
Abstract
Triple A syndrome is a rare autosomal recessive disorder characterised by alacrimia, achalasia and adrenal failure. It was first reported by Allgrove in 1978 and 100 cases have been reported worldwide. This case report concerns a 24-year-old woman who was referred for evaluation of dysphagia and was finally diagnosed as such a case. A high degree of suspicion enables all the components of this syndrome to be searched for, as early diagnosis can reduce the morbidity and mortality.
Case report
A 24-year-old woman came to the outpatient department with complaints of dysphagia to liquids and solids since the age of five years. Dysphagia was gradually progressive, more to liquids and was associated with regurgitation, vomiting and cough. She was also having salt cravings, pica, darkening of skin, darkening inside the mouth and gums, and generalised weakness since childhood. Her parents also gave a history of an absence of tears since birth. She was born full-term by normal vaginal delivery from non-consanguineous parents and her perinatal period was unremarkable. She has one younger sibling who is healthy and asymptomatic.
On examination she was thin with a body mass index of 16.0 kg/m2. Her blood pressure was 100/70 mmHg with no postural drop. She had hyperpigmentation of the skin, knuckles, tongue, gums and buccal mucosa (Figure 1). Her systemic examination including detailed sensory and motor neurological examination was normal.
Pigmentation of skin, knuckles, tongue, buccal mucosa and gums.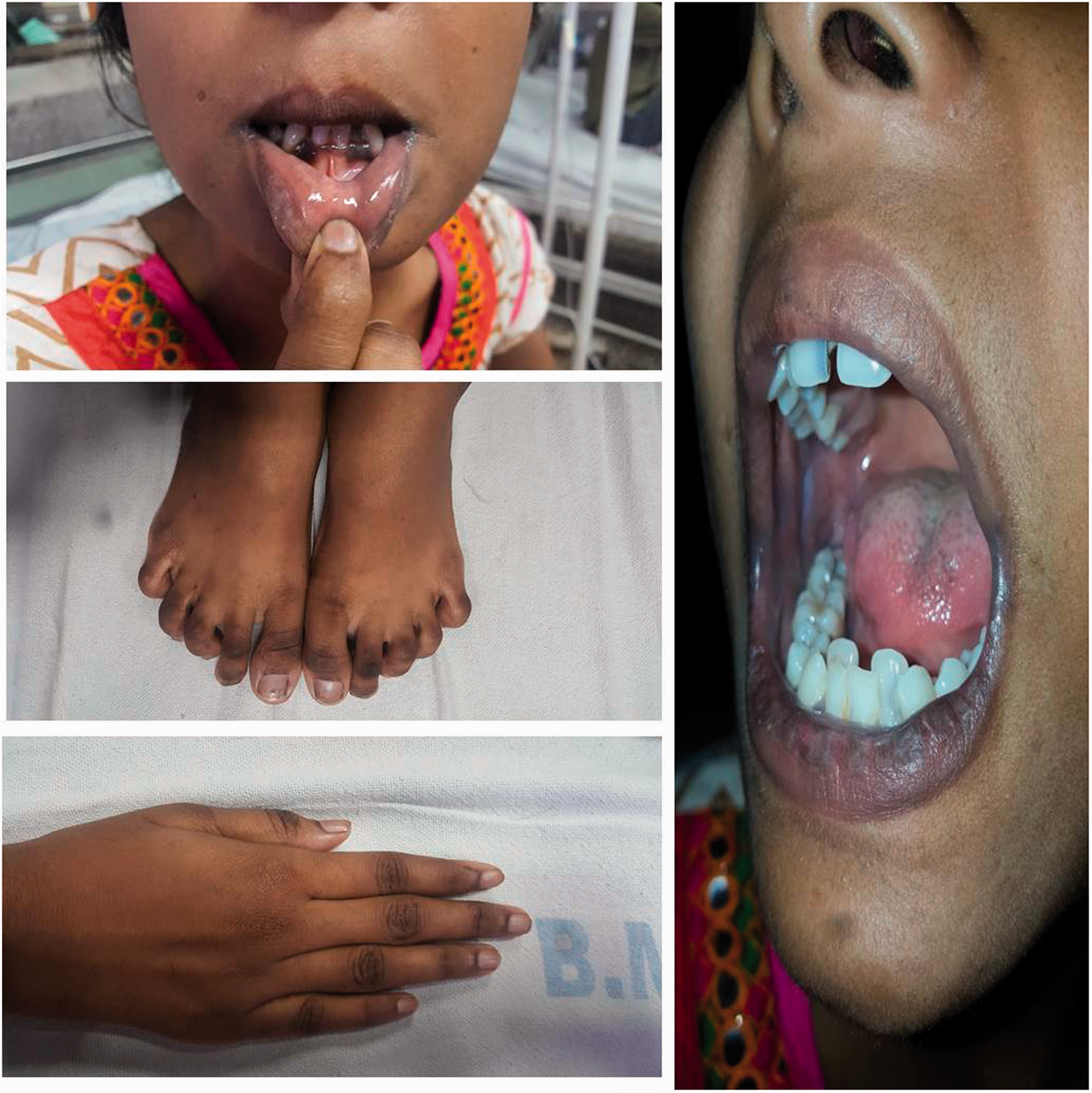
