
Abstract
The present research work was performed to study the properties of siloxane-based polyurethane (SPU) elastomers using aliphatic diisocyanate. SPU samples constituting of hexamethylene diisocyanate, hydroxyl-terminated polybutadiene, and polydimethylsiloxane (PDMS) were synthesized by two-step polymerization technique. Molecular engineering and surface characterization were carried out, and the outcome of the results was discussed. The conventional spectroscopic characterization of the synthesized samples using Fourier transform infrared spectroscopy confirms the existence of the proposed SPU structure. Surface properties of the synthesized material were studied determining the percentage of water absorption and equilibrium of the degree of swelling. It was found that by increasing the mole ratio of PDMS, the synthesized PU samples showed hydrophobic behavior while placing them in water and in dimethylsulfoxide solvent.
Keywords
Introduction
Today, polyurethanes (PUs) are one of the most versatile materials in the world. They represent a class of synthetic polymers that have found a widespread use in the medical, automotive, and industrial fields. PUs can be found in products such as furniture, coatings, adhesives, constructional materials, fibers, padding, paints, elastomers, and synthetic skins. 1 The repeating unit in PUs is the urethane linkage produced from the reaction of an isocyanate (–NCO) with an alcohol (–OH). The basic reaction of urethane formation is shown in Figure 1. Although PUs contain repeating urethane groups, other moieties such as urea, ester, ether, and aromatic may also be present in the structure. 2,3
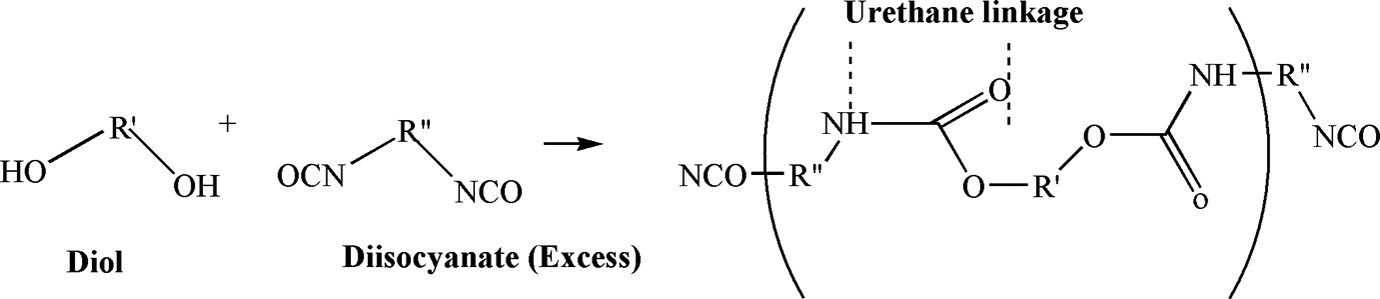
Basic reaction scheme for urethane formation.
Polyurethane elastomers (PUEs) are possibly the most versatile class of polymers since they can be molded, injected, extruded, and recycled. 4 Synthetic PUEs are widely utilized as engineering materials in various industries and are well known for their outstanding properties. 5 From a generic point of view, it is accepted that better properties are achieved as the microphase segregation is increased between the soft and hard blocks. The existence of the phase segregation caused by the clustering of hard and soft segments into separate domains has been a subject of continued research interest. 6 It is important to consider that efficient packing in hard domains is largely driven by the amenability to strong hydrogen bonding between the hard segments of adjacent chains. Varieties of polyols, diisocyanates, and chain extenders have been used in the synthesis of PUs and their effect on the properties has been investigated. 6,7 The thermoplastic PUEs are linear block (AB) n copolymers, the “A” segments, which built from alternating sequences of chain extender and NCO molecules, are called “hard” segments. The “B” segments, known as the soft segment, originate from the intermediate chain polyol. The constitution of A and B in this linear block copolymer and their sequence length play an important role in the physical properties of thermoplastic PUEs. Their versatile physical properties are usually attributed to both their microphase- and their macrophase-separated structures deriving from the thermodynamic incompatibility between the soft and hard segments. 8,9 The silicon-based polymer such as polydimethylsiloxane (PDMS) is the most frequently used polymer in many lithographic methods. It has several appealing features including optical transparency, chemical and biological inertness, nontoxicity, and gas permeability. Furthermore, PDMS has been used to bond other materials such as glass and polymers. Despite these advantages, the surface of PDMS cannot be used in many other applications and requires functionalization, because the surface of PDMS is naturally hydrophobic and a number of efforts have been made to refine the surface of PDMS. 10 PDMS have been used in many applications due to their unique properties that arise mainly from the nature of the siloxane bond (Si–O). These properties include extremely low glass transition temperature (−123°C), low surface energy, and high permeability to gases, good insulating properties, and very good thermal stability. Moreover, many physical properties remain relatively unchanged over a wide range of temperature. However, the mechanical properties of PDMS are usually very poor at room temperature, unless reinforced with silica and vulcanized. In order to develop useful properties, very high-molecular-weights are required. Even for chemically cross-linked PDMS, the tensile strength is still relatively low compared with other elastomers. 