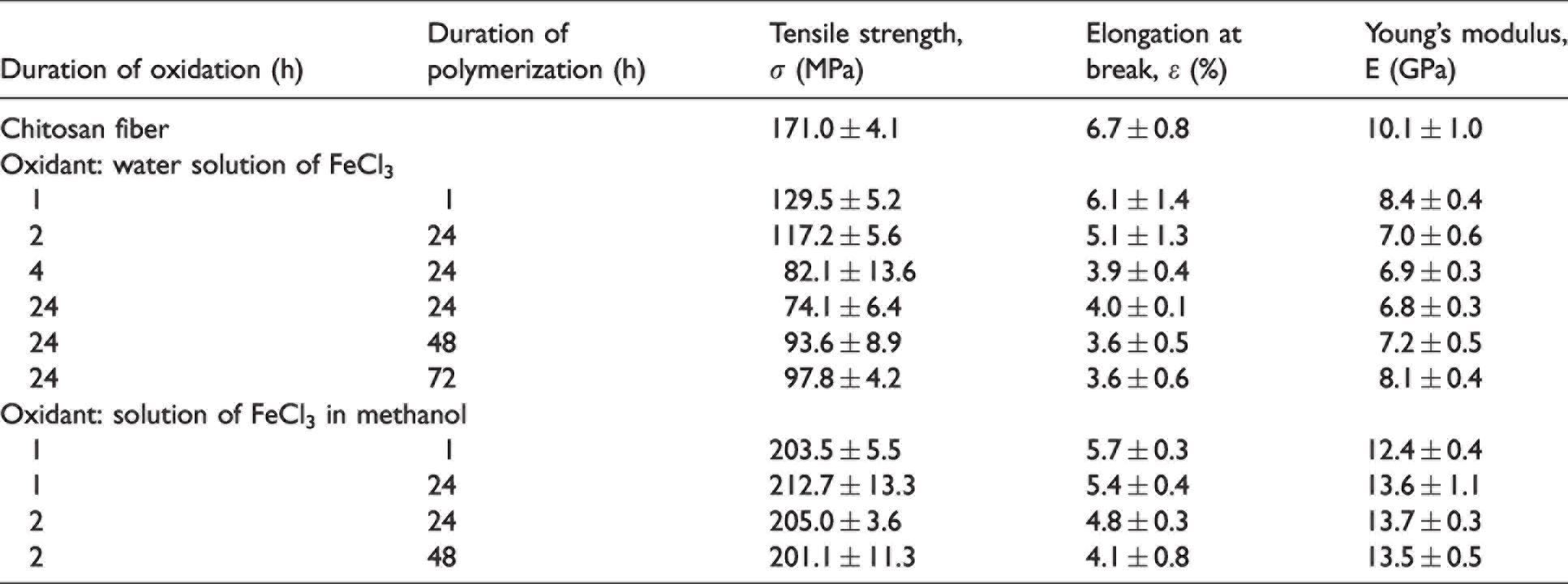
The obtained data show that treatment of the fibers with an aqueous solution of the oxidant for 1 h followed by polymerization of pyrrole for 1 h yields the composites with relatively poor mechanical characteristics (compared with those of the initial chitosan fibers). This result is caused by changes in fiber structure and an increase in the diameter of the treated fiber. Chitosan fibers possess high water absorption ability (Figure 4); drying of fibers (upon exposure to the oxidant, and before polymerization) leads to shrinkage, modification of surface structure (Figure 7(b)) and an increase in fiber diameter (Figure 6). The increase in the time of exposure of chitosan fibers to the oxidant (polymerization times being equal) causes further drops in both tensile strength and breaking elongation, whereas the elastic module decreases slightly. When exposure time of chitosan fibers to the oxidant was 24 h, the following values of σ, ε, and E were obtained: σ = 71.4 ± 6.4 MPa, ε = 4.0 ± 0.1%, and E = 6.8 ± 0.3 GPa. At the same time, upon increase in the pyrrole polymerization time (the oxidant treatment time remaining similar (24 h)), the resulting fibers have enhanced strength and elastic modulus, whereas elongation at break continues to decrease gradually. This result can be explained by the formation of the layer of rigid chain PPy on the surface of chitosan fibers.

Scanning electron microscopy images of the initial chitosan fibers (a) and chitosan fibers after their exposure to water (b) and methanol (c) followed by drying.
Exposure of chitosan fibers to ferric chloride solution in methanol for 1 h followed by polymerization for 1 h is sufficient to form a thin homogeneous layer on the fiber surface. This is due to insignificant swelling of the fiber in methanol and, correspondingly, to the absence of any noticeable changes in its structure and surface characteristics (Figure 7(c)). Formation of the homogeneous PPy layer on the surface of chitosan fiber leads to an increase in strength and elastic modulus accompanied by a drop in breaking elongation, regardless of oxidation and pyrrole polymerization times.
