
Abstract
New interventions that may have some advantage over standard treatments by being less invasive, less toxic, or less costly are often compared to standard therapy. Noninferiority trials aim to demonstrate that the new intervention is almost as good as, or even better than, the standard. A commonly used paradigm makes numerous assumptions to obtain a statistical and clinical margin of noninferiority, many of which are difficult to prove and may be based on subjective assessments. We discuss these assumptions and give examples where they are not met. Regardless of the methods that are used, the margin of noninferiority must reflect what patients and health care providers are willing to balance for the possible benefits of the new intervention for fewer adverse events or less invasiveness or cost.
Introduction
Traditionally, most clinical trials were superiority trials. That is, they were designed to demonstrate that a new treatment or intervention was better than some standard, either an existing treatment or placebo. However, recently, many trials have a different goal 1,2 since there is effective standard therapy, making it neither ethical nor of medical interest to show that a new intervention is better than a placebo. The new interventions may have little or no superiority to standard therapy with respect to clinical outcomes but still may be of interest because they are less toxic, more easily administered, cheaper, or have some other feature of value to patients. Here, the trial aims to demonstrate that the new intervention is not worse than the standard treatment by some predefined margin of noninferiority. In this article, we assume that all trials are conducted with a background of usual care.
A key consideration is that the standard therapy is known to be better than no treatment or placebo by some amount. If the new intervention is not worse than the standard, one would want to conclude, therefore, that if it had been compared against a placebo, it too would have beaten the placebo. As discussed below, however, unless clear guidelines are established in the design, implementation, and analysis of noninferiority trials, such a conclusion is far from certain.
To illustrate the concepts around noninferiority designs, consider the trials represented in Figure 1, showing relative risk estimates for the intervention effect with 95% confidence intervals. 3 The vertical line on the right specifies the amount of worse effect of the intervention compared relative to the control that could be tolerated, defined as δ, and the other line represents a relative risk of unity, or no intervention difference. Trial A is everyone’s hope that the new intervention will, in fact, be superior to the control (ie, the upper confidence interval excludes a relative risk of unity). Trial B indicates a favorable estimate of intervention effect, but the upper limit of the confidence interval does not exclude unity. However, it is less than the margin of indifference and thus meets the criterion of being noninferior. Trial C is also noninferior, but the estimate is slightly in favor of the control. Trial D is not conclusive for superiority or noninferiority, probably because the trial is too small. Trial E indicates inferiority for the new intervention.
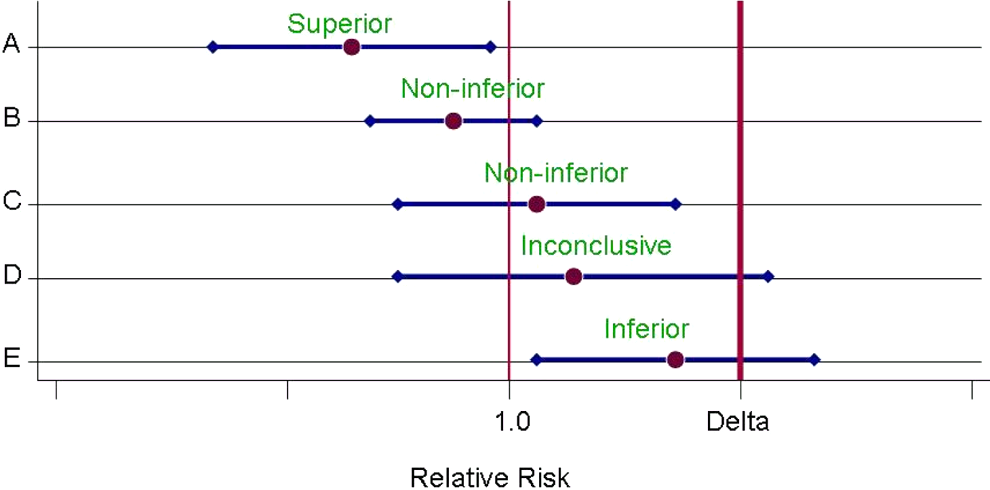
Trial potential results. Estimates of relative risks and 95% confidence intervals for 5 hypothetical trials.