11 PDMS is one of the most widely utilized polymeric materials for the manufacture of biomedical devices including oxygenators, blood pumps, and contact lenses because of its superior biocompatibility, low toxicity, oxidative, and thermal stability as well as wide service temperature. 12 These advantages also allow its use as a soft segment in PU for biomedical devices, and these polymers have displayed a high phase-separated morphology caused by its incompatibility with polar hard segment. They also exhibit poor mechanical properties but better blood contact properties when compared with commercial PUs. 13 Several strategies have also been applied to modify the surface as well as the interfacial properties of siloxane-based PUs (SPUs). 14 Increase in the mole ratio of chain extender/cross-linker resulting in final PU is considered to be the most useful method as chemical modification. 15 Depending on the types, the content, and the methods, the increase in the mole ratio of chain extender/cross-linker can affect not only the bulk properties but also the surface properties. 16,17 The structure of the microphase separation depends not only on a function of the system thermodynamics but also greatly on the ability of the hard segments to pack itself correctly to form a hard domain. Attempts have been made to study the crystallinity and hydrophilicity by determining water absorption and swelling behavior of different polymeric materials. 18 Due to the availability of limited literature on the synthesis of SPU, the present report has been presented. The effect of increase in mole ratio of PDMS on the thermal behavior and hydrophilicity is investigated and discussed in this article. For this investigation, PUs were synthesized via a standard two-step reaction procedure, using hydroxyl-terminated polybutadiene (HTPB, molecular weight 2950 g/mol), 1,6-hexamethylene diisocyanate (HDI), and PDMS. The effect of chain extender/cross-linker contents on thermal behavior and surface morphology was studied and discussed.
Experimental
Chemicals
Aliphatic diisocyanate (i.e. HDI) was purchased from Sigma-Aldrich Chemical Co. (St Louis, Missouri, USA) and polyol (i.e. HTPB, molecular weight 2950 g/mol) from NESCOM (Islamabad, Pakistan). Chain extender/cross-linker (i.e. PDMS) was obtained from Merck Chemicals Co. (Darmstadt, Germany). The HTPB and chain extender used in this study were dried at 80°C under vacuum for 24 h before use to ensure the removal of all air bubbles and water vapors that may otherwise interfere with the NCO reactions. Molecular weight of HTPB was confirmed by applying the procedure reported in ASTM D-4274C standard. All the reagents used in this work were of analytical grade.
Synthesis of PU
For this investigation, prepolymer was synthesized according to a recommended procedure. 8,9 Following the procedures, HDI was reacted with diol, for example, HTPB with mole ratio 3:1 to obtain −NCO-terminated PU prepolymers. For this purpose, a four-necked reaction kettle equipped with mechanical stirrer, heating oil bath, reflux condenser, dropping funnel, and nitrogen (N2) inlet and outlet (HTPB) was placed. The temperature of the oil bath was increased to 60°C. Then, HDI was added and the temperature was then increased to 100°C. It almost took 1.0 h to obtain NCO-terminated PU prepolymer. Conversion of the PU prepolymer into the final PU was carried out by stirring the prepolymer vigorously and then adding a previously degassed chain extender/cross-linker, for example, PDMS with different mole ratio (Table 1). When homogeneity was obtained in the reactant mixture, the dispersion of chain extender/cross-linker was considered complete, and the liquid polymer was casted into a Teflon material to form a uniform sheet. The synthesized polymer samples were first placed under vacuum for 15 min to ensure the removal of air bubbles before casting and then cured for 24 h in hot air circulating oven at 100°C. Schematic illustration of chemical route for synthesis of PU is shown in Figure 2.
