
Abstract
Ionising radiation induces various types of DNA damage, which, if not repaired correctly, lead to mutations. Nonetheless, the association between radiation exposure and hereditary effects has not yet been confirmed in humans. In this study, we used WGS to study low dose rate radiation–induced mutations in cultured human cells. Normal human fibroblast NB1RGB cells were seeded as single cells by limiting dilution and grown under irradiation with 137Cs γ-irradiation of 1 or 20 mGy day-1 for 21 days. The genomic DNA prepared from respective clones or the bulk culture was sequenced using the Illumina NGS platform with paired-end 150-bp reads with reference to the standard human genome assembly GRCh38. By subtracting the calls in the bulk culture and applying several filters, approximately 750–1200 single-nucleotide polymorphisms (SNPs) and 2500 small insertions/deletions (indels) per clone were obtained on average. The number was not substantially different between unirradiated and irradiated cultures. Spectral analysis revealed that the C > A transversion, which is associated with guanine oxidation, was dominant.
1. INTRODUCTION
The constant threat of ionising radiation (IR) and its potential to induce DNA damage has long been a subject of scientific scrutiny (Snyder and Morgan, 2004). From nucleotide modifications to DNA strand breaks, the range of IR-induced damage poses a significant risk to cellular genomic integrity. Unrepaired DNA damage can lead to mutations, with implications ranging from cancer to hereditary effects, contingent upon the magnitude of radiation exposure. Remarkably, a conclusive association between radiation exposure and hereditary damage in humans has yet to be established, and the statistical impact of low doses of radiation on the human body remains elusive (Paunesku and Woloschak, 2018).
Historically, mutation detection methods, such as the specific locus test, were constrained by the need for an extensive number of samples and limited to specific genomic regions, typically around 100 kilobases. This limitation hindered the comprehensive evaluation of mutation induction effects. However, recent advancements in high-throughput sequencing technologies have revolutionised our ability to probe the entire genome, approximately 6 gigabases, with remarkable accuracy (Satoh et al., 2020). Whole-genome DNA sequencing (WGS) has emerged as a powerful tool, offering a holistic approach to detect mutations and investigate hereditary effects in human genes (Milholland et al., 2017; Cagan et al., 2022). Despite the promise of WGS, its implementation has historically required a substantial number of subjects, hindering its widespread application.
In light of these challenges, our research aims to address critical gaps in our understanding of cellular mutations induced by ionising radiation. First and foremost, we sought to determine mutation frequencies in human primary fibroblasts, a pivotal cell type often used in radiation studies. Additionally, we attempted to establish a novel method for studying cellular mutations, leveraging the capabilities of whole-genome sequencing (WGS) through next-generation sequencing (NGS) technologies. Our investigation aimed to provide a comprehensive and detailed understanding of the mutational landscape in human cells exposed to ionising radiation, paving the way for enhanced radiation risk assessment and potential interventions to safeguard genomic integrity.
2. MATERIALS AND METHODS
2.1 Cell culture and irradiation
Normal human skin fibroblast NB1RGB cells at the population doubling time of 15 were obtained from RIKEN BioResource Center (Tsukuba, Ibaraki, Japan) and was cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Nacalai Tesque, Kyoto, Japan) supplemented with 10% v/v fetal bovine serum (HyClone, GE HealthCare, Chicago, IL, USA), 100-U mL-1 penicillin, 100-μg mL-1 streptomycin (Nacalai Tesque), and 1% v/v MEM non-essential amino acid solution (100×) (Nacalai Tesque) at 37°C in a humidified atmosphere with 5% CO2. Cells were dissociated from the dishes by treatment with 0.05% w/v trypsin and 0.2-mM EDTA.
Descriptive summary of the variant calling pipelines included in the comparative analyses.

For the measurement of the surviving fraction, cells were irradiated in the Chiyoda Technol Cobalt 60 Irradiation Facility at the Tokyo Institute of Technology. For low dose rate irradiation, cells were seeded as single cells by limiting dilution, i.e. at the rate of 0.1 cell on average in each well of 96-well plates, and were grown in a CO2 incubator in a facility equipped with 137Cs gamma-ray irradiation source in Institute of Environmental Science at the dose rates of 1 and 20 mGy day-1 for 21 days (the cumulative dose of 21 and 420 mGy, respectively).