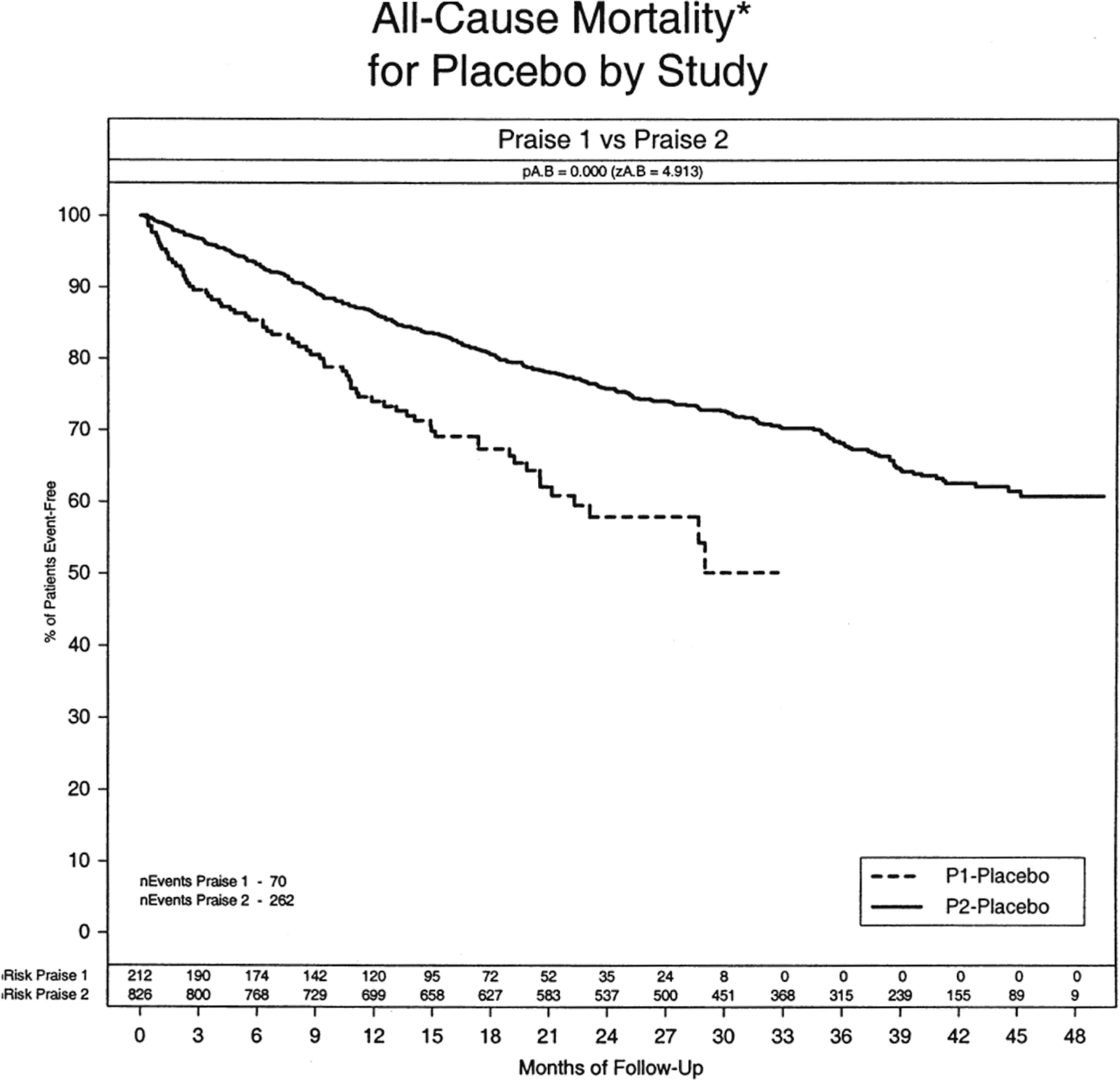
PRAISE-I and -II comparison of placebo arms. 17
The paradigm in Figure 1 is relatively straightforward conceptually, but special challenges exist in noninferiority trial design regarding the margin of indifference δ. These include choosing the active control against which to compare the new intervention, the size of the margin of indifference, the availability of relevant historical control data to document effectiveness of the standard therapy, the relevance of the historical studies to current practice, and features of the noninferiority trial such as size, power, quality and completeness of data, and adherence. If superiority trials are underpowered, have poor quality data, or poor adherence to the protocol, important intervention effects could be missed. For noninferiority trials, however, it is even more critical that the trial be of high quality since poor execution favors the new intervention, which may be falsely claimed as noninferior to the standard therapy. Much has been written about noninferiority designs, 4 –14 and the FDA has developed guidelines for designing and conducting such trials. 12 These guidelines suggest a 2-step approach: first, obtaining a statistical margin of noninferiority based on historical data, and second, determining a clinical margin of noninferiority based on the statistical margin plus what clinical trade-offs can be made. That is, how much worse can a new intervention be compared to a control or standard intervention before its advantages are no longer of interest? We think, however, that certain aspects of noninferiority trials are underappreciated and that guidelines such as those from the FDA require assumptions that are often difficult to establish. Ignoring those assumptions may lead to inappropriate conclusions.