To summarize, an increase in duration of treatment of chitosan fibers with aqueous solution of oxidant and in polymerization time leads to growth in the PPy content in the composite. The times of oxidant treatment and the subsequent polymerization do not have any influence on mechanical characteristics of the composites. On the whole, the composite fibers are more rigid and brittle than the initial chitosan samples. It should be noted that upon treatment with methanol solution of FeCl3, the PPy layer is formed only on the fiber surface, whereas treatment of chitosan fibers with aqueous solution of the oxidant leads to formation of the PPy layer both on the surface and in the near-surface area of chitosan. These results agree with the data obtained by other authors.48,49 The PPy layer shows good adhesion to chitosan matrix; no flaking of PPy was registered even during mechanical tests and destruction of samples. The observed adhesion can be explained by formation of strong hydrogen bonds between chitosan and PPy. 50 This type of interaction was also revealed by Fourier transform infrared spectroscopy in the chitosan–PPy nanocomposites containing 5 and 10 wt.% of PPy.50–53
Electrical conductivity of the chitosan–PPy composite fibers
Electrical conductivity of the obtained composites depends on the amount of the conductive PPy present on the surface of chitosan fiber as well as on uniformity and homogeneity of the formed PPy layer.
It was demonstrated that electrical conductivity increases with increasing polymerization time (up to 48 h) for the whole series of samples, regardless of oxidant type, due to an increase in the PPy amount on the surface of chitosan fibers. The obtained data (Table 2) show that the sample treated with ferric chloride solution in methanol (exposure time 2 h, polymerization time 48 h) and the sample treated with ferric chloride solution in water (exposure time 24 h, polymerization time 48 h) have the highest values of electrical conductivity (3 × 10−2 S/cm and 2 × 10−2 S/cm, respectively).
Electrical conductivity of the chitosan–polypyrrole composite fibers
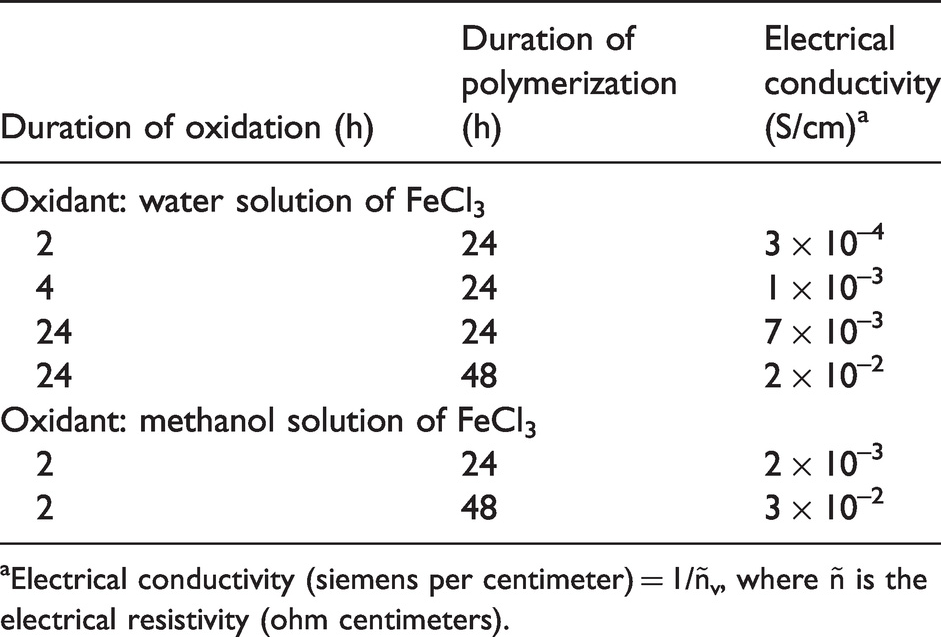
aElectrical conductivity (siemens per centimeter) = 1/ñv, where ñ is the electrical resistivity (ohm centimeters).
Meanwhile, the duration of oxidation that facilitates formation of the homogeneous PPy layer on the fiber surface is determined by the type of oxidant used (Table 2). Thus, when ferric chloride solution in water is used, the optimal oxidation time is 24 h due to the high swelling ability of chitosan in water. When ferric chloride solution in methanol (in which chitosan does not swell) is used, 2 h of oxidation is sufficient to provide formation of the homogeneous conducting layer.
It was not possible to measure electrical conductivity of the samples obtained after polymerization for 72 h because the samples became brittle.
Adhesion and proliferation of fibroblasts
The developed composite fibers are promising materials for preparing tissue engineering scaffolds and hemostatic materials; thus, biological properties of the obtained composites were investigated as well. Fibroblasts are among the key effector cells in wound healing, fibrotic processes, and in the initial inflammatory phase that begins after implantation of a biomaterial. Therefore, the initial adhesion, spreading, and the following proliferation and outgrowth of fibroblasts (which are crucial for potential wound healing and development of fibrosis) were studied.
Human dermal fibroblasts were cultured on chitosan, chitosan–PPy film matrices, and cultural polystyrene surface (control). The studies of cellular morphology and attachment were carried out using an inverted light microscope; the images were obtained using a digital camera. The cell viability and proliferation were evaluated using the MTT test.
