
Abstract
Accelerating the development and approval of novel therapeutics has emerged as a key public health priority given the mortality, morbidity, and economic costs associated with infections caused by drug-resistant bacteria. However, there is limited empirical evidence to guide policymaking, such as the factors that may disadvantage antibiotics compared to other classes of drugs. In this Article, we empirically examine characteristics of the key clinical trials underpinning FDA's approval of antibiotics and other drugs over the past decade. Despite perceptions that antibiotic trials are larger and more difficult to conduct, we find that antibiotic trials are no larger than those conducted for drugs approved in other disease areas with high unmet medical needs, suggesting that policymakers may need to target other levers to meaningfully stimulate innovation. We discuss the risks and benefits of harnessing new and existing regulatory pathways to speed the approval of new drugs, particularly those intended to treat patients with serious and life-threatening infections, and we evaluate ways that proposals for new regulatory pathways could be improved to better prioritize and expedite the approval of therapies with the greatest potential for patient health benefits.
I. INTRODUCTION
Antimicrobial resistance is a growing public health concern. 1 The Centers for Disease Control and Prevention (“CDC”) estimates that infections due to antibiotic-resistant organisms cause at least 23,000 deaths in the United States annually. 2 Antibiotic-resistant infections are more difficult and expensive to treat than antibiotic-susceptible infections, and the spread of antibiotic-resistant pathogens may disproportionately affect the global poor. 3 In 2014, the World Health Organization (“WHO”) warned that every region had national reports of resistance to third-generation cephalosporins among E. coli and K. pneumoniae, meaning that clinicians would need to rely on costly, broad-spectrum carbapenems, which are less likely to be available in resource-poor settings, for treatment of severe infections caused by these pathogens. 4 The United Kingdom government's Review on Antimicrobial Resistance estimates that a continued rise in resistance by 2050 would lead to ten million deaths globally per year and a reduction of 2.0-3.5% in global gross domestic product, imposing a cost of $100 trillion. 5
To address the threat of antibiotic resistance, society will need both new antibiotics and better conservation of existing (and future) antimicrobial resources. 6 Recently, legislators have focused on developing incentives to promote the development of novel antibiotics. For example, in 2012, Congress passed the Generating Antibiotic Incentives Now Act (“GAIN”), which established five years of additional market exclusivity for qualified infectious disease products, in addition to the five to seven years already guaranteed by law to all new drugs. 7 In addition, in 2012, the President's Council of Advisors on Science and Technology (“PCAST”) proposed the creation of an accelerated regulatory approval process for antibiotics intended to be used in a “specific subpopulation at high risk from the disease” that would curtail the size and scope of required late-stage clinical trials. 8 PCAST also recommended that the Food and Drug Administration (FDA) use existing mechanisms to facilitate the approval of new drugs intended for patients infected with antibiotic-resistant bacteria. 9 In July 2015, the United States House of Representatives passed the 21st Century Cures Act, which included provisions that would codify this new regulatory pathway for approval of antibiotics to be used in limited patient populations. 10
However, there is limited empirical evidence on antibiotic approvals (such as the size of antibiotic trials and their design) to inform ongoing policymaking. In addition, policymakers will need to ensure that new regulatory incentives are truly novel, and therefore additive to, existing approval mechanisms.
In this Article, we explore the risks and benefits of harnessing new and existing regulatory pathways to speed the approval of new drugs, particularly those intended to treat patients with serious and life-threatening infections, including infections caused by multidrug-resistant bacteria. We focus on the development of systemic anti-bacterial agents (and use the terms “antimicrobial” and “antibiotic” interchangeably here), but our results may also apply to other classes of antimicrobial agents, including antifungal and antiparasitic drugs. Part II describes the typical clinical development process for new drugs and reviews characteristics of the key trials underpinning FDA's approval of antibiotics and other drugs over the past decade. Part III discusses how existing expedited approval programs, as well as two additional pathways (505(b)(2) and the Animal Rule) that may help streamline the development of promising new antibiotics. Finally, Part IV concludes that a legislative proposal for a new regulatory pathway for antibiotics could be improved to better prioritize and expedite the approval of therapies with the greatest potential for patient health benefits.
II. FDA APPROVAL OF NEW ANTIBIOTICS
A. Background on Clinical Development of New Drugs
Clinical trials are essential to evidence-based drug development because they allow patients, investigators, and regulators to assess the safety and efficacy of new drugs before they are used in general practice. 11 In turn, the data generated from well-designed preapproval trials help patients and clinicians weigh the risks and benefits of new treatments and, ultimately, determine whether to use a newly approved therapeutic. 12 Traditionally, investigational drugs are tested in three phases of clinical trials. 13 During Phase 1, the sponsor recruits healthy volunteers to assess the drug's pharmacokinetic and safety characteristics. 14 After demonstrating in Phase 1 some sense of how a drug is metabolized and what the right dose range might be, the sponsor carries the drug forward into Phase 2 trials, the first trials that involve a population of patients with the disease. 15 Phase 2 trials are intended to establish a proper dose for the drug, to provide information on the drug's safety profile, and potentially to provide the first signs of a drug's efficacy. 16 If the results of the Phase 2 study suggest that the drug may be safe to administer to patients and may provide clinical benefit for the intended patient population, Phase 3 trials are then organized to establish a drug's efficacy by demonstrating that an observed benefit is attributable to the drug, as well as to provide more evidence of the drug's safety profile. 17 Phase 3 trials may enroll hundreds or thousands of patients. 18
The trial design influences the level of confidence with which investigators can assess a drug's cause-effect hypothesis.
19
Design features, including randomization, blinding, and controls, help elucidate the risk-benefit balance of a drug by isolating the specific treatment effect due to the drug from non-specific effects such as the placebo effect and the natural progression of the disease.
20
Some of the earliest randomized controlled trials were developed to evaluate treatments for infectious diseases.
21
For example, in a seminal trial of streptomycin for the treatment of tuberculosis, Marshall and colleagues noted: The natural course of pulmonary tuberculosis is in fact so variable and unpredictable that evidence of improvement or cure following the use of a new drug in a few cases cannot be accepted as proof of the effect of that drug. The history of chemotherapeutic trials in tuberculosis is filled with errors due to empirical evaluation of drugs; the exaggerated claims made for gold treatment, persisting over 15 years, provide a spectacular example.
22
Thus, without adequate and rigorous data collection before these drugs are used, there is greater risk that patients may be exposed to therapies that are not effective or that are associated with serious safety issues.
The primacy of randomized controlled trials in the hierarchy of research designs is statutorily defined. By law, FDA must certify that proposed new drugs demonstrate “substantial evidence” from “adequate and well-controlled studies” evaluating the safety and efficacy of investigational agents. 23 While rigorous evidence from pivotal trials is crucial for patients, clinicians, and regulators needing to make decisions about the risks and benefits of potential treatments, conducting these pivotal trials (defined as the primary clinical studies that demonstrate safety and efficacy and that serve as the basis for regulatory approval) is also a resource-intensive process. 24 Surveys of pharmaceutical companies indicate that clinical trials comprise between 40-60% of total research and development expenditures, 25 and the results of these studies often have an immediate and material impact on financial returns. 26 The size of the pivotal trial is an important determinant of the overall cost to the developer, and previous studies found that the quality of clinical trial evidence varied widely within and between indications. 27 Additional features of clinical trial design can affect its size. For example, a non-inferiority design measures whether a treatment is no worse than another within a predetermined margin; in contrast, the better known superiority design tests whether a drug is better than a comparator. 28 Reducing the margin of non-inferiority typically results in a larger required patient population. 29
The need to enroll large numbers of patients in pivotal trials has been noted as a cause for the declining number of antibiotic approvals. 30 In response, recent proposals to accelerate antibiotic development have centered on reducing the size and number of required preapproval trials. 31 However, there is limited empirical evidence about the size and design of antibiotic pivotal trials, crucial data points that could illuminate ways to appropriately incentivize antibiotic development. In this Part, we review characteristics of the trials underpinning FDA's approval of antibiotics over the past decade (2002-2012), and we compare the evidentiary standards for new antibiotics with those for two other categories of disease (cancer and HIV/AIDS), which are also characterized by high unmet medical need.