A Common Paradigm for the Statistical Margin
Superiority trials are conducted to show that a treatment is better than the control, when both are given on top of a background of usual care. Such trials are designed to have power to detect a specified difference between the treatment (S) and the placebo or no treatment control (P). That is, the trial showed that S > P (or S + P > P).
In contrast, for noninferiority trials, we have to define, in advance, a margin of indifference (noninferiority), or δ, such that the new intervention, I, is no worse than the standard treatment, S, by δ. As discussed below, the margin of indifference can be defined in either absolute or relative terms.
In a noninferiority trial, not only should the new intervention be no worse than the standard therapy, S, by δ, but it should also be better than the control, P, used in the original trials that demonstrated the effect of the standard therapy. Three relevant comparisons, 2 direct and 1 indirect, provide an estimate of effect, such as relative risk (RR).
Direct: new intervention (I) versus standard therapy (S): estimate: RR (I/S)
Direct: standard therapy (S) versus placebo (P): estimate: RR (S/P)
Indirect: new intervention (I) versus placebo (P): estimate: RR (I/P) = RR (I/S) × RR (S/P)
Comparison 2 requires access to relevant historical data where the standard therapy was compared to a placebo or usual care. These data may not always be available or, if available, not entirely relevant in the setting of the planned noninferiority trial. Using the direct estimates of effect for I versus S and S versus P, we can then get the imputed or indirect estimate of I versus P (comparison 3).
Selecting the margin of indifference or noninferiority, δ, is a major challenge. Some have suggested that any new intervention approved as being noninferior to a standard therapy should retain at least 50% of the effect of that standard therapy in the original superiority trials. 5,8 This, of course, is an arbitrary goal and may not always be clinically reasonable.
Consider a noninferiority design using a relative risk (RR) scale. (The arguments are similar for other scales.) Assume a goal of retaining 50% of the effectiveness of the standard therapy. Historical estimate method: RR (S/P) RR (P/S) = 1/RR (S/P) If RR (P/S) – 2 SE > 1, the standard S is considered effective relative to placebo. Point estimate method for δ Let δ = 1 + .5(RR(P/S) – 1) Confidence interval method for δ Let δ = 1 + .5((RR(P/S) – 2 SE) – 1) If RR (I/S) + 2 SE < δ, then the new intervention, I, is considered noninferior to the standard S.
The estimate of δ may be based on a single key study of S versus P or a meta-analysis of a series of studies. This paradigm has appeal if the assumptions necessary for determining δ are valid or can be established. One critical assumption is constancy of effect. It is assumed that the effect of the standard therapy, S (the control in the new noninferiority trial), compared to placebo, P, for example, remains the same over time and that the participants being enrolled in the noninferiority trial are sufficiently similar in disease or condition and concomitant treatments to those in the original superiority trials. A second assumption is that the outcome measure or endpoint is sensitive to the effects of both the new intervention and the standard therapy. Third, there may be more than one currently accepted or commonly used standard therapy. The therapy selected as the standard control for the noninferiority trial must be the best one that is available, implemented in standard fashion, and not an inferior alternative. Fourth, access to key relevant historical data from the superiority trials is necessary. However, the estimate of intervention effect from historical studies is based on a nonrandom and nonrepresentative sample of participants or patients whose characteristics we can measure only to a limited extent.