Her baseline investigations revealed a haemoglobin of 101 g/L, total leucocyte count of 7500/mm3, platelet count of 350 × 109/L, urea 11.4 mmol/L, creatinine 0.08 mmol/L, sodium 128 mmol/L and potassium 5.9 mmol/L. Her basal serum cortisol level was 13.8 nmol/L (normal range = 110–600 nmol/L) which failed to increase after a stimulation test. Serum adrenocorticotropic hormone (ACTH) levels were markedly raised at 209 pmol/L (normal range <10 pmol/L). A contrast-enhanced computed tomography scan showed atrophy of both adrenal glands. Thus, a diagnosis of primary adrenal insufficiency was made and a Shirmer test confirmed bilateral alacrimia. A barium swallow (Figure 2) followed by oesophageal manometry confirmed the diagnosis of achalasia.
Barium swallow showing dilated oesophagus with bird’s beak deformity.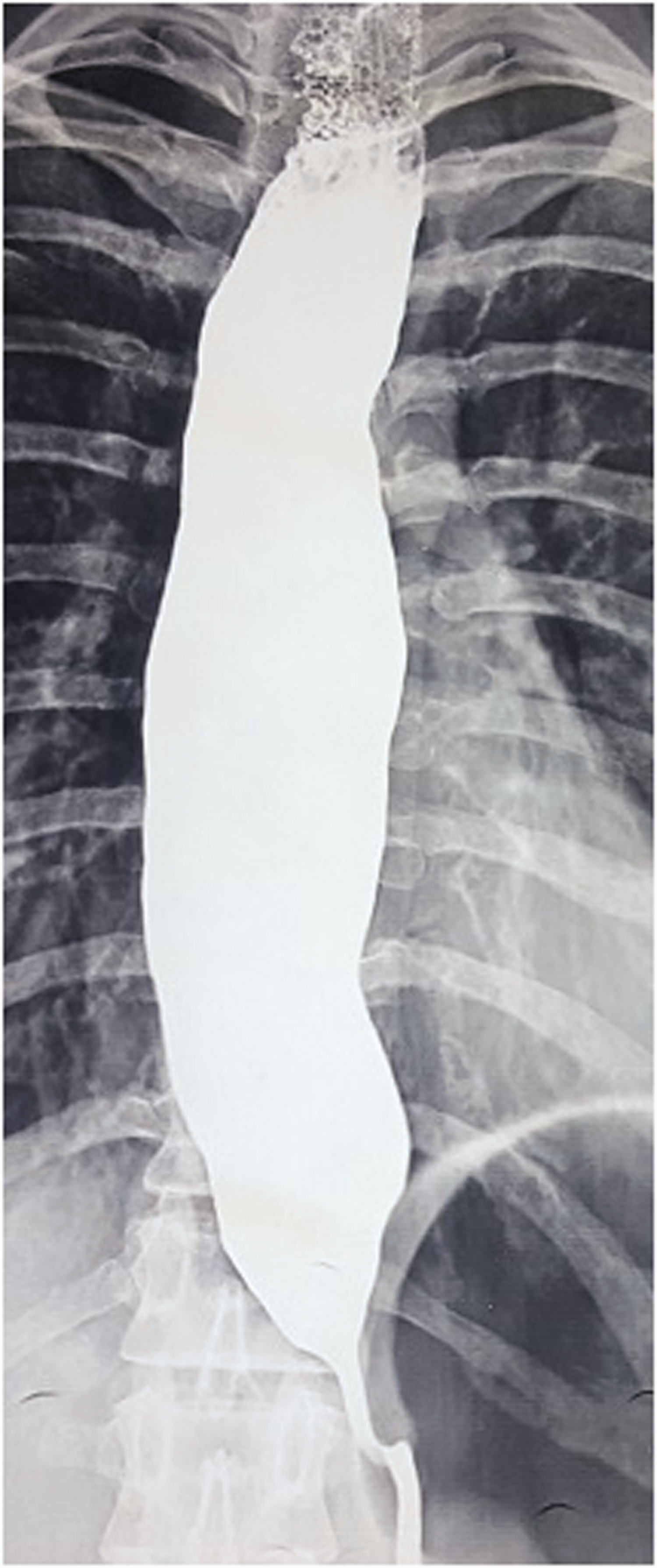
Thus, the diagnosis of triple A syndrome was made. Glucocorticoid and mineralocorticoid replacement therapy and artificial tears were introduced. A pneumatic balloon dilatation was performed. With this, her dysphagia improved significantly, the darkening of her skin reduced and her salt cravings ceased; she gained >10 kg in weight.
Discussion
Triple A syndrome is a hereditary autosomal recessive disorder characterised by alacrimia, adrenal failure and achalasia. It should not be confused with an abdominal aortic aneurysm. It occurs due to a mutation in the AAAS gene on chromosome 12q13 which codes for ALADIN protein. This mutated product protein fails to localise nuclear pore complexes which forms a selective channel between cytoplasm and nucleus and so it remains mostly in cytoplasm. 1 Its exact prevalence is unknown as case reports are a rarity. It usually manifests in the first decade of life.