2.2 Whole-genome sequencing
DNA was extracted from the cells using NucleoSpin (Takara Bio). DNA concentration was measured first using NanoDrop and then using Qubit (HS) Assay. Extracted DNA samples were stored at −30°C. WGS analysis was conducted in Kurabo Co. Ltd. (Neyagawa, Osaka, Japan). The library was prepared using the TruSeq DNA Nano Library kit (Illumina). Clustering and sequencing were conducted in Macrogen Co. Ltd. using the Illumina Hi-Seq Platform. Median fragment size was 400–500 base pairs (bp). Sequences of paired ends were read for a length of 151 bases at a coverage of >30×. Paired-end sequences were mapped to the human genome using the iSAAC aligner (iSAAC-04.18.11.09) using the standard human genome assembly hg38 from UCSC, the size of which is 2934 Mbp, as the reference. Strelka (2.9.10) was applied to identify single-nucleotide variants (SNVs) and small insertions and deletions (indels). Below, we focused on autosomes: X and Y chromosomes were not examined due to the difference in copy number and, therefore, expected variant allele frequency. Initial calls were distilled through a pipeline consisting of three filters. The first filter, which is denoted ‘Germline Filter’, subtracted calls, which were present in the bulk culture, from those present in clones. The second filter, which is denoted ‘1st Quality Filter’, removed calls present in too many (>100) or too few (<10) reads, in the proximity (<10 bp) of other calls and in reads with low mapping quality (<20). The third filter, which is denoted ‘2nd Quality Filter’, selected variant allele frequency between 0.35 and 0.65, on the assumption that de novo mutations would be heterozygous and theoretically appear at the frequency of 0.5 and removed calls with strand bias (p < 0.05 in Fisher’s exact test) and those registered in the databases of single-nucleotide polymorphism (SNP), i.e. dbSNP138 and dbSNP154.
3. RESULT
3.1 Normal human fibroblast NB1RGB cells
In our initial attempt to establish single-cell cultures, as illustrated in Fig. 1a, we chose normal human skin fibroblast NB1RGB cells, which had been used in our laboratory because of their ease of proliferation and adaptability to the culture’s objectives, including the establishment of induced pluripotent stem cells (Miyake et al., 2019; Shimada et al., 2019, 2023). The IR (gamma-ray) sensitivity of NB1RGB cells was assessed using a colony formation assay, a standard method for measuring the independent growth ability of cells. Cells were subjected to gamma-irradiation after which colony formation was observed. The average plating efficiency was determined to be 50%, indicating the baseline ability of cells to form colonies under normal conditions. Following gamma-irradiation at a dose of 2 Gy, approximately 40% of the cells were able to survive as shown in Fig. 1b.
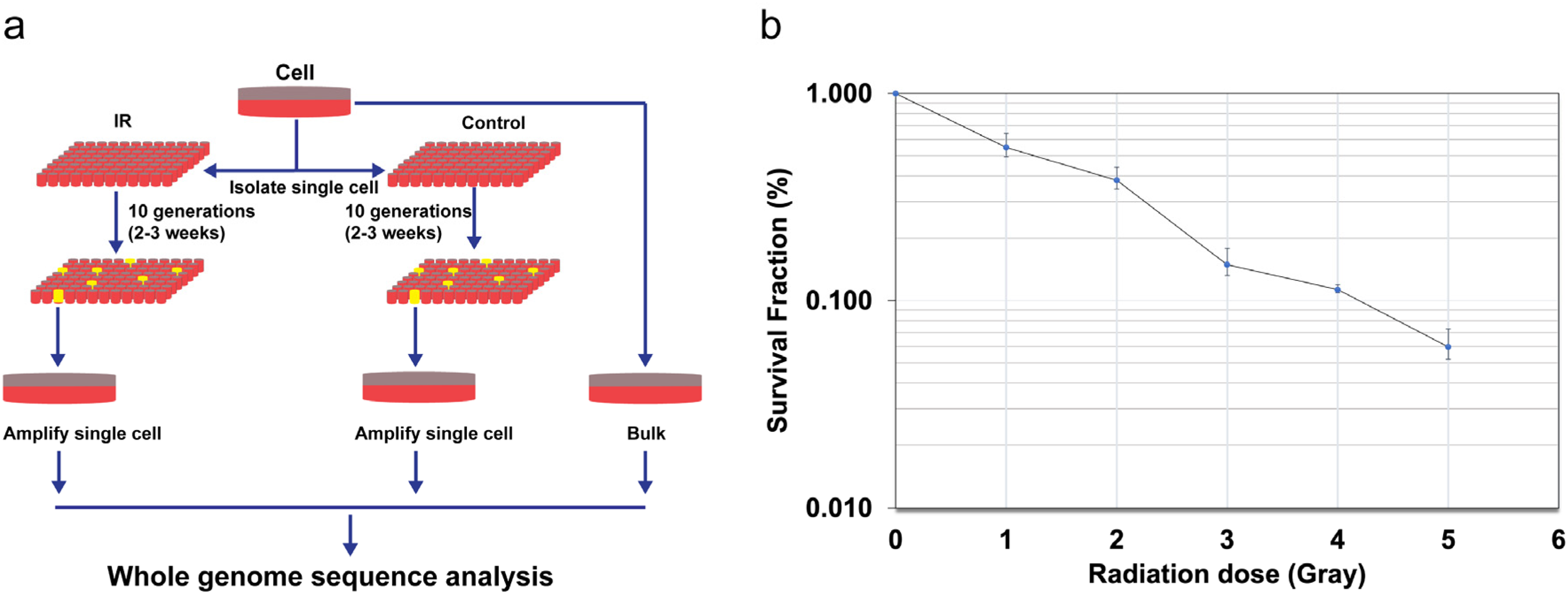
Experimental design (a) and radiosensitivity (b) of NB1RGB.