The Paradigm Challenge
Although the usual noninferiority design paradigm may appear to be relatively straightforward, the assumptions may not be valid or easy to validate for several reasons.
First, the constancy assumption that the standard therapy versus placebo effect has not changed over time is often not correct. The population being studied may have changed or the effect of the intervention lessened because of different or evolving background therapy. This can be seen, for example, in 2 trials of the same intervention and control, conducted back to back with essentially the same protocol and investigators. These trials, the Prospective Randomized Amlodipine Survival Evaluation I (PRAISE-I) 15 and PRAISE-II, 16 compared amlodipine against a placebo in patients with chronic heart failure. In PRAISE-I, randomization was stratified according to whether the etiology of the heart failure was ischemic or nonischemic. Most of the apparent favorable effect on mortality was seen in the nonischemic stratum, contrary to the initial expectation. To validate this observation, PRAISE-II was conducted using the same drug dose, the same protocol and design, and most of the same investigators and sites, but only in nonischemic heart failure patients. This time, no benefit of amlodipine was observed. 16 Comparison of the placebo arms in PRAISE-I and PRAISE-II is shown in Figure 2. 17 The 2 populations of nonischemic heart failure patients were at substantially different risk, despite having been enrolled close in time, with the same entry criteria and many of the same investigators. No covariate analyses could explain this difference in risk. For whatever reason, the enrolled populations differed in the 2 trials, challenging the constancy assumption.
As background therapy changes, the effect of the standard therapy may also change. In the PRAISE trials described above, the event rates for the placebo arms changed from the first to the second trial. 15 –17 However, if the effect size was constant, the relative treatment effect seen in the second PRAISE trial should have been the same as in the first trial, regardless of change in the placebo rates. Although the treatment effect in PRAISE-I was nearly 40% for the nonischemic stratum, there was essentially no effect in PRAISE-II in that same stratum. Clearly, constancy of effect size was not seen.
A series of studies were conducted in the 1990s evaluating the effects of beta blockers on chronic heart failure patients. 18 –21 Three of these trials ended early with remarkably consistent and highly significant 30% to 35% reductions in mortality. The fourth trial terminated largely due to the results of the first three, but with consistent trends. These beta blocker trials were conducted with relatively few effective concomitant therapies. Today, there are several other beneficial treatments for heart failure. While it is very likely that beta blockers still contribute to mortality reduction, the absolute benefit is almost certainly less than what was seen in the early trial. Data from one or more historical studies are necessary to estimate the statistical margin. Ideally, the raw or original data would be available to conduct a meta-analysis. However, some studies may not have been published or even presented at scientific meetings. Often, trials of the same agent are not of the same intervention duration or follow-up, have different major entry criteria, and likely have different definitions of outcomes. Some may have used composite outcomes, making it even more difficult to combine the specific outcome of interest. Which studies are selected can easily influence the size of the intervention effect and perhaps even its direction (Figure 3). This figure summarizes a series of studies on the apparent effects of fibrinolytic intervention on mortality. 22 As shown, the size and direction of the effect vary, while the overall effect indicates a significant benefit. However, if a subset of trials were selected based on size, date, or some other criterion, results could change substantially. These limitations make the meta-analysis difficult and of limited value for precise estimates of a margin of indifference.