Alacrimia is the most early and persistent finding. It occurs due to structural abnormalities in the lacrimal gland and autonomic dysregulation. 2 The diagnosis of alacrimia is confirmed by Shirmer’s test. Small lacrimal glands and reduced secretion cells on lacrimal gland biopsy have been found in three patients with triple A syndrome. 3 Adrenal failure due to ACTH resistance is the second component. It occurs owing to progressive adrenal destruction at a variable time after birth, 4 but usually in the first decade of life and may produce hypoglycaemic seizures and shock. Mineralocorticoid deficiency occurs in only 15% of cases. Achalasia is the third component and usually occurs in the first to second decades of life. It usually occurs along with adrenal failure or 1–4 years later. It occurs due to lack of neuronal nitric oxide synthase in autonomic plexuses of the oesophagus. 5 Progressive neurological impairment also constitutes another component of this syndrome and can occur in up to 60% of patients with age, manifesting with autonomic dysregulation and orthostatic hypotension, pupillary abnormalities, sexual impotence, palpitation and abnormal intradermal histamine reactions.
There is no cure for triple A syndrome. Treatment focuses on managing individual conditions. Prognosis depends on identification and treatment of adrenal failure. Lifelong glucocorticoid replacement therapy is needed for adrenal insufficiency. Mineralocorticoid replacement is also needed whenever this deficiency is also present. Alacrimia is managed by applying topical lubricants. Achalasia can be treated with pneumatic balloon dilatation, Heller’s or endoscopic myotomy.
By actively searching for the other elements of triple A syndrome, their effects may be minimised early.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.