Sample code designation and different formation of SPU.a
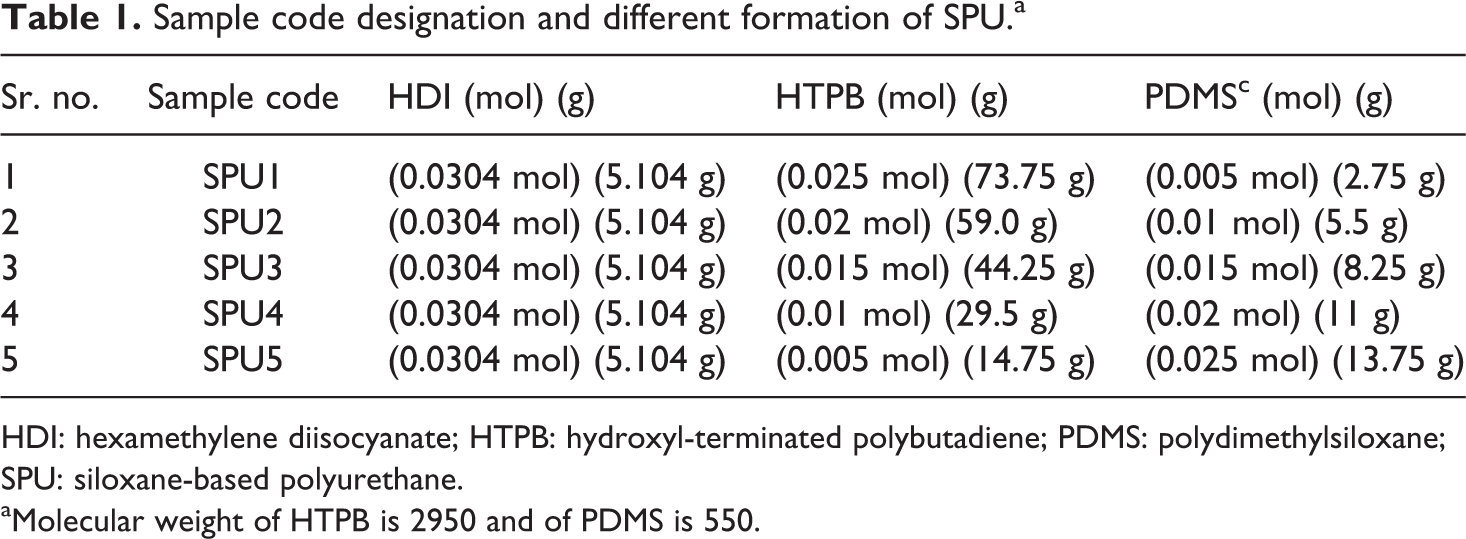
HDI: hexamethylene diisocyanate; HTPB: hydroxyl-terminated polybutadiene; PDMS: polydimethylsiloxane; SPU: siloxane-based polyurethane.
aMolecular weight of HTPB is 2950 and of PDMS is 550.
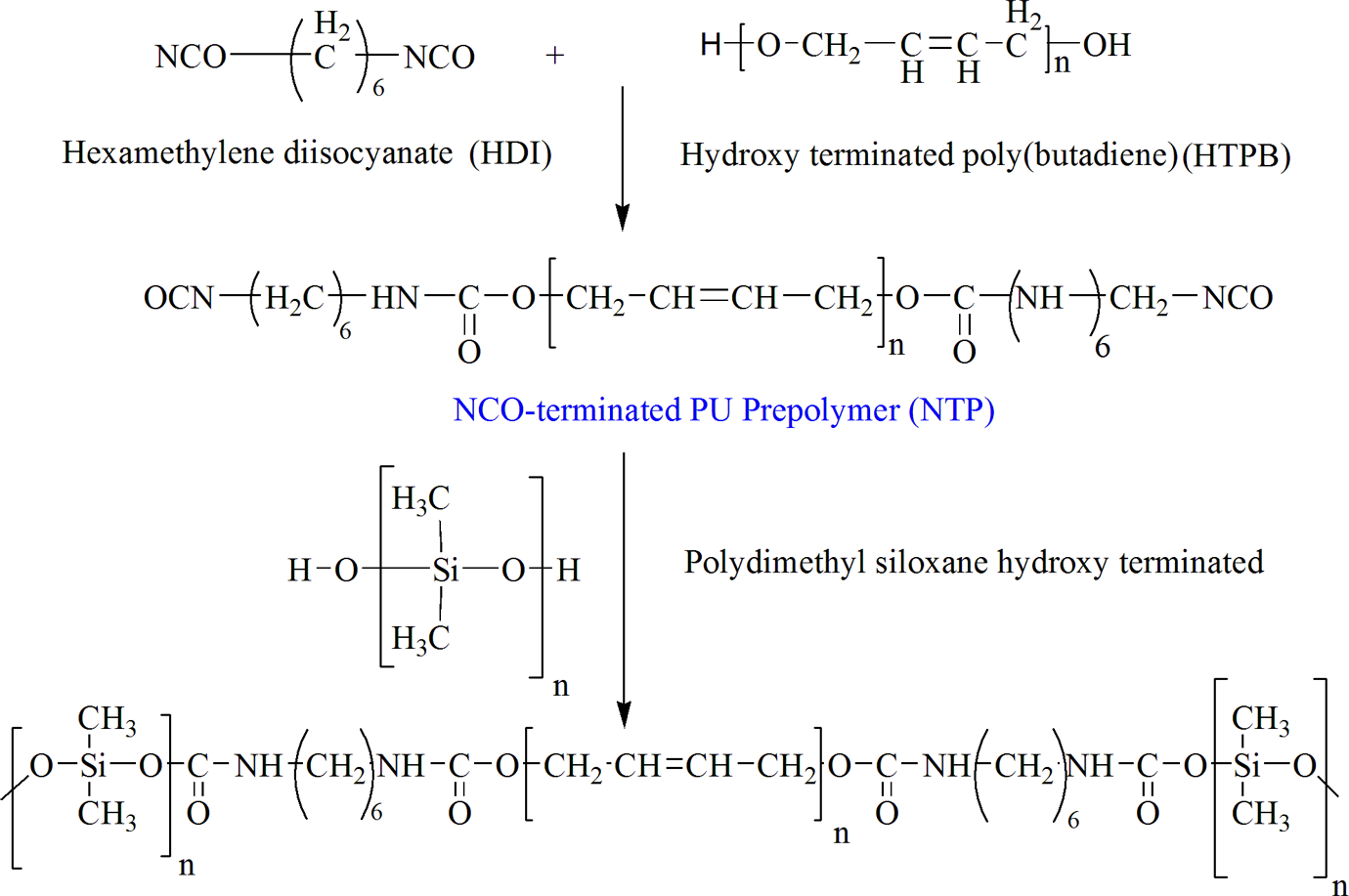
Synthesis of PDMS-based PU. PDMS: polydimethylsiloxane; PU: polyurethane.
After optimizing the experimental condition, five samples of siloxane-based PUE were finally prepared varying the mole ratio of PDMS. All the synthesized samples were then stored for 1 week at ambient temperature (25°C) and 40% of relative proposed humidity before testing.
Characterization
Fourier transform infrared (FTIR) spectrum of the PU polymer films were obtained in the transmission mode using an FTIR spectrometer (model NICOLET 6700; Thermoscientific, West Palm Beach, Florida, USA) at Quaid-i-Azam University (QAU), Islamabad, Pakistan. FTIR scans were collected on completely dried thin films cast on potassium bromide disks from N,N′-dimethylformamide solution. The spectra covered the infrared region 4000–500 cm−1, the number of scans per experiment was 16, and resolution was 4 cm−1. Thermogravimetric analysis (TGA) of the synthesized PU sample was recorded on a thermogravimetric analyzer (TGA/SDTA 851e; Mettler Toledo, Columbus, Ohio, USA) at QAU under N2 atmosphere from room temperature up to 650°C with a heating rate of 20°C min−1.
Surface morphology
Bulk hydrophilicity of the PUs was quantified by measuring the amount of water that each polymer absorbed at 37°C following the method reported elsewhere. 16 The equilibrium degree of swelling was calculated using the mathematical relations. 19,20 Density of the dry polymer samples (ρR) was measured according to the method reported in ASTM D-1817 standard. 21
Results and discussion
The main aim of this research work was to study the effect of mole ratio of chain extender/cross-linker PDMS using aliphatic diisocyanate in the backbone of the PU. So, the synthesis of siloxane-based PUEs was considered following the synthesis route as outlined in Figure 2. The reaction of one equivalent of polyol (HTPB) with three equivalents of HDI leads to NCO-terminated PU prepolymer that was subsequently extended with different mole ratio of chain extender/cross-linker to prepare final PU.