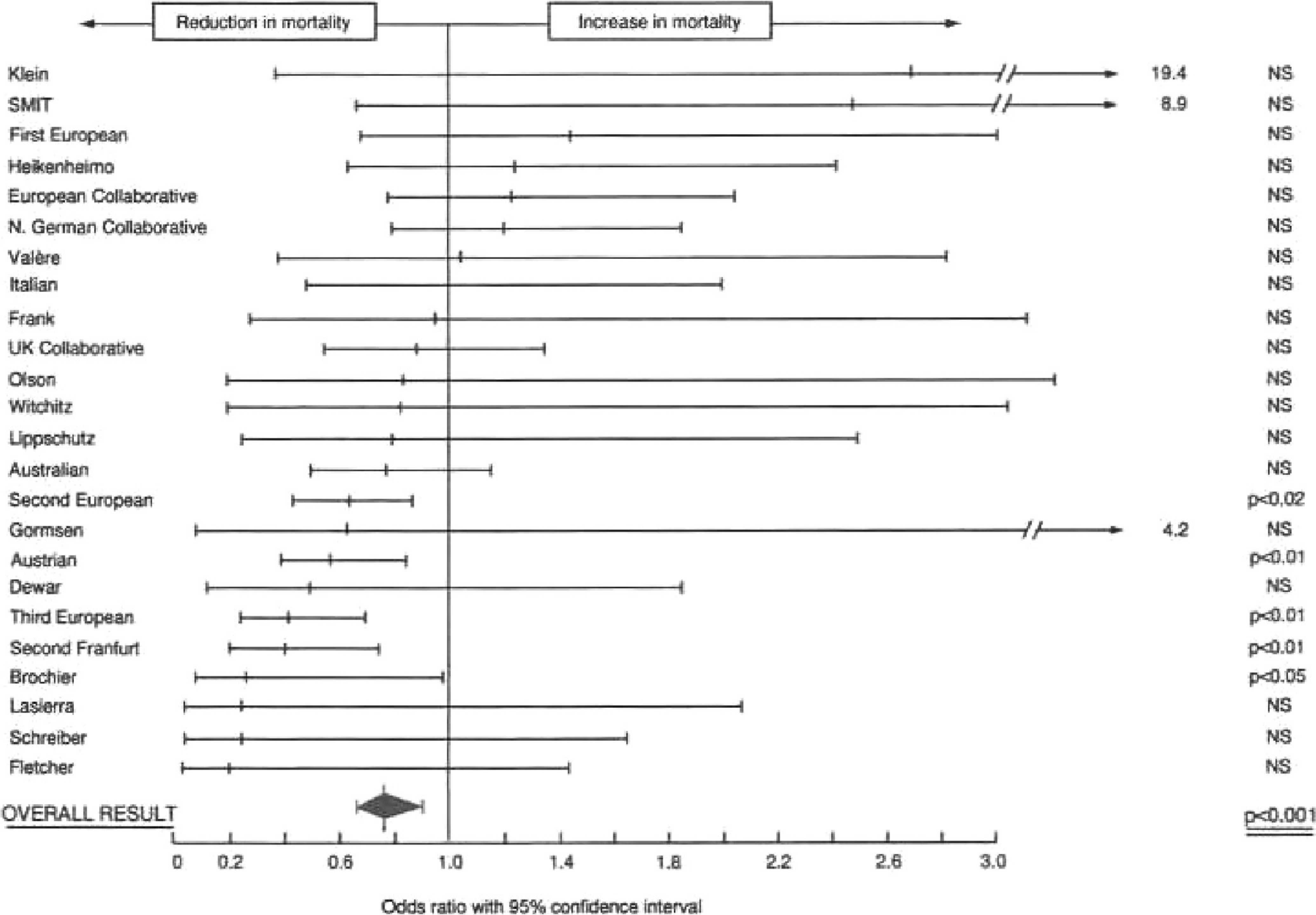
Meta-analysis of 24 thrombolytic trials, representing the odds ratio for intervention effect and 95% confidence interval plus an overall estimate. 22
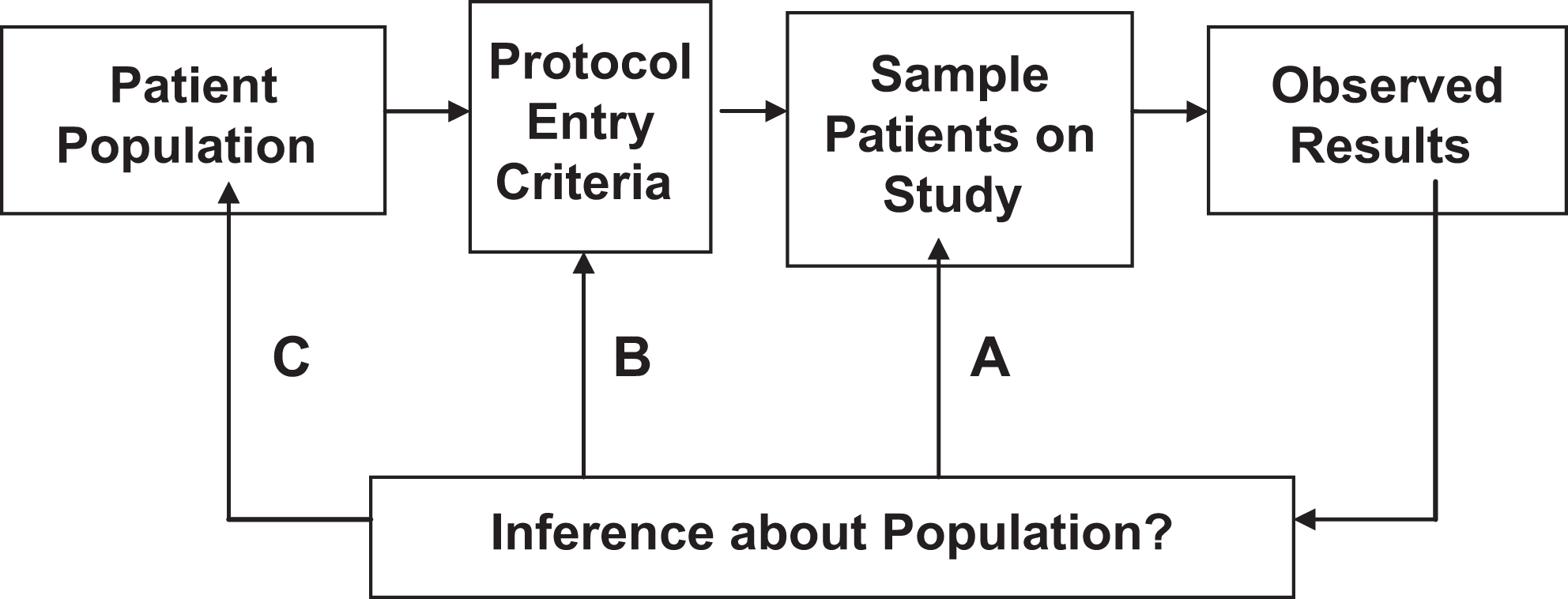
Populations for statistical inference. 17
Even if the historical data are available, the estimates of effect of the treatments may not be entirely representative. As in all trials, any treatment effect is based on the sample of participants who volunteered. This effect estimate is not likely to reflect exactly the effect in some other clinical trial. In the PRAISE example, 15 –17 it was not possible with standard baseline covariates to explain the difference in risk between 2 populations studied closely in time. The participants were volunteers who were influenced by the opinions and expertise of their health care providers in their decision to join the study.
The estimate of the noninferiority margin is often based on historical data, with its limitations, and the percentage of the effect (eg, 50%) assumed to be retained by the standard treatment (the control in the new noninferiority trial) is somewhat arbitrary. Using the lower confidence interval as the value of the control effect to be retained is even more conservative than using the point estimate. Yet, some of the margins set for noninferiority seem very generous, such as a relative risk, δ, of 1.5.