3.2 Whole-genome sequencing profile
As summarized in Table 1, the number of initially called SNVs and indels averaged around 4.5 million, which agreed with the estimated personal difference in the human genome, i.e. 0.1% (The 1000 Genomes Project Consortium, 2015). Applying the Germline Filter reduced the calls by approximately 15,000-fold. The 1st and 2nd Quality filter further reduced the calls by approximately 2-fold and 5-fold, respectively. The average number of de novo substitutions and small indels were 888 and 2539, respectively. The numbers were not substantially different between unirradiated and irradiated cultures.
Considering that NB1RGB cells were obtained at the population doubling time of 15 and used for experiments after two population doublings, the lower limit of the number of divisions that each clone has undergone is 17. The upper limit can be considered approximately 50, which is the general life span of human fibroblasts. The size of hg38 excluding sex chromosomes is 2.88 × 109 bp. Then, the lower and upper limits of the mutation rate are estimated to be 3.1 × 10−9 bp/division and 9.1 × 10−9 bp/division, respectively. The lower limit is close to the somatic mutation rate, i.e. 2.66 × 10−9 bp/division, obtained by Milholland et al (2017).
3.3 Qualitative analysis of SNVs
The difference in the spectrum of the mutation between unirradiated and irradiated clones was not discernible (Figure 2). In all clones, C–A transition, which is associated with guanine oxidation, was the most frequent, followed by T–C transition, which is associated with pyrimidine dimer formation.
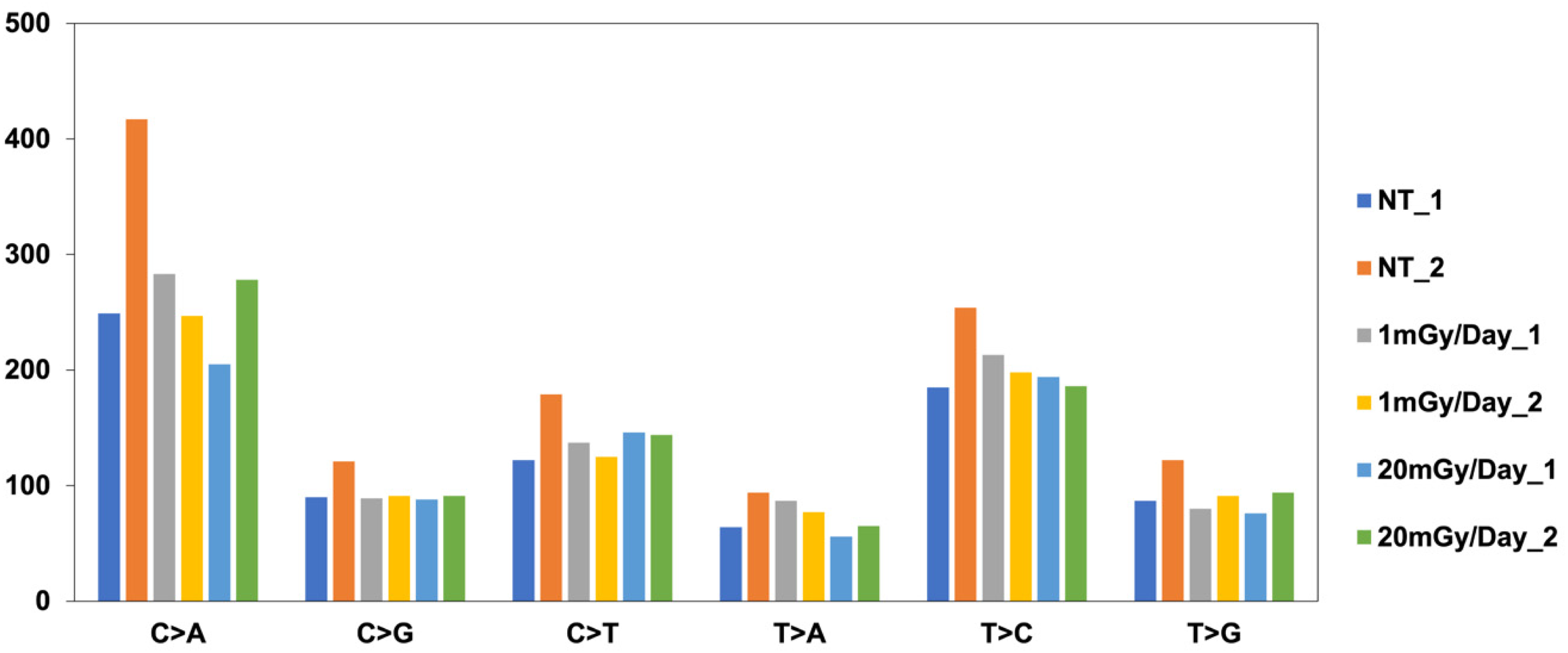
SNP mutational signature.