FTIR spectral study
FTIR spectra of HDI, HTPB, NCO-terminated PU prepolymer (obtained by the reaction of HDI with HTPB), chain extender/cross-linker (i.e. PDMS), and PU extended with PDMS is shown in Figure 3(a) to (e). The FTIR spectrum of HDI (Figure 3(a)) showed sharp peaks at 2935.6 cm−1, which is due to the -CH2 antisymmetric stretching and at 2860.0 cm−1, which is due to the -CH2 symmetric stretching. FTIR spectrum of HDI showed very strong peak at 2249.0 cm−1 attributed to the −NCO group attached to HDI. The observed peaks in the FTIR spectrum of HTPB (Figure 3(b)) were assigned as 3506.74 cm–1 (OH stretching vibration), 2963.75 cm−1 (antisymmetric -CH2 stretching), 1726.36 cm−1 (C=O stretching that may be due to oxidation of C=C), 1459.21 cm−1 (C-O-H bending), 1382.06 cm−1 (OH in out of plane deformation), and 1139.98 cm−1 (C-O and C-C stretching in the amorphous phase). FTIR spectrum of NCO-terminated PU prepolymer has also been presented in Figure 3(c). It is clearly observed that the signal for the OH groups disappeared and that of the intensity of NCO groups has been reduced to some extent resulting that NCO group has entirely reacted and a signal for NH units appeared at 3324 cm–1 suggesting that PU prepolymer had been formed (Figure 3(c)). The other observed peaks in the FTIR spectrum of PU prepolymer (Figure 3(c)) were assigned as 3072.2 cm−1 (C−H stretching of C=C−H), 2913.8 cm−1 (CH2 antisymmetric stretching), 2842.5 cm−1 (CH2 symmetric stretching), 2264.1 cm−1 (−NCO group), 1639.5 cm−1 (C=O stretching), 1513.7 cm−1 (–NH deformation), and 1435.0 cm−1 (CH2 bending). Peaks corresponding to –NH at 3324.0 cm−1 indicate the chemical reaction of the aliphatic diisocyanate with HTPB and observed sharp peak at 2264.5 cm−1, which is due to −NCO group, indicates the product contain the NCO group. These peaks provide strong evidence for the formation of NCO-terminated PU prepolymer. The chain extender/cross-linker (i.e. PDMS) was added in the final step to end the polymerization reaction. FTIR spectrum of PDMS (Figure 3(d)) showed that the antisymmetric and symmetric stretching vibrations of CH3 groups were observed at 2900.9 and 2962.0 cm−1, respectively. The other peaks observed at 1413.0 cm−1 and 1256.8 cm−1 are attributed to antisymmetric and symmetric deformation of Si-CH3 groups, respectively. Another sharp peak appears at 1008.5 cm−1 attributed to Si–O–Si stretching. To provide clear information about the vibrational mode changes due to involvement of PDMS into the PU backbone during the polymerization reaction, FTIR spectrum obtained from the cast film is shown in Figure 3(e). In the FTIR analysis obtained for the PU sample (Figure 3), the disappearance of the -NCO peak at 2249.0 cm−1 and the appearance of N-H peak at 3333.3 cm−1, confirmed the completion of polymerization reaction. The observed peaks in the spectrum imply that the reaction was completed and the predesigned PU was formed. The obtained FTIR spectra support the proposed structure of the final polymer. FTIR spectra showed characteristic bands of urethane groups at 3333.3 