Another assumption is that of assay sensitivity. That is, the primary outcome variable must be sensitive to the interventions being tested in the population of interest. However, it may not be feasible to use the same outcome variable that was used in the initial standard versus placebo trial(s). For example, due to prevention and treatment efforts, the rates of events such as cause-specific death, nonfatal myocardial infarction, and nonfatal stroke may have decreased to such an extent that extremely large, perhaps infeasible sample sizes are needed if they are used as primary outcomes. Thus, composite outcomes are created that may add events or procedures, such as hospitalization, cause-specific hospitalization, or coronary revascularization, in order to increase the control group event rate. Simple quality of life metrics (improved vs not improved) may be included. In principle, this should reduce the target sample size. However, if the control and the new intervention do not have a similar impact on the added outcome components, the newly derived composite outcome may not have assay sensitivity, biasing the comparisons towards the null in favor of a noninferior result. For example, a superiority trial, Carvedilol Post Infarction Survival Control in Left Ventricular Dysfunction (CAPRICORN), compared carvedilol against a placebo in patients with left ventricular heart failure. The composite outcome of all-cause mortality plus hospitalization for cardiovascular reasons was not significantly different, but the comparison of mortality by itself (the original primary outcome) was. 23 The composite outcome of death plus cardiovascular hospitalization was not sensitive to the drug.