B. Study Sample and Data Extraction
To shed light on the state of the regulatory environment for antibiotics, we examined the pivotal trials for all new antibiotics (antibacterials, antifungals, and antiparasitics) approved by FDA between January 1, 2002 and December 31, 2012. The names and indications of all antibiotics were identified from public domain master lists of approvals published by FDA. Reformulations, previously approved agents, and generic drugs were excluded from the study cohort.
For all new drugs, FDA publishes key information relating to the novel therapeutic on its website. 32 Each online dossier comprises the approval order, product label, and drug approval package; the latter includes summaries of the agency's medical, chemistry, pharmacology, statistical, and microbiology reviews for that particular therapeutic. 33 Using methods described previously, 34 we extracted information on the design, including number of participants, number of pivotal trials, and non-inferiority versus superiority, of the pivotal studies from the summary reviews published by FDA, which contain condensed reports of the clinical evidence submitted by the manufacturer in support of the new drug, as well as analysis by FDA reviewers. For drugs approved for more than one indication, we considered each indication separately.
We then compared the characteristics of these trials with those conducted for antiviral drugs and oncology products, two therapeutic areas characterized by unmet medical needs and high public health importance. We used the nonparametric Wilcoxon rank sum test to compare the numbers of pivotal trials and patient enrollment in antibiotic versus antiviral and antibiotic versus cancer pivotal trials. 35 We compared both the total number of patients, including those on the comparator arm(s), if any, with the number of patients exposed to active treatment (hereinafter “efficacy patients”) across disease areas. Statistical analyses were performed using the Stata software package, with a two-sided α=0.05. 36
C. Number of Pivotal Trials
From 2002 to 2012, FDA approved fifteen new antibiotics, fifteen antiviral drugs, and twenty-two cancer drugs. FDA approvals for the fifty-two drugs in our sample were based on ninety-one pivotal trials, enrolling a total of 47,939 patients and 28,692 efficacy patients.
Traditionally, FDA prefers sponsors to conduct two pivotal trials before approval to ensure the validity and reproducibility of any treatment effect observed in the study. 37 We found that there were 2.1 pivotal trials on average per approved antibiotic and 1.9 per antiviral (Wilcoxon rank sum test, P = 0.71), though only 1.2 per cancer drug approval (Wilcoxon rank sum test, P < 0.001).
D. Clinical Trial Size
We evaluated the total number of patients enrolled in pivotal trials, as well as the number of efficacy patients. We hypothesized that antibiotic trials would enroll significantly greater numbers of patients than those for antiviral and cancer drugs. For example, previous studies have suggested that cancer drugs are often approved on the basis of small-sized trials. 34 Similarly, drugs intended to treat HIV/AIDS have historically been granted accelerated approval, which allows FDA reviewers to approve drugs on the basis of surrogate endpoints, such as biomarkers, that are only reasonably likely to predict clinical benefit. 38
However, we found no evidence for a significant difference in trial size between approved antibiotics and drugs used in other disease areas. The pivotal trials for antibiotic drugs enrolled a median of 990 patients (interquartile range (“IQR”): 518-1105), including 509 efficacy patients (IQR: 260-605). Pivotal trials for antibiotic drugs enrolled significantly fewer total and efficacy patients than those for antiviral drugs (1,203 versus 990 total patients, Wilcoxon rank sum test, P = 0.03; 680 versus 509 efficacy patients, Wilcoxon rank sum test, P = 0.008). There was no difference in the size of antibiotic and cancer pivotal trials (990 vs. 731 total patients, Wilcoxon rank sum test, P = 0.12; 509 vs. 402 efficacy patients, Wilcoxon rank sum test, P = 0.31). In a sensitivity analysis, we repeated the comparisons with only anti-bacterial drugs, which enrolled an average of 1,057 total patients and 538 efficacy patients, and similarly found no relationship between trial size and disease area (Wilcoxon rank sum test, P > 0.07).
E. Trial Design
Finally, we explored what proportion of pivotal preapproval studies for antibiotics were non-inferiority studies. While highly dependent on the selected margin, non-inferiority studies often require a larger sample size than superiority trials and may explain observed differences in trial size between disease areas. We found that most antibiotic pivotal trials (78%), and all pivotal trials for systemic antibacterial drugs, used a non-inferiority design (Table 1). By contrast, only four antiviral drugs (27%) were approved on the basis of non-inferiority trials.
III. EXISTING PATHWAYS FOR EXPEDITED DEVELOPMENT AND APPROVAL
Our study showed that over the past decade, most new antibiotics were approved on the basis of pivotal non-inferiority trials that enrolled fewer participants compared to trials conducted for antivirals, and that were no different than trials for cancer drugs. Even though trials for antibiotics do not appear to be more difficult to conduct than those for drugs in other disease areas, over the past decades, few antibiotics with novel mechanisms of action have been developed, and few new agents have demonstrated the potential to meaningfully improve patient health outcomes in comparison to existing therapies. Given the proliferation of antibiotic-resistant bacteria and the growing burden of infections caused by these pathogens, fostering greater innovation in this field remains a key public health priority. Our findings suggest that policymakers should focus their attention on other factors that influence antibiotic development. In this Part, we explore alternative strategies to ensure that promising antibiotics intended for treatment of serious diseases with few alternative treatment options are approved efficiently, and we consider the risks and benefits associated with these various approval pathways.