cm−1 (N–H stretching). The other peaks observed were assigned as 3072.1 cm−1 (C-H stretching of C=C-H), 2960.7 cm−1 (antisymmetric stretching vibrations of CH3), 22912.8 cm−1 (antisymmetric stretching vibrations of CH2 groups), and 2843.1 cm−1 (symmetric stretching vibrations of CH2 groups). By extending prepolymer with PDMS, the FTIR spectra showed a very strong, new peak at 1693.6 cm−1, which was assigned to C=O. Another new peak was also observed at about 1519.4 cm−1, which was assignable to urethane (–NH deformation). The other sharp peaks observed at 1435.6, 1259.2, and 1016.9 cm−1 correspond to CH2 bending vibration, symmetric deformation of Si-CH3, and Si–O–Si stretching, respectively. Peaks corresponding to the absorption of –NH, –C=O, and –CHN were observed at 3333.3, 1693.6, and 1519.4 cm−1, respectively, which indicate the new synthesized product having –NHCOO group. It is worthwhile mentioning that the N–H group in PU could form hard–hard segment H-bonding with the carbonyl oxygen and hard–soft H-bonding with the ether oxygen. The stronger hard–hard segment H-bonding acts as physical cross-links leading to difficult segmental motion of the polymer chain that results in a more significant phase separation between hard and soft segments. The phase separation improves mechanical properties of PUs but reduces the flexibility and solubility. 22
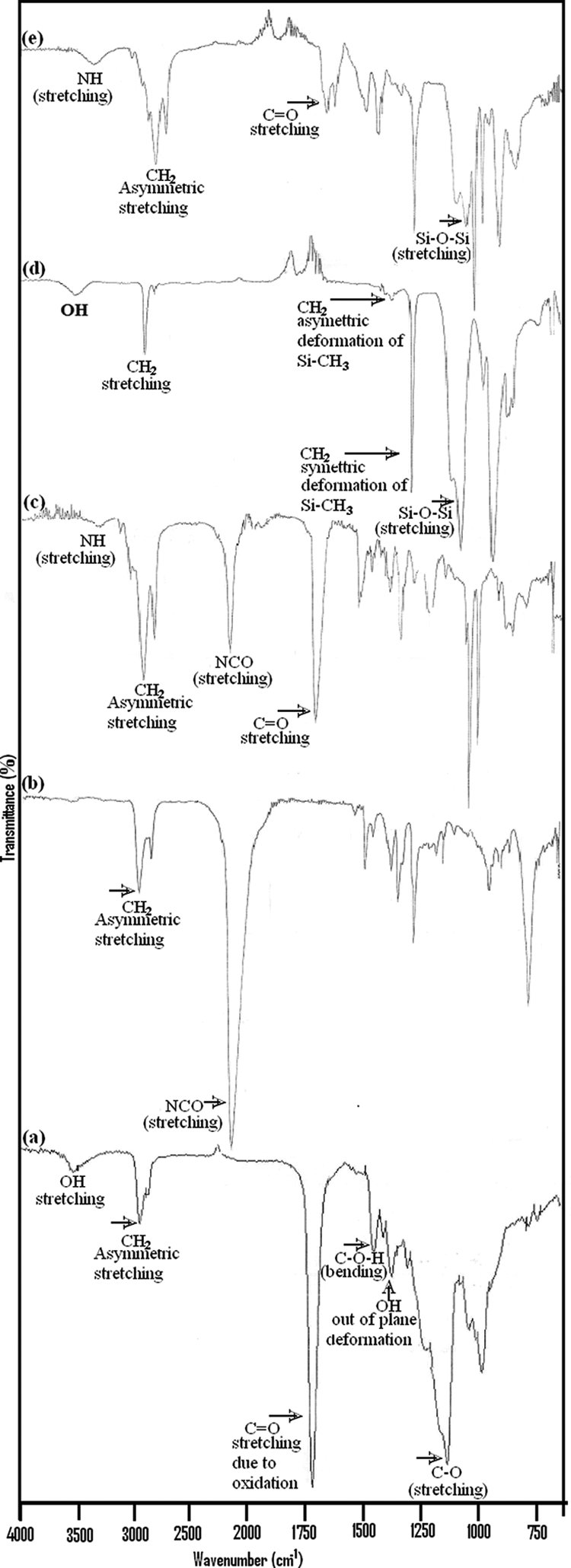
FTIR spectra of (a) HTPB, (b) HDI, (c) NCO-terminated PU prepolymer, (d) HTPB, and (e) siloxane-based final PU. FTIR: Fourier transform infrared; HTPB: hydroxyl-terminated polybutadiene; HDI: hexamethylene diisocyanate; NCO: isocyanate; PU: polyurethane.
Thermogravimetric analysis
TGA technique was used to evaluate the thermal stability of prepared PU samples. The thermal decomposition of all the SPU samples was evaluated at different percentages of weight loss (i.e. 10, 20, 30, 50, 80%, and at the maximum), and the results are presented in Table 2. For the prepared final PU, the decomposition temperature for 10% weight loss was in the range of 350–360°C. The maximum decomposition temperature was in the range from 595 to >600°C. It is quite clear that a sample extended with PDMS (0.5 mol; SPU1) is more thermally stable than all the other samples that were extended with 1.0, 1.5, 2.0, and 2.5 mol. It can be seen that by increasing the mole ratio of PDMS, thermal stability of PU films decreased. It means the contents of PDMS used as chain extender have a considerable effect on the thermal stability of the prepared PU samples. TGA studies of the PU films showed that the degradation of PU films starts at about 25°C (T onset) and ended at >600°C (T max). It can be concluded that increase in PDMS contents results to decrease in thermal stability of the final PU. This lower thermal stability is probably due to the significantly lower HDI mass fraction and therefore a lower inter-urethane H-bond concentration. It is considered that increase in PDMS contents of chain extender leads to weaken the urethane linkages that results lower in inter-urethane H-bond concentration.