Viable cells grown on the chitosan–PPy composite matrices and cultural polystyrene surface (control) displayed a normal spindle-shaped morphology and were spread across the whole surface (Figure 8(i)). Growing cells on pure chitosan matrices led to the appearance of irregularly shaped cells, spherical cell conglomerates, and considerably elongated rod-like cells. The cells grown on the chitosan matrices were poorly spread and distributed over the surface (Figure 8).
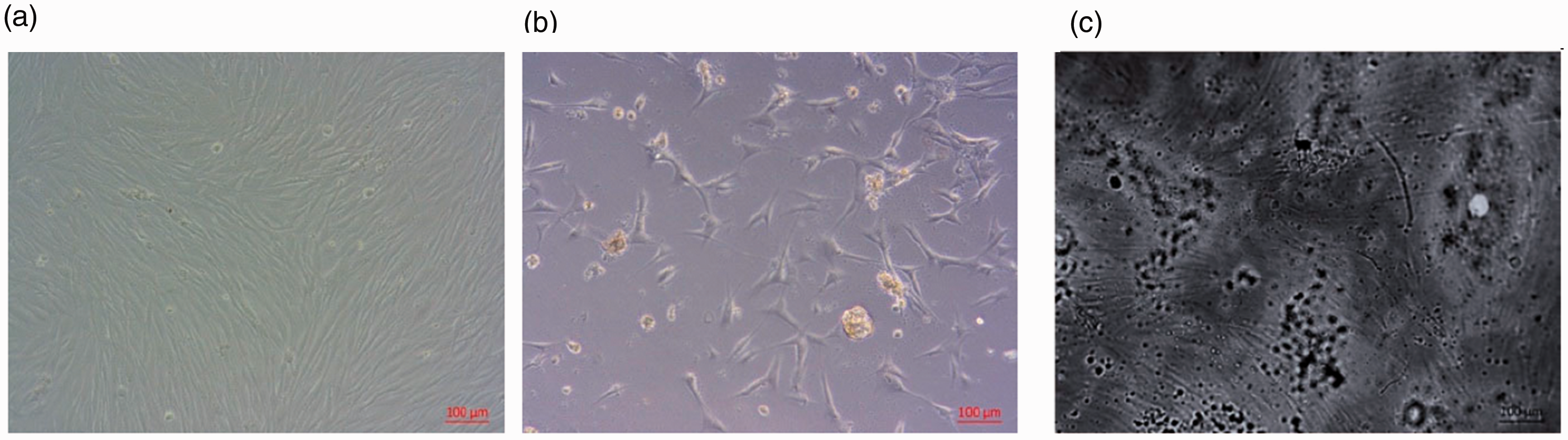
(i) Human dermal fibroblasts cultured on (a) cultural polystyrene surface (control), (b) chitosan, and (c) chitosan–polypyrrole film matrices for 4 days. Original magnification ×100, phase-contrast light microscopy. (ii) Human dermal fibroblasts viability on film matrices and cultural polystyrene surface (control) as determined by MTT assay (absorbance 570 nm).
The proliferation rate of cells (for 4 days) on chitosan–PPy was found to be higher than that in the case of the chitosan matrices (Figure 8).
Thus, we can conclude that use of chitosan–PPy composite films makes it possible to obtain more viable cells and achieve higher biocompatibility.
The information about biocompatibility and bioactivity of the obtained materials can be compared with the results of previous studies. Biocompatibility of various types of PPy-containing matrices was revealed by cultivation of cells on these matrices and monitoring their vital activity. The PPy-containing matrices demonstrated significantly better cell adhesion as compared with pure chitosan scaffolds. It was also shown that use of PPy-containing scaffolds in combination with electrical stimulation made it possible to adjust cell behavior according to therapeutic purposes. 54
Considerable disagreement between the data on biocompatibility and bioactivity of materials obtained by different research groups is also related to the used types of tissues or cells. The use of conductive PPy-containing scaffolds can be a promising strategy in tissue engineering, especially when electroactive cells and tissues are involved.
Conclusions
A new tissue engineering composite scaffold based on chitosan fibers has been prepared by polymerizing pyrrole on the surface of oxidized chitosan fibers. Pyrrole polymerization was carried out in two different media: (a) solution of ferric chloride in water; and (b) solution of ferric chloride in methanol. Experiments showed that the pyrrole polymerization carried out in the methanol solution yielded the samples with much more continuous and homogeneous PPy coating as compared with that prepared by polymerization in water solution. This result can be explained by the fact that chitosan fibers do not swell in methanol, whereas their swelling degree in water is relatively high. Therefore, pyrrole polymerization in methanol solution was found to be the method of choice in fabricating the experimental scaffolding composites. Studies of electrical conductivity, mechanical properties (tensile strength, elongation at break, and elastic modulus), and biocompatibility testing involved chitosan–PPy composites prepared by two methods and demonstrated advantages of PPy polymerization in methanol. MTT tests showed better biocompatibility of the chitosan–PPy composite films compared with that of unmodified chitosan films. It is concluded that the developed chitosan–PPy composite fibers can be further used as structural elements in bioactive matrices.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Russian Science Foundation (Project No. 19-73-30003).