When designing a superiority trial, we rarely assume that treatment will be better than the control by 50%, particularly if the outcome of interest is mortality or major morbidity. More commonly, 20% to 30% is used for the sample size calculations. However, if we accept differences of 20% as being clinically relevant for superiority trials, it does not seem reasonable to allow a new intervention to be as much as 50% worse and yet be called noninferior. Obviously, the selection of the final noninferiority margin must take into account the clinical importance of the outcome being assessed as well as the known toxicity of the standard therapy.
Regardless of the margin of indifference, whether it is 50% of the historical effect or some other amount, how is it calculated? One could choose either the point estimate from the standard therapy versus placebo comparison or the lower confidence interval estimate of that comparison. The metric or scale must also be selected. Is it a relative risk, an odds ratio, a hazard ratio, or an absolute difference? The Stroke Prevention using an Oral Thrombin Inhibitor in Atrial Fibrillation (SPORTIF) trials 24 –26 chose an absolute difference in event rates for establishing the margin of noninferiority, but the observed event rate for the standard treatment arm was approximately half of what was expected based on historical data. This rendered the predefined margin based on absolute risk irrelevant. 26
Connected with this is the choice of the standard treatment control. There may be several possible standards or control interventions. Additionally, there may be more than one acceptable dose or way of administering the treatment. Current treatment A may have been shown beneficial when compared with a placebo. However, current treatment B may not have been shown beneficial when compared with a placebo but may have been shown noninferior to treatment A. Similarly, current treatment C may have been shown noninferior to A or even B. Which of these treatments should be used as the control for the new intervention in the planned trial? Choosing B or C as the control for the new trial could result in selecting a comparator for which noninferiority of a new intervention is readily demonstrated and for which superiority to a placebo cannot be guaranteed. This problem is sometimes referred to as “control creep.” 27
Another issue is whether the historical data are relevant at all, even if the assumption of constancy is reasonably valid. As depicted in Figure 4, the population at large is not the population that is defined by the protocol eligibility criteria and eventually studied. 27 Moreover, the recruitment process does not completely reflect the inclusion or exclusion criteria since individuals entered must volunteer and consent and the participant’s personal caregiver must also be supportive. Thus, the population actually studied is a very nonrandom sample of the target population and cannot be easily described, as many of the risk factors remain unknown or unmeasured. Our statistical methods are designed to make inferences on participants like the ones we studied (ie, inference A in Figure 4). To make inferences about the protocol-defined population (ie, inference B) or the target population (ie, inference C) is a leap. While it may be reasonable to conclude that the intervention is beneficial, it may not be so reasonable to infer the precise size of the benefit. Yet, the common noninferiority paradigm places considerable emphasis on the size of the observed historical effects.
In addition to the challenges of these key assumptions, historical trials need to be selected and combined using statistical methods for meta-analyses. Many methods exist, and there is no universal agreement as to which one to use. 28 The metrics that can summarize the outcomes of trials include relative risks, odds ratios, hazard ratios, and rate differences.
Meta-analyses may use a fixed effects or a random effects model, depending on whether the effects across studies are homogenous or not. 28,29 Even within one of these classes of analytic approaches, there are varying statistical methods and models. These may differently weigh studies depending on sample size, with the random effects model putting less weight on large trials. The important point here is that even the method for estimating the noninferiority statistical margin δ using meta-analysis is not simple or straightforward.