Thermal stability data of the SPU samples (from TGA curves).

SPU: siloxane-based polyurethane; TGA: thermogravimetric analysis; T onset: initial decomposition temperature; T 10: temperature of 10% weight loss; T 20: temperature of 20% weight loss; T 30: temperature of 30% weight loss; T 50: temperature of 50% weight loss; T 80: temperature of 80% weight loss; T Max: maximum decomposition temperature.
Evaluation of water absorption (%) and swelling behavior
Physical properties of PU such as density, water absorption (%), and equilibrium degree of swelling were determined (Tables 3 and 4). Also, while these polymers can be used to perform and degrade in biological environments, HTPB can be used as a soft segment into the PU backbone. So, bulk water absorption ability plays an important role on degradation rate of them. Water absorption as a function of time and type of SPU samples is presented in Table 3. There was no considerable difference in the amount of absorbed water as a function of time. The chain extender/cross-linker mole ratio was the main factor that can control the amount of absorbed water. The results presented clearly showed that water absorption of samples decreased with increasing mole ratio of PDMS (used as chain extender/cross-linker) into the final PU samples. In addition to this, swelling behavior was also in accordance with water absorption presented in Table 4. The swelling ability of final PU steadily decreased as the contents of PDMS used as chain extender/cross-linker increases. It was found that SPU5 has better solvent resistance when compared with SPU1, and this resistance continuously decreases as the contents of PDMS into the backbone of PU decrease. This effect can be elucidated by the degree of physical cross-linking and hydrogen bonding in PU. It is worthwhile mentioning that crystallinity decreased by changing the aliphatic to aromatic character of the diisocyanate in synthesized PU samples. 16 The increases in crystalline pattern ultimately result to increase in hydrophobic character of the synthesized PU samples. Therefore, using the aliphatic diisocyanate, the synthesized material has shown water- and solvent-repellent behavior. This clearly indicates that the PDMS contents have no effect on the surface characteristics; however, the nature of the diisocyanate (aliphatic or aromatic) is more important. In a similar study using the aromatic diisocyanate, the adverse effect has been observed, which also supports the above-discussed results. 23
Water absorption (%) of PU samples.
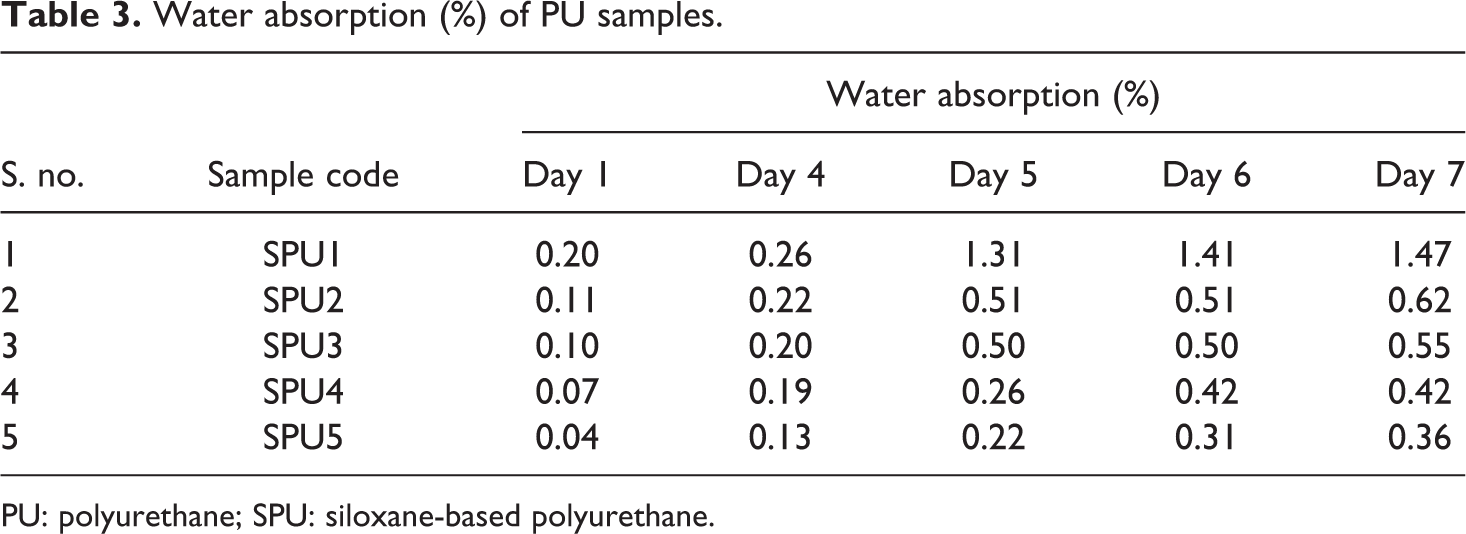
PU: polyurethane; SPU: siloxane-based polyurethane.