A. Expedited Development and Approval
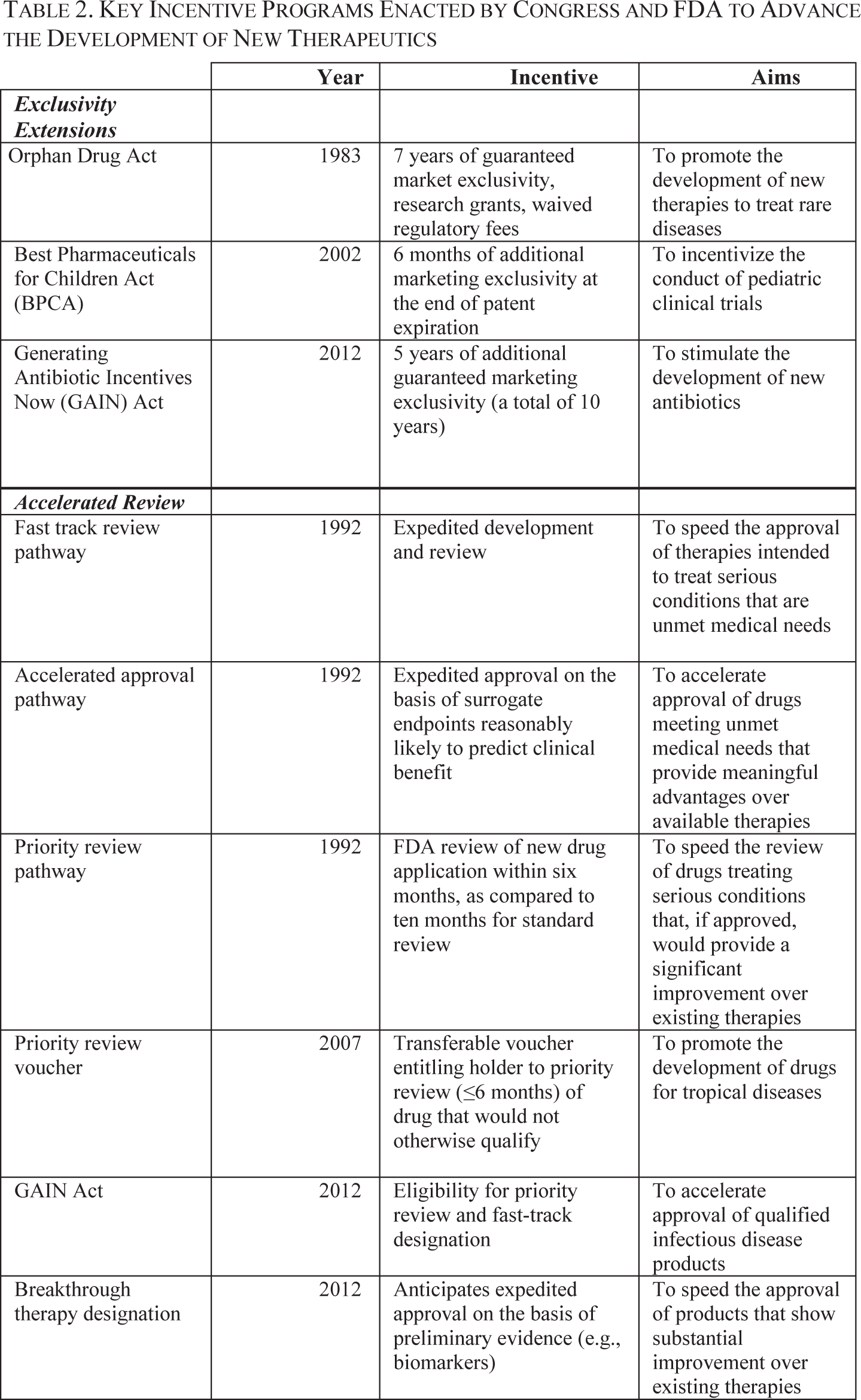
Existing federal incentives could be marshaled to advance the development of medicines for serious unmet medical needs. 39 FDA currently oversees four expedited development and approval pathways: the fast track (1988), accelerated approval (1992), priority review (1992), and breakthrough therapy designation programs (2012). 40 Key features of these programs are summarized in Table 2.
Many of these expedited approval programs were institutionalized following public controversy surrounding access to new medicines for HIV/AIDS and cancer. 41 As described above, the accelerated approval pathway grants conditional approval for drugs that are intended to treat serious conditions, and that provide a meaningful advantage over available therapies. 42 For drugs considered under this pathway, the FDA is directed to consider as “substantial evidence” those results from trials using a surrogate endpoint reasonably likely to predict a clinical benefit or an intermediate clinical endpoint that can be measured earlier than traditional endpoints like mortality and irreversible morbidity. 43 Although data can be submitted after, instead of as a precondition for, approval, confirmatory trials are required to verify and describe the effect on traditional endpoints of clinical benefit, and drugs approved through this pathway may be subject to withdrawal if those endpoints are not met. 44
More recently, in 2012, Congress approved the creation of a new program, the breakthrough therapy designation, to streamline the development of drugs intended to treat serious conditions for which preliminary clinical evidence indicates that the drug may demonstrate “substantial improvement” for a clinically significant endpoint over available therapies. 45 Several anti-infective therapies have gained this “breakthrough” designation. For example, a microbiome therapeutic and a microbiota restoration therapy received breakthrough therapy designation for the treatment of recurrent Clostridium difficile infection. 46 Similarly, FDA granted breakthrough therapy designation for a novel monoclonal antibody intended to treat Staphylococcus aureus infections. 47
These existing pathways for expedited development and approval offer developers the possibility of truncated timelines and lower development costs. For example, sponsors of drugs receiving fast track or breakthrough therapy designations would be more likely to gain FDA approval on the basis of a single Phase 2 trial, instead of needing to conduct a pivotal Phase 3 trial before approval. 48
B. 505(b)(2) Approval
The 505(b)(2) pathway was established by the Hatch-Waxman Amendments of 1984 to facilitate approvals comprising changes to previously approved drugs, such as the formulation, route of administration, dosage, or strength. 49 In contrast to the approval of new molecular entities (for which the application criteria are codified at 505(b)(1)), sponsors can use the 505(b)(2) pathway to rely on existing data, either from a prior FDA approval or from the published literature, to show that the reference drug is safe and effective, thereby reducing the amount of original data that would need to be submitted. 50 In a guidance document released in 1999, FDA clarified the intent of this regulatory pathway: “This use of section 505(b)(2), described in the regulations at 21 CFR 314.54 [sic], was intended to encourage innovation without creating duplicate work and reflects that … it is wasteful and unnecessary to carry out studies to demonstrate what is already known about a drug.” 51 The use of 505(b)(2) approvals has increased over time. According to data collected by Life Sciences Connect, a Thomson Reuters company, in 2002, FDA reviewed twenty applications through the 505(b)(2) pathway compared to fifty-eight through 505(b)(1), and in 2014, FDA reviewed fifty-six applications through 505(b)(2) and thirty-eight through 505(b)(1). 52
Recently, FDA has used the 505(b)(2) pathway to approve antibiotics intended for limited patient populations with unmet medical needs. In February 2015, FDA approved ceftazidime-avibactam (Avycaz; Forest Laboratories) through the 505(b)(2) pathway for the treatment of complicated intra-abdominal infections (“cIAI”), used in combination with metronidazole, and complicated urinary tract infections (“cUTI”), including pyelonephritis. 53 Ceftazidime-avibactam is an intravenous combination of the cephalosporin ceftazidime (approved by FDA in 1985) and a novel β-lactamase inhibitor (avibactam). 54 Avibactam alone has no meaningful antibacterial activity; rather, it helps protect ceftazidime from degradation by bacterial β-lactamase enzyme production, a commonly acquired bacterial resistance mechanism. 55 Clinical trials of the combination began in 2008, but ownership of the drug changed hands three times: from Novexel to AstraZeneca in 2010, AstraZeneca to Forest Laboratories in 2011, and Forest Laboratories to Actavis in 2014. 56
In 2013, citing the urgent unmet medical need for antibiotics in the treatment of multidrug-resistant Gram-negative bacterial infections, FDA met with the sponsor to discuss a revised development strategy that would allow approval of ceftazidime-avibactam prior to the completion of any Phase 3 clinical studies via the 505(b)(2) pathway. 57 Specifically, FDA advised that animal data, pharmacokinetic and pharmacodynamics analyses, and Phase 2 clinical data, coupled with the agency's prior findings and published literature on the efficacy and safety of ceftazidime alone, could support approval of ceftazidime-avibactam. 58 Summaries of FDA's internal decision-making suggest that the 505(b)(2) pathway was chosen instead of other expedited approval programs because there was no “surrogate endpoint that [was] reasonably likely to predict clinical benefit for [ceftazidime-avibactam].” 59
Even though FDA and the sponsor were agreeing on a streamlined development plan, the agency stressed that approval would be limited to patients with complicated infections caused by susceptible organisms and with limited or no alternative treatment options.
