
Abstract
Despite enormous advances in translational biomedical research, there remains a growing demand for improved animal models of human disease. This is particularly true for diseases where rodent models do not reflect the human disease phenotype. Compared to rodents, pig anatomy and physiology are more similar to humans in cardiovascular, immune, respiratory, skeletal muscle, and metabolic systems. Importantly, efficient and precise techniques for genetic engineering of pigs are now available, facilitating the creation of tailored large animal models that mimic human disease mechanisms at the molecular level. In this article, the benefits of genetically engineered pigs for basic and translational research are exemplified by a novel pig model of Duchenne muscular dystrophy and by porcine models of cystic fibrosis. Particular emphasis is given to potential advantages of using these models for efficacy and safety testing of targeted therapies, such as exon skipping and gene editing, for example, using the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated system. In general, genetically tailored pig models have the potential to bridge the gap between proof-of-concept studies in rodents and clinical trials in patients, thus supporting translational medicine.
Keywords
Genetically Engineered Pig Models of Human Monogenic Diseases
Rare monogenic diseases are an attractive market for the pharmaceutical industry, since they provide—once the underlying mutation is identified—validated targets for drug development (Brinkman et al. 2006) or genetic treatment approaches (O’Connor and Crystal 2006). Development of drugs for these orphan diseases frequently has higher success rates and shorter times to approval and may—in spite of much smaller target patient populations—generate potential lifetime revenues comparable to nonorphan drugs (Fagnan et al. 2014). Currently the molecular etiology for more than half of the estimated 7,000 rare monogenic human diseases is known, and marked acceleration of disease gene discovery is expected from the dramatic improvements in DNA-sequencing technologies and associated analyses (Boycott et al. 2013). Model organisms are required to dissect the biological consequences of a particular mutation and to provide proof of concept for therapeutic intervention. The mouse is the most widely used model organism in mammalian genetics, and powerful platforms/networks for large-scale systematic mutagenesis and standardized phenotyping have been established (Bradley et al. 2012; Infrafrontier Consortium 2015). However, mutant mouse models do not always reflect the phenotypes of the corresponding human genetic diseases. Moreover, translation of findings in mouse models into clinical studies and applications may be difficult. Thus, large animal models mimicking human anatomy and physiology more closely are additionally needed. In this respect, the pig as a monogastric omnivore is an attractive model organism (Aigner et al. 2010). A detailed discussion of advantages of miniature swine for use as relevant translational animal model is provided by Tellez and Shanmuganayagam (in press).
Over the last three decades, a broad spectrum of techniques for genetic engineering of pigs has facilitated the generation of large animal models tailored for studying mechanisms of and testing treatment options for human genetic diseases. A major breakthrough was the establishment of somatic cell nuclear transfer (SCNT) in pigs, which provided for the first time a technological basis for introducing targeted genetic modifications in this species (Lai et al. 2002; Dai et al. 2002). The low rate of gene targeting/homologous recombination (HR) in somatic cells was overcome by positive/negative selection (Jin et al. 2003) or gene-trapping strategies (Lai et al. 2002), by using adeno-associated virus (AAV) targeting vectors (Rogers, Hao, et al. 2008) or large targeting constructs based on modified bacterial artificial chromosomes (BACs; Klymiuk, Mundhenk, et al. 2012; Klymiuk et al. 2013). SCNT also facilitated the establishment of other sophisticated modifications, such as inducible transgene expression based on the binary Tet-On system (Klymiuk, Bocker, et al. 2012).
The availability of porcine whole genome sequences (Groenen et al. 2012) and the adaptation of efficient gen(om)e editing technologies, such as zinc finger nucleases (ZFNs; Hauschild et al. 2011; Whyte et al. 2011), transcription activator-like effector nucleases (TALENs; Carlson et al. 2012), and RNA-guided endonucleases derived from the bacterial clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated (Cas) system (Hai et al. 2014), to this species will further increase the potential to develop tailored pig models of human monogenic diseases. While ZFNs and TALENs tremendously improved upon the efficacy and specificity of gene editing, the complexity of the redesign and construction of the entire protein for each target limits the use of these technologies (Sander and Joung 2014). The emergence of CRISPR/Cas9 gene editing technology provides an easier, faster, and less expensive genetic engineering approach for generating targeted disease models in pigs. A novel pig model of Duchenne muscular dystrophy (DMD) and pig models of cystic fibrosis (CF) is discussed in detail subsequently.