Swelling data of PU samples.
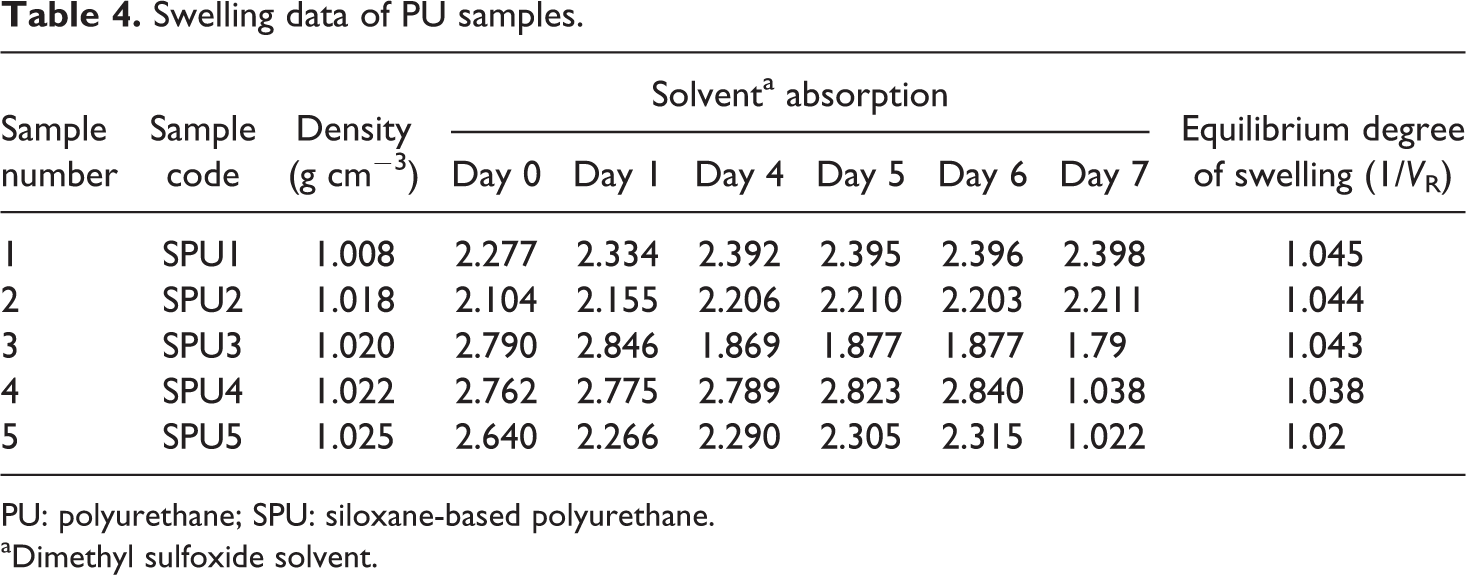
PU: polyurethane; SPU: siloxane-based polyurethane.
aDimethyl sulfoxide solvent.
Conclusion
Following prepolymer method, PDMS-extended PUs was prepared using HDI and HTPB in the prepolymer step. Structural, thermal, and surface characterization were carried out and discussed. The conventional spectroscopic characterization of the synthesized samples using FTIR confirms the existence of the proposed SPU structure. Surface properties of the synthesized material were studied determining the water absorption (%) and equilibrium degree of swelling. The results revealed that the synthesized PDMS-based PU samples showed hydrophobic behavior while placing them in water and or in dimethyl sulfoxide solvent.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.