60
Preliminary results from the randomized, double-blind Phase 3 clinical trial (which was completed after approval and not reviewed as part of FDA's decision), assessing the non-inferiority of ceftazidime-avibactam plus metronidazole versus meropenem, raised questions about the drug's safety and efficacy profile.
61
Subgroup analyses indicated that in patients with moderate renal impairment, those treated with the combination had significantly lower cure rates than those in the control group (45% vs. 74%; P=0.02).
62
Moreover, in this subgroup, there were more deaths in the treatment group in comparison to the control group (eight versus three, respectively).
63
Commenting on these data, FDA's statistical reviewer concluded: [A]bsent reliance on the 505(b)(2) approval pathway, the evidence of efficacy of [ceftazidime-avibactam] is scant and uncertain …. It is tempting but probably prudent, from a rigorous scientific standpoint, to withhold the decision on the limited use in cIAI until all new data have been completely analyzed…. By no means conclusive as previously stated, the [ceftazidime-avibactam] subgroup is probably the only result that gives clue that somehow the [ceftazidime-avibactam] works alongside comparability of [ceftazidime-avibactam] treatment response, from published literature, and supportive data from in vitro microbiology, PK/PD models, and animal studies. With this and with all the reservations mentioned, I support approval of this product for limited use in the indications sought.
64
Reflecting the concerns raised by reviewers, the product label for ceftazidime-avibactam now indicates that only limited clinical safety and efficacy data are available and that its use should be reserved for patients with few treatment options and with infections caused by susceptible bacteria (i.e., those likely to respond to treatment). 65 The label also features modified dosage recommendations for patients with renal impairment, and in September 2015, FDA released a Drug Safety Communication that warned about the risk of dosing errors. 66 Still, the safety risks are not fully understood and may not be until patient-level data from the pivotal trial are fully analyzed.
The experience of ceftazidime-avibactam shows that FDA can use already existing regulatory pathways to streamline regulatory approval of particular antibiotic products intended to treat limited patient populations with high unmet medical needs. The 505(b)(2) pathway may be appealing for antibiotic developers because many new antibiotics have similar mechanisms of action to previously approved products. 67 505(b)(2) approval would, in many cases, also reduce preapproval data collection, given the ability to reference prior agency findings and published literature. However, the approval of ceftazidime-avibactam also illustrates the challenges inherent in expedited development and approval programs. Despite sophisticated pharmacokinetic and pharmacodynamics simulations, trial investigators were unable to predict the negative safety signal or mortality imbalance in patients with moderate renal impairment. 68 Without appropriate safeguards to ensure judicious use of this drug in patients with truly limited treatment options, overutilization may expose patients to undue harms while also fueling the development of antibiotic resistance.
C. Animal Rule
In 2002, FDA issued a final rule (known simply as the “Animal Rule”) allowing approval of drugs on the basis of animal efficacy studies if these agents were developed to ameliorate or prevent serious or life-threatening conditions caused by exposure to lethal or permanently disabling toxic substances. 69 The rule was intended to support the development of medical countermeasures against biological, chemical, radiological, or nuclear threats. 70 For drugs approved, or biologics licensed, through this pathway, FDA recognized that traditional human efficacy studies may not be ethical or feasible (e.g., they would require exposing patients to a lethal agent not otherwise present in the population). 71 For drugs or biologics considered through this pathway, the agency will consider as substantial evidence the results from “adequate and well-controlled” animal efficacy studies, as well as human safety trials. 72
To qualify for the Animal Rule, sponsors must demonstrate that four criteria are met: There is a reasonably well-understood pathophysiological mechanism of the toxicity of the substance and its prevention or substantial reduction by the product; The effect is demonstrated in more than one animal species expected to react with a response predictive for humans, unless the effect is demonstrated in a single animal species that represents a sufficiently well-characterized animal model for predicting the response in humans; The animal study endpoint is clearly related to the desired benefit in humans, generally the enhancement of survival or prevention of major morbidity; and The data or information on the kinetics and pharmacodynamics of the product or other relevant data or information, in animals and humans, allows selection of an effective dose in humans.
73
Approvals under the Animal Rule are subject to postmarketing study requirements (e.g., field studies if an emergency arises and the drug is used), restrictions to ensure safe use, and labeling that indicates efficacy studies were conducted in animals alone. 74
Since 2002, FDA has used its authority under the Animal Rule to approve a number of anti-infective products. 75 For example, raxibacumab is a monoclonal antibody intended to treat inhalational anthrax, a form of the infectious disease caused by breathing in the spores of the bacterium Bacillus anthracis, by neutralizing the toxins produced by the bacterium. 76 The sponsor conducted four studies in two animal species (one study in monkeys and three studies in rabbits), which demonstrated a survival benefit for animals treated with raxibacumab (64% of treated monkeys [P=0.003] and 44% of treated rabbits [P<0.001] survived, compared to 0% of animals in the placebo groups). 77 Additionally, in the rabbit study, 82% of animals treated with a combination of raxibacumab and antibiotics survived exposure to anthrax, compared with 65% of animals treated with antibiotics alone, although this numerical advantage was not statistically significant (P=0.09). 78 FDA reviewers noted that a study with sufficient statistical power to detect a difference would have needed to expose over 550 animals to anthrax, which was not feasible and may have raised animal use concerns. 79 The therapy was also reasonably well-tolerated in healthy human volunteers. On the basis of this animal efficacy and human safety data, in 2012, FDA approved raxibacumab for treatment of inhalational anthrax. 80 To prepare for a potential bioterrorism event, the Department of Health and Human Services has also committed to purchasing doses of raxibacumab for the Strategic National Stockpile. 81
FDA has also approved new indications under the Animal Rule for already marketed products. In 2012, levofloxacin received approval under the Animal Rule for the treatment of plague and to reduce the risk of plague after exposure to Yersinia pestis. 82 Levofloxacin's approval was based on an efficacy study conducted in twenty-four African green monkeys that were infected with the plague bacterium. 83 The monkeys were randomly allocated either to the treatment group receiving a ten day course of levofloxacin (N=17) or to the placebo control group (N=7). 84 Of the seventeen treated animals, sixteen survived (94%), compared with no surviving animals in the placebo group. 85 In its judgment that the drug was likely to be effective in the treatment of pneumonic plague in humans, FDA also referenced existing human data from levofloxacin's prior approval for other respiratory infections, including nosocomial and community-acquired pneumonia. 86 In May 2015, another antibiotic with a similar mechanism of action (moxifloxacin) received approval under the Animal Rule for the same indication. 87
Recently, the Animal Rule has been invoked in debates about the use of experimental therapies for emerging infectious disease threats, such as Ebola. 88 The 2015 epidemic of Ebola virus disease in West Africa resulted in over 11,000 deaths. 89 For one product, a recombinant vesicular stomatitis virus-based vaccine candidate, the sponsor was able to conduct clinical trials at or near peak infection rates during the recent epidemic. 90 However, because the epidemic has gradually been contained, and because substantially fewer new cases have been reported, any new clinical trials for therapies or vaccines may face difficulty recruiting sufficient numbers of infected patients to demonstrate safety and efficacy. 91 Approval or licensure through the Animal Rule could be based on a combination of animal data as well as surrogate endpoints of effectiveness in humans. For example, at a May 2015 advisory committee on licensure of Ebola vaccines, FDA cited preliminary data indicating that, “vaccinated humans may achieve immune responses comparable in magnitude to those associated with protection in [non-human primates],” suggesting the feasibility of an immunogenicity-based approval. 92
Thus, the Animal Rule may be useful for developers of new products for emerging bacterial threats that are either too uncommon or too unpredictable to permit human clinical testing of efficacy. Other features of the Animal Rule process could be applied to proposed regulatory pathways for the expedited development and approval of new antibiotics. For example, under the Animal Rule, the animal study endpoint must be clearly linked to outcomes of human clinical benefit, namely survival or prevention of major morbidity. 93 Using surrogate endpoints that are less strongly associated with clinical improvement may heighten uncertainty around the true benefits and harms, thereby compounding the risks associated with a new therapy. Moreover, this focus on clinically meaningful outcomes can help ensure that regulators prioritize the approval of products with the greatest potential for patient benefits.