Regardless of the methodology employed, the margin, δ, must be defined in advance. The Ongoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial (ONTARGET) investigators 30 did so, but this margin was not agreed to in advance by the FDA. The investigators determined that the margin should be a relative risk of 1.13, using a fixed effects model. In its evaluation of the trial, the FDA used a random effects model and calculated a margin of 1.08. 31 The observed relative risk was 1.0 with an upper 95% confidence level of 1.09. Thus, the methodology selected was important in the evaluation process.
Importance of the Clinical Margin
The paradigm described above for the statistical margin is based on assumptions that are either not likely valid or difficult, if not impossible, to validate for many common diseases. Failure to consider all of the assumptions could lead to the erroneous conclusion that a precise statistical estimate of the margin of noninferiority has been obtained when in fact it is only approximate. Therefore, rather than focus heavily on an assumption-filled model to obtain the statistical margin, we propose that more emphasis be given to the clinical margin in cases where the assumptions cannot be validated or seem quite likely not to be true. 27
The trial should use the best current therapy as the control. Choice and implementation of control might be set by regulatory agencies or others independent of the trial sponsor and investigators, perhaps considering practice-based guidelines established by professional medical societies. This approach should avoid, or at least minimize, the temptation to use controls that make it easier for the new intervention to show noninferiority.
Historical data (ie, data from the trials that originally showed benefit for the standard treatment) should be combined via meta-analysis to get an initial estimate of the effect of the treatment but should not be viewed as a precise statistical value. Rather, using the historical data to get an approximate statistical margin should aid in setting the clinical margin of noninferiority.
Trial investigators and regulators, possibly with input from others who are independent of the sponsor, should determine what margin of clinical noninferiority would support using a new intervention when balanced against presumed benefits. This decision would depend on the nature of the medical condition being studied, the clinical importance of the outcome being assessed, the known risks of the standard intervention, and the trade-offs that might be achieved with a new alternative intervention. The investigators and others must also take into account the practicality of trial size.
A high-quality trial, with good adherence and as complete data collection as possible, must be properly conducted, analyzed, interpreted, and reported. This goal of conducting a very high-quality trial is more within the control of the investigators than defending the assumptions about historical data.
Even with the choice of the best control, the noninferiority trial must implement this control in an optimal way or at least consistent with best clinical practice. If any noninferiority trial is conducted such that the control arm is not optimally implemented or compliance is poor, the trial is of little value. This must be a primary focus in the conduct of noninferiority trials. We must be intolerant of sloppily done noninferiority trials.
Analysis cannot correct for poor compliance. The intention-to-treat (ITT) principle for analysis is the standard because to not include all patients as randomized and all events observed opens the door for bias, and the direction of that bias cannot be predicted. 27 If compliance is poor, the comparison of the 2 interventions is pushed towards the null in favor of noninferiority. “On treatment” or “per protocol” analyses are often proposed to address noncompliance, but such approaches violate the ITT principle and leave open the door for serious bias.
The proposed approach is challenging, but it does not make unverifiable assumptions and focuses more on what clinical margin would be reasonable in a trade-off between a control or standard and a new competitor intervention.
Conclusions
Noninferiority trials are becoming more common. If conducted without adequate attention to the validity of the various assumptions that go into the design, serious false conclusions may be drawn. We have suggested that more consideration be given to recognizing the critical assumptions used in designing noninferiority trials, that statistical estimates of the historical effect be used to guide establishing the clinical noninferiority margin, and that considerations for the clinical noninferiority margin need much more discussion. While this approach perhaps requires more medical judgment, it may be more defensible and implementable than relying heavily on the statistical margin in the more commonly described noninferiority design.
Footnotes
Acknowledgments
The preparation of this article was facilitated by Sue Parman from the Department of Biostatistics and Medical Informatics, University of Wisconsin–Madison.
Declaration of Conflicting Interests
The author(s) declared the following potential conflict of interest with respect to the research, authorship, and/or publication of this article: Dr DeMets is a consultant for the National Institutes of Health, the FDA, and the pharmaceutical and medical device industry on the design, monitoring, and analysis of clinical trials. He receives compensation for serving on industry-sponsored data and safety monitoring committees. Dr Friedman consults with the National Institutes of Health, as a former employee, and receives standard federal compensation for consultants.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.