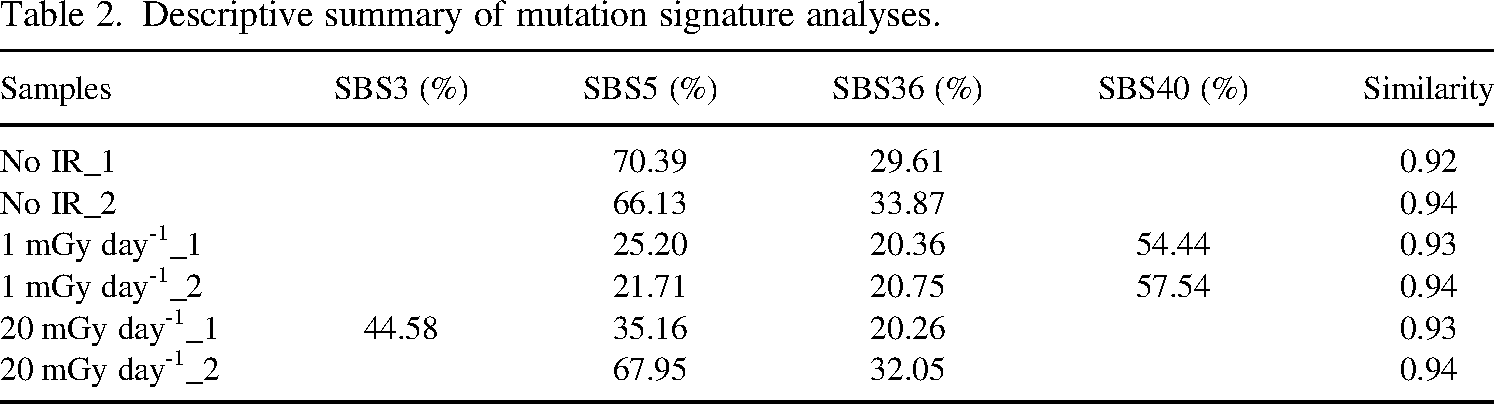
Mutation signature analysis exhibited high content of SBS5, SBS36, and SBS40 (Table 2). Although there were some differences in the relative content of these signatures among samples, no consistent differences could be found between unirradiated and irradiated clones.
Descriptive summary of mutation signature analyses.
4. CONCLUSION AND PERSPECTIVE
We analysed naturally occurring and radiation-induced mutations in cultured human cells by WGS. A discernible quantitative or qualitative difference has not been found so far between unirradiated and irradiated clones, possibly due to low cumulative radiation dose, i.e. 21 and 420 mGy. More detailed analyses of SNVs, like sorting the small indels by the length of homopolymers, are expected to reveal some change in the quantity or quality of mutations by low dose/low dose rate radiation. In addition, in the pipeline used in this study, only the mutation caused in the first generation, if any, could have passed the variant allele frequency criterion, i.e. 0.35 to 0.65, in the 2nd Quality Filter. If the single-cell–derived clones are obtained after irradiation, accumulated mutations will be detected.
Footnotes
ACKNOWLEDGEMENTS
We thank Isao Yoda (Institute of Science Tokyo) for cooperation in 60Co γ-ray irradiation.
FUNDING
The authors disclosed receipt of the following financial support for the research, authorship, and/ or publication of this article: This study was conducted as Research Project on the Health Effects of Radiation, the Ministry of the Environment of Japan.