IV. DISCUSSION AND POLICY IMPLICATIONS
To accelerate the development of novel antibiotics, policymakers are considering a slate of new incentives for pharmaceutical manufacturers, including modification of the regulatory process that antibiotics, like all other drugs, must go through to obtain marketing approval. One of these proposals, which would create an approval pathway for certain antibiotics for use in a limited population of patients, was passed in 2015 by the House of Representatives as part of the 21st Century Cures Act (“Cures Act”). 94 In this Part, we discuss how this legislative proposal could be refined to better balance the need for innovative antibiotics with the imperative for scientifically valid information available for clinicians and patients to make informed decisions after the drug is approved.
Under this pathway, antibacterial or antifungal drugs can benefit from a streamlined development and regulatory approval process if they are intended for a limited population of patients for which there is an unmet medical need. 95 The bill also permits FDA to approve drugs through this pathway based on limited datasets: preclinical, pharmacologic, or pathophysiologic evidence; data from Phase 2 clinical trials; and other “confirmatory evidence” determined appropriate. 96 Sponsors of qualifying drugs would also benefit from early consultation and close coordination with FDA. 97 New antibiotics approved through this pathway would be required to have labeling clearly indicating that the approval was for a limited population. 98
As the Senate considers various proposals also found in the Cures Act, 99 the experience of FDA's existing expedited development and approval programs could help legislators refine this proposed pathway for new antibiotics. First, the approval of ceftazidime-avibactam through the 505(b)(2) pathway illustrates the continued importance of well-controlled clinical trials to detect critical safety signals and to adequately test whether a drug yields meaningful patient benefit. If such rigorous trials are not conducted prior to gaining approval, they should be required post-approval. However, there is considerable literature indicating that post-approval trials, even when they are mandated, are often delayed or not conducted. Legislators could explore instituting stronger provisions to incentivize the timely completion of any required post-approval studies. For example, the approval of an antibiotic could automatically lapse (or “sunset”) if data from the post-approval study are not submitted according to pre-agreed milestones. 100
Second, if the public health needs merit approval based on less rigorous evidence, policymakers and FDA should ensure that such evidence is of the highest possible quality and is likely to predict clinical benefit in the disease population. In the case of the Animal Rule, FDA's guidance specifies that trial endpoints in the animal studies should ideally be related to survival benefit or prevention of major morbidity. 101 The priority review, accelerated approval, and breakthrough therapy programs all require evidence suggesting the potential for improvement in safety or clinical benefit over available therapies. Any new regulatory pathway for antibiotics should have a similar stipulation that scientific evidence, even if it is from limited datasets or Phase 2 trials, should investigate the drug's impact on clinically meaningful endpoints and potentially in comparison to available treatments. Trials of new therapies for unmet medical needs do not need to be large or overly burdensome for the sponsor, and our study showed that antibiotic trials are already no larger than those conducted for drugs approved in other disease areas with high unmet medical needs. Generally, when there is greater expected magnitude of improvement of the new therapy, fewer patients would need to be enrolled. For example, if a disease has a survival rate of 40% (consider severe infections caused by carbapenem-resistant Enterobacteriaceae), a clinical trial would only need to enroll 190 total patients to demonstrate a statistically significant survival rate of 60% in the treatment group. 102
Finally, there are many unanswered questions about how this new regulatory pathway would impact antibiotic innovation and health outcomes. To ensure that patients are not exposed to undue risk and to stem the proliferation of resistance, public health officials will need to ensure that utilization of these new antibiotics is limited. Legislators could commission an independent body, such as the Government Accountability Office (in cooperation with FDA and the CDC) to actively monitor uptake of products approved through this new regulatory pathway. It would also be useful to determine whether products approved through this pathway would, or would not, have qualified for approval under FDA's other expedited approval programs. In time, these data could help policymakers make necessary modifications to the pathway.
It is worth noting that regulatory efficiency is an important, but certainly not the only, factor in the clinical or commercial success of a new drug. For example, one study found that more antibiotics were withdrawn from the market, many due to poor commercial performance, compared to other drug classes. 103 Simply approving antibiotics faster does not guarantee that those drugs should be used by patients or prescribed by physicians. For example, without parallel efforts to improve conservation, the marketing of new antibiotics could increase overall utilization, thereby contributing to the proliferation of resistance. Similarly, regulatory approval does not necessarily mean that payors will pay for these therapies. 104 To sustainably combat antibiotic resistance, stakeholders will also need to address market and stewardship challenges.
V. CONCLUSION
In conclusion, FDA has considerable scope to exercise regulatory flexibility to approve new therapies for serious diseases and unmet medical needs. Over the past decade, antibiotics have been approved on the basis of pivotal trials that are not necessarily larger, or more costly by implication, than trials for antiviral or cancer drugs—two other therapeutic areas that have been a priority for policymakers. Any new regulatory pathway should ensure that patients have access to the highest quality data, including, when possible, studies investigating clinically meaningful endpoints, so that patients and clinicians can make informed treatment decisions about the risks and benefits of newly approved products.
Footnotes
Acknowledgements
Dr. Kesselheim is supported by a Greenwall Faculty Scholarship in Bioethics, the Harvard Program in Therapeutic Science, and the Laura and John Arnold Foundation. This work was also supported by unrestricted grants from the Interfaculty Initiative in Health Policy (Cordeiro Family Research Fellowship), the Center for American Political Studies, and the Dunwalke Fund, all at Harvard University. Mr. Hwang was previously employed by Bain Capital, which has invested in healthcare companies but did not have any involvement in this study. The authors' funders, grantors, and employers had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or the decision to submit the manuscript for publication.
APPENDIX
K
| Year | Incentive | Aims | |
|---|---|---|---|
|
|
|||
| Orphan Drug Act | 1983 | 7 years of guaranteed market exclusivity, research grants, waived regulatory fees | To promote the development of new therapies to treat rare diseases |
| Best Pharmaceuticals for Children Act (BPCA) | 2002 | 6 months of additional marketing exclusivity at the end of patent expiration | To incentivize the conduct of pediatric clinical trials |
| Generating Antibiotic Incentives Now (GAIN) Act | 2012 | 5 years of additional guaranteed marketing exclusivity (a total of 10 years) | To stimulate the development of new antibiotics |
|
|
|||
| Fast track review pathway | 1992 | Expedited development and review | To speed the approval of therapies intended to treat serious conditions that are unmet medical needs |
| Accelerated approval pathway | 1992 | Expedited approval on the basis of surrogate endpoints reasonably likely to predict clinical benefit | To accelerate approval of drugs meeting unmet medical needs that provide meaningful advantages over available therapies |
| Priority review pathway | 1992 | FDA review of new drug application within six months, as compared to ten months for standard review | To speed the review of drugs treating serious conditions that, if approved, would provide a significant improvement over existing therapies |
| Priority review voucher | 2007 | Transferable voucher entitling holder to priority review (≤6 months) of drug that would not otherwise qualify | To promote the development of drugs for tropical diseases |
| GAIN Act | 2012 | Eligibility for priority review and fast-track designation | To accelerate approval of qualified infectious disease products |
| Breakthrough therapy designation | 2012 | Anticipates expedited approval on the basis of preliminary evidence (e.g., biomarkers) | To speed the approval of products that show substantial improvement over existing therapies |
1
See Anthony S. Fauci & Hilary D. Marston, The Perpetual Challenge of Antimicrobial Resistance, 311 JAMA 1853, 1853-54 (2014) (“In fact, the challenge of antimicrobial resistance is an enduring threat that likely will never be eliminated. The threat is due, in part, to the inherent ability of microbes to replicate rapidly and mutate, offering them an evolutionary advantage in fending off hazards to their survival. Addressing the threat of antimicrobial resistance is a never-ending challenge.”)
2
CDC, U.S. D
3
See, e.g., id.; Matthew J. Neidell et al., Costs of Healthcare- and Community-Associated Infections with Antimicrobial-Resistant Versus Antimicrobial-Susceptible Organisms, 55 C
4
W
5
U.K. R
6
James Beardsley et al., Show Me the Money: Long-Term Financial Impact of an Antimicrobial Stewardship Program, 33 I
7
See Food and Drug Administration Safety and Innovation Act, Pub. L. No. 112-144, 126 Stat. 1077, §§ 801-06 (2012). The GAIN Act, or GAIN provisions, was passed as part of this larger statute. See id. Since existing patents may provide for longer exclusivity periods, these periods effectively serve as bare minimum guarantees and are most applicable for approved new drugs lacking much remaining patent protection. See Bo Wang et al., Variations in Time of Market Exclusivity Among Top-Selling Prescription Drugs in the United States, 175 J
8
PCAST, R
9
See id.
10
21st Century Cures Act of 2015, H.R. 6, 114th Cong. § 2121 (2015); see also Norm Ornstein, A Bipartisan Victory for Medical Research in Congress, A![]() ].
].
11
See S
12
See Donna T. Chen et al., U.S. Physician Knowledge of the FDA-Approved Indications and Evidence Base for Commonly Prescribed Drugs: Results of a National Survey, 18 P
13
See Stephan A. Billstein, How the Pharmaceutical Industry Brings an Antibiotic Drug to Market in the United States, 38 A
14
Id. at 2680.
15
Id.
16
Id.
17
Id. at 2680-81.
18
Id.
19
See Bonnie Sibbald & Martin Roland, Understanding Controlled Trials: Why Are Randomised Controlled Trials Important? 316 B
20
See, e.g., Karin Meissner et al., Differential Effectiveness of Placebo Treatments: A Systematic Review of Migraine Prophylaxis, 173 JAMA I
21
See S
22
See Geoffrey Marshall et al., Streptomycin Treatment of Pulmonary Tuberculosis: A Medical Research Council Investigation, 2 B
23
See 21 C.F.R. § 314.126 (2015).
24
See Robert Kocher & Bryan Roberts, The Calculus of Cures, 370 N
25
See Hagop M. Kantarjian et al., Cancer Drugs in the United States: Justum Pretium—The Just Price, 31 J. C
26
See Thomas J. Hwang, Stock Market Returns and Clinical Trial Results of Investigational Compounds: An Event Study Analysis of Large Biopharmaceutical Companies, P
27
See Nicholas S. Downing et al., Clinical Trial Evidence Supporting FDA Approval of Novel Therapeutic Agents, 2005-2012, 311 JAMA 368, 372 (2014) (describing the variation in clinical trial evidence across indications); Ruben G. Duijnhoven et al., Number of Patients Studied Prior to Approval of New Medicines: A Database Analysis, 10 PL
28
See Ralph B. D’Agostino, Sr. et al., Non-Inferiority Trials: Design Concepts and Issues, 22 S
29
See John H. Powers & Thomas R. Fleming, Noninferiority Trials: Clinical Understandings and Misunderstandings, 3 C
30
See Joseph A. DiMasi et al., Cost of Innovation in the Pharmaceutical Industry, 10 J. H
31
See Sandeep Kumar Gupta & Roopa P. Nayak, Dry Antibiotic Pipeline: Regulatory Bottlenecks and Regulatory Reforms, 5 J. P
33
See id.
34
See Thomas J. Hwang et al., Assessment of US Pathway for Approving Medical Devices for Rare Conditions, 348 B
35
See Frank Wilcoxon, Individual Comparisons by Ranking Method, 1 B
36
See History, S![]() ].
].
37
Warner-Lambert Co. v. Heckler, 787 F.2d 147, 151 (3d Cir. 1986) (“Because the [FDCA] requires ‘investigations,’ the FDA requires drug manufacturers to submit at least two … studies showing the effectiveness of the drug.”); FDA, Final Decision on Benylin, 44 Fed. Reg. 51,512, 51,518 (Aug. 31, 1979) (explaining that the two study requirement is premised on the notion that a study must be capable of reproduction for the results to be reliable). Although the FDA generally requires two adequate and well controlled studies to establish evidence of a drug’s effectiveness, the federal Food, Drug, and Cosmetic Act was amended in 1997 to explicitly allow only one pivotal trial to meet the substantial evidence standard. See Food, Drug, and Cosmetic Act of 1938, 21 U.S.C. § 355(d) (2012) (“If the Secretary determines, based on relevant science, that data from one adequate and well-controlled clinical investigation and confirmatory evidence (obtained prior to or after such investigation) are sufficient to establish effectiveness, the Secretary may consider such data and evidence to constitute substantial evidence for purposes of the preceding sentence.”).
38
21 C.F.R. § 312.84 (2015).
39
See Jonathan J. Darrow et al., New FDA Breakthrough-Drug Category—Implications for Patients, 370 N
40
Kesselheim et al., supra note 39, at 1772-73.
41
See Investigational New Drug, Antibiotic, and Biological Drug Product Regulations; Procedures for Drugs Intended to Treat Life-Threatening and Severely Debilitating Illnesses, 53 F
42
See 21 C.F.R § 312.500 (2015).
43
Id. § 312.510.
44
Id. §§ 312.530, 312.540.
45
Food and Drug Administration Safety and Innovation Act of 2012, Pub. L. No. 112-144, 126 Stat. 1087, § 902 (2012) (codified as amended at 21 U.S.C. § 356).
46
See Press Release, Rebiotix, Receives Breakthrough Therapy Designation for RBX2660 (Oct. 12, 2015) http://www.rebiotix.com/news-media/press-releases/rebiotix-receives-breakthrough-therapy-designation-for-rbx2660-recurrent-c-diff/ [http://perma.cc/77XP-SBHR]; Press Release, Seres Therapeutics, Seres Therapeutics Receives FDA Breakthrough Therapy Designation for Its Lead Product Candidate, SER- 109 (June 12, 2015), http://www.serestherapeutics.com/pipeline/ser-109 [![]() ].
].
47
See Press Release, XBiotech, XBiotech Receives FDA Fast Track Designation for its Novel True Human™ Therapeutic Antibody for Treating Serious Infections Due to Staphylococcus aureus (Oct. 1, 2015), http://www.xbiotech.com/about/news/xbiotech-receives-fda-fast-track-designation-for-its-novel-true-human-therapeutic-antibody.html [![]() ].
].
48
See generally FDA, G
49
See Drug Price Competition and Patent Term Restoration (Hatch-Waxman) Act of 1984, Pub. L. No. 98-417, 98 Stat. 1585, § 505(b)(2) (1984) (codified as amended at 21 U.S.C. § 355(b)(2)). This statute is commonly referred to as the Hatch-Waxman Act or the Hatch-Waxman Amendments, and the relevant subsection is still referred to as § 505(b) even though it is codified in § 355(b). Accordingly, it will be referred throughout this article simply by § 505(b).
50
Compare § 505(b)(1), with § 505(b)(2); see also FDA, G![]() ] (describing the types of evidence that can be accepted as part of a 505(b)(2) application).
] (describing the types of evidence that can be accepted as part of a 505(b)(2) application).
51
See FDA, supra note 50, at 3.
52
See Albane D’Argent, The 505(b)(1) and 505(b)(2) Application Process in the US, L![]() (“Since its introduction, the 505(b)(2) path has become slowly but surely a more and more appealing and lucrative pathway for manufacturers. Not only is this pathway shortening development time and mitigating costs, but it may also qualify for 3 to 5 years of market exclusivity, unlike generics for which exclusivity can be held for only 180 days.”).
(“Since its introduction, the 505(b)(2) path has become slowly but surely a more and more appealing and lucrative pathway for manufacturers. Not only is this pathway shortening development time and mitigating costs, but it may also qualify for 3 to 5 years of market exclusivity, unlike generics for which exclusivity can be held for only 180 days.”).
53
See Press Release, FDA, FDA Approves New Antibacterial Drug Avycaz (Feb. 25, 2015), http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm435629.htm [![]() ].
].
54
See Robert K. Flamm et al., Ceftazidime-Avibactam and Comparator Agents Tested against Urinary Tract Isolates from a Global Surveillance Program, 80 D
55
Premavathy Levasseur et al., In Vitro Antibacterial Activity of the Ceftazidime-Avibactam (NXL104) Combination Against Pseudomonas aeruginosa Clinical Isolates, 56 A
56
Ctr. for Drug Evaluation & Research, FDA, Application Number: 206494O
57
See id. (“In December 2013, the Applicant and the Agency agreed that a New Drug Application (NDA) covered under Section 505(b)(2) of the Food Drug and Cosmetic Act relying in part on the Agency’s previous finding of safety and efficacy of ceftazidime (one of the components of the drug product, ceftazidime-avibactam), could be submitted.”).
58
See id. (“Additional data would include nonclinical data, Phase 1 data, data from two Phase 2 trials, and published ceftazidime data. The application also includes safety data on avibactam, including data from patients who received ceftazidime-avibactam. The contribution of the avibactam component is being assessed primarily in in vitro studies and in animal models of infection, where the addition of avibactam restored the activity of ceftazidime against ceftazidime-nonsusceptible bacteria.”).
59
Bridget Silverman, Flexibility Or Formal Pathway? Avycaz Suggests FDA Doesn't Need Congress To Expedite Limited Use Antibiotics, P![]() ] (“FDA floated the idea of using accelerated approval regulations to expedite Avycaz’s approval but rejected it in favor of the 505(b)(2) strategy. Minutes of the Dec. 19, 2013 pre-NDA meeting report that, after ‘several internal discussions,’ FDA concluded that ‘there is no surrogate endpoint that is reasonably likely to predict clinical benefit for [ceftazidime-avibactam].’”).
] (“FDA floated the idea of using accelerated approval regulations to expedite Avycaz’s approval but rejected it in favor of the 505(b)(2) strategy. Minutes of the Dec. 19, 2013 pre-NDA meeting report that, after ‘several internal discussions,’ FDA concluded that ‘there is no surrogate endpoint that is reasonably likely to predict clinical benefit for [ceftazidime-avibactam].’”).
60
See Ctr. for Drug Evaluation & Research, supra note 56, at 2 (“Since submission of the NDA, the Applicant clarified that they were seeking all the above indications when limited or no alternative treatments are available.”).
61
Ctr. for Drug Evaluation & Research, FDA, Application Number: 206494O
62
Id. at 8, 20 (2015) (“Clinical cure rates were lower in a subgroup of patients with baseline creatinine clearance (CrCL) of 30 to 50 ml/min compared to those with CrCL >50 ml/min. The reduction in clinical cure rate was pronounced in [ceftazidime-avibactam]-treated patients (85% to 45%) compared to meropenem-treated patients (86% to 74%).”).
63
Id. at 20.
64
Id. at 10-11.
65
See AVYCAZ: Indications and Usage, A![]() ] (“As only limited clinical safety and efficacy data for AVYCAZ (ceftazidime-avibactam) are currently available, reserve AVYCAZ for use in patients who have limited or no alternative treatment options …. To reduce the development of drug-resistant bacteria and maintain the effectiveness of AVYCAZ and other antibacterial drugs, AVYCAZ should be used only to treat infections that are proven or strongly suspected to be caused by susceptible bacteria.”).
] (“As only limited clinical safety and efficacy data for AVYCAZ (ceftazidime-avibactam) are currently available, reserve AVYCAZ for use in patients who have limited or no alternative treatment options …. To reduce the development of drug-resistant bacteria and maintain the effectiveness of AVYCAZ and other antibacterial drugs, AVYCAZ should be used only to treat infections that are proven or strongly suspected to be caused by susceptible bacteria.”).
66
See FDA, Avycaz (ceftazidime and avibactam): Drug Safety Communication – Dose Confusion and Medication Errors (Sept. 22, 2015), http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm463595.htm [![]() ] (“FDA is warning health care professionals about the risk for dosing errors with the intravenous antibacterial drug Avycaz (ceftazidime and avibactam) due to confusion about the drug strength displayed on the vial and carton labels ….”).
] (“FDA is warning health care professionals about the risk for dosing errors with the intravenous antibacterial drug Avycaz (ceftazidime and avibactam) due to confusion about the drug strength displayed on the vial and carton labels ….”).
67
See Anthony Coates et al., The Future Challenges Facing the Development of New Antimicrobial Drugs, 1 N
68
C
69
See 21 C.F.R. § 314.600 (2015).
70
FDA, G
71
See 21 C.F.R. § 314.600.
72
Id. § 314.610.
73
Id.
74
See id.
75
See Gigi Kwik Gronvall et al., Letter to the Editor, The FDA Animal Efficacy Rule and Biodefense, 25 N
76
See Press Release, FDA, FDA Approves Raxibacumab to Treat Inhalational Anthrax (Dec. 14, 2012), http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm332341.htm [![]() ].
].
77
See Thi-Sau Migone et al., Raxibacumab for the Treatment of Inhalational Anthrax, 361 N
78
See Ctr. For Drug Evaluation & Res., Application Number: 125349Orig1s000, Summary Review, at 6 (Dec. 14, 2012) (“The results of the primary analysis showed survival in 24/37 (65%) NZW rabbits treated with levofloxacin alone, compared to 32/39 (82%) NZW rabbits treated with raxibacumab plus levofloxacin. The 17% difference in survival rates did not reach statistical significance (p=0.0874).”).
79
Id.
80
Id.
81
See Press Release, Human Genome Sciences, Inc., Human Genome Sciences Announces New Order for Raxibacumab (ABthrax™) from U.S. Government (July 22, 2009, 7:00 PM), http://www.prnewswire.com/news-releases/human-genome-sciences-announces-new-order-for-raxibacumab-abthraxtm-from-us-government-62250457.html [![]() ] (“Human Genome Sciences, Inc. today announced that the U.S. Government has exercised its option to purchase an additional 45,000 doses of raxibacumab (ABthrax™) for the Strategic National Stockpile, to be delivered over a three-year period, beginning near the end of 2009. HGS expects to receive approximately $151 million from this award as deliveries are completed.”).
] (“Human Genome Sciences, Inc. today announced that the U.S. Government has exercised its option to purchase an additional 45,000 doses of raxibacumab (ABthrax™) for the Strategic National Stockpile, to be delivered over a three-year period, beginning near the end of 2009. HGS expects to receive approximately $151 million from this award as deliveries are completed.”).
82
See Press Release, FDA, FDA Approves New Antibacterial Treatment for Plague (Apr. 27, 2012), http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm302220.htm [![]() ].
].
83
See Ctr. For Drug Evaluation & Res., Application Numbers: 020634O
84
See id. at 8-9 tbl.3.
85
See id.
86
See id. at 10.
87
See Press Release, FDA, FDA Approves Additional Antibacterial Treatment for Plague (May 8, 2015), http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm446283.htm [![]() ].
].
88
See Marion Gruber, Dir., Office of Vaccines Research & Review, Presentation at the Inst. of Med.’s Fast Track Dev. of Ebola Vaccines (Mar. 26, 2015), http://www.nationalacademies.org/hmd/~/media/Files/Activity%20Files/PublicHealth/MedPrep/Gruber%20IOM%20March%2026.pdf [![]() ] (“Ebola vaccines could be licensed based on clinical endpoint efficacy studies, studies that show an effect on a marker reasonably likely to predict clinical benefit, or animal studies. Accelerated Approval and approval under the Animal Rule considered if Ebola infection rates do not permit direct assessment of efficacy in clinical trials, or for vaccines not being evaluated in current efficacy trials.”).
] (“Ebola vaccines could be licensed based on clinical endpoint efficacy studies, studies that show an effect on a marker reasonably likely to predict clinical benefit, or animal studies. Accelerated Approval and approval under the Animal Rule considered if Ebola infection rates do not permit direct assessment of efficacy in clinical trials, or for vaccines not being evaluated in current efficacy trials.”).
89
See World Health Org., Ebola Situation Report (Oct. 21, 2015), http://apps.who.int/iris/bitstream/10665/190067/1/ebolasitrep_21Oct2015_eng.pdf [![]() ].
].
90
See Ana Maria Henao-Restrepo et al., Efficacy and Effectiveness of an rVSV-vectored Vaccine Expressing Ebola Surface Glycoprotein: Interim Results from the Guinea Ring Vaccination Cluster- Randomised Trial, 386 L![]() ] (“On the basis of the data presented here and additional clinical and preclinical data, the rVSV-ZEBOV vaccine (at the dose of 20 million PFU) was selected for inclusion in the Partnership for Research on Ebola Vaccines in Liberia trial, a recently initiated phase 3 efficacy study in Guinea, and the soon-to-be-initiated phase 3 Sierra Leone Trial to Introduce a Vaccine against Ebola.”)
] (“On the basis of the data presented here and additional clinical and preclinical data, the rVSV-ZEBOV vaccine (at the dose of 20 million PFU) was selected for inclusion in the Partnership for Research on Ebola Vaccines in Liberia trial, a recently initiated phase 3 efficacy study in Guinea, and the soon-to-be-initiated phase 3 Sierra Leone Trial to Introduce a Vaccine against Ebola.”)
91
See Steven Joffe, Evaluating Novel Therapies During the Ebola Epidemic, 312 JAMA 1299, 1299- 1300 (2014) (describing key considerations for conduct of clinical trials for Ebola); Kai Kupferschmidt, As Ebola Wanes, Trials Jockey for Patients, 348 S
92
See FDA, Briefing Document: Licensure of Ebola Vaccines: Demonstration of Effectiveness 10 (May 12, 2015), http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/VaccinesandRelatedBiologicalProductsAdvisoryCommittee/UCM445819.pdf [![]() ] (“Preliminary data presented at a December 12, 2014, US government- sponsored workshop indicated that vaccinated humans may achieve immune responses comparable in magnitude to those associated with protection in [non-human primates], suggesting the feasibility of an immunogenicity-based approval, although the thresholds associated with protection may differ for each vaccine candidate.”).
] (“Preliminary data presented at a December 12, 2014, US government- sponsored workshop indicated that vaccinated humans may achieve immune responses comparable in magnitude to those associated with protection in [non-human primates], suggesting the feasibility of an immunogenicity-based approval, although the thresholds associated with protection may differ for each vaccine candidate.”).
93
See 21 C.F.R. § 314.610 (2015) (“FDA will rely on the evidence from studies in animals to provide substantial evidence of the effectiveness of these products only when … the animal study endpoint is clearly related to the desired benefit in humans, generally the enhancement of survival or prevention of major morbidity.”).
94
21st Century Cures Act of 2015, H.R. 6, 114th Cong., § 2121 (2015).
95
See id.
96
See id.
97
See id.
98
See id.
99
In April 2016, a companion bill covering this proposal passed out of the Senate Health, Education, Labor and Pensions committee. See Zachary Brennan, Senate Committee Advances Five More Bills as Part of Medical Innovation Package, R![]() (“S. 185, Promise for Antibiotics and Therapeutics for Health Act, which requires FDA to establish a program to approve as a limited population antibacterial drug intended to treat a serious medical condition and to address an unmet medical need within an identifiable limited population, among other things.”).
(“S. 185, Promise for Antibiotics and Therapeutics for Health Act, which requires FDA to establish a program to approve as a limited population antibacterial drug intended to treat a serious medical condition and to address an unmet medical need within an identifiable limited population, among other things.”).
100
See, e.g., Thomas J. Hwang et al., Accelerating Innovation in Rapid Diagnostics and Targeted Antibacterials, 33 N
101
FDA, supra note 70, at 3.
102
This estimate assumes that the study has 80% power to detect, at a significance level of 5%, and a survival rate of 60% in the treatment group versus 40% in the control group. Readers can run their own sample size calculations using online tools. See, e.g., Power (Sample Size) Calculators, S![]() .
.
103
See Kevin Outterson et al., Approval and Withdrawal of New Antibiotics and Other Antiinfectives in the U.S., 1980-2009, 41 J.L. M
104
See Thomas J. Hwang et al., Paying for Innovation: Reimbursement Incentives for Antibiotics, S