A Pig Model of DMD Resembles Biochemical, Clinical, and Pathological Hallmarks of the Human Disease
In humans, the severe X-linked disease DMD is caused by loss-of-function mutations of the DMD gene (∼2.5 Mb, 79 exons) and affects 1 in 3,500 males. Characteristic mutations are losses of complete exons with hot spots in the regions of exons 3–7 and exons 45–55. These may lead to shifts in reading frame, out-of-frame transcripts, and loss of the essential muscle cytoskeletal protein dystrophin (Hoffman, Brown, and Kunkel 1987). DMD is characterized by progressive muscle weakness and wasting: most patients die of respiratory or heart failure between the second and fourth decade of life (reviewed in Spurney 2011). Genetic and pharmacological treatment approaches are in different phases of clinical testing (reviewed in Fairclough, Wood, and Davies 2013). Existing animal models of DMD provided insights into disease mechanisms but have limitations related to the type of DMD mutation and/or the clinical phenotype (Nakamura and Takeda 2011).
The original X-linked muscular dystrophy mouse (mdx) occurred spontaneously in the C57BL/10 strain and has a nonsense mutation in exon 23 of the Dmd gene. Four further mdx mice strains have been identified with different mutations. In addition, an mdx mouse lacking Dmd exon 52 has been generated by gene targeting (Araki et al. 1997). However, mdx mice do not develop overt muscle wasting except for the diaphragm and have a near-normal life span. The disparity in pathological consequences of dystrophin loss in DMD patients and the mdx mouse has been attributed to different patterns of muscle growth and regeneration (Partridge 2013).
In addition, mutations in the DMD gene have been identified in several dog breeds, with golden retriever muscular dystrophy (GRMD) being the most extensively examined (reviewed in McGreevy et al. 2015). GRMD dogs are more severely affected than mdx mice, but display a highly variable phenotype, and are difficult to breed. Further, the DMD mutation of the GRMD model (point mutation at the intron 6 splice acceptor site, leading to skipping of exon 7, and a premature stop codon in exon 8) does not reflect the situation in the majority of human DMD patients with a hot spot for deletions between exons 45 and 55 of the DMD gene. In addition to GRMD, DMD mutations have been identified in 8 other dog breeds, but most studies are limited to case reports (reviewed in McGreevy et al. 2015). A severe feline muscular dystrophy with dystrophin deficiency is caused by a large deletion in the promoter region of the DMD gene, but it has not been used as a model for testing therapeutic approaches (reviewed in Nakamura and Takeda 2011). Recently generated rat models of DMD have either CRISPR/Cas-induced deletions between exons 3 and 16 (Nakamura et al. 2014) or a TALEN-induced 11-bp deletion in exon 23 of the Dmd gene (Larcher et al. 2014), resulting in dystrophin deficiency. The DMD rats showed muscle weakness and histological signs of muscular dystrophy. However, no treatment studies were reported so far and findings from such studies may be—due to the small size of rats—difficult to extrapolate to humans. Very recently rhesus monkeys with mutant DMD alleles were generated by using CRISPR/Cas for injection into fertilized oocytes (Y. Chen et al. 2015). Although partial dystrophin depletion and hypertrophic myopathy were observed, the monkeys were mosaic, resulting in genetic and phenotypic variability, which limits their value as translational animal models.
To establish a tailored large animal model of DMD, we deleted DMD exon 52 in male pig cells by gene targeting using a modified BAC and generated DMD mutant pigs by nuclear transfer (Klymiuk et al. 2013). Cloned DMD pigs lacked dystrophin in skeletal muscles and exhibited increased serum creatine kinase levels, impaired movement and muscle weakness, and a maximum life expectancy of 14 weeks. Pathological analysis of DMD pigs demonstrated pale moist skeletal muscles with multifocal areas of pale discoloration. Histological examination revealed a myopathy with excessive fiber size variation, numerous large rounded hypertrophic fibers, branching fibers and fibers with central nuclei, as well as scattered clusters of segmentally necrotic fibers, next to hypercontracted fibers and groups of small regenerating muscle fibers (Figure 1). These lesions were accompanied by interstitial fibrosis and mononuclear inflammatory cell infiltration, mimicking the hallmarks of the human disease. The severity and extent of these alterations progressed with age (Klymiuk et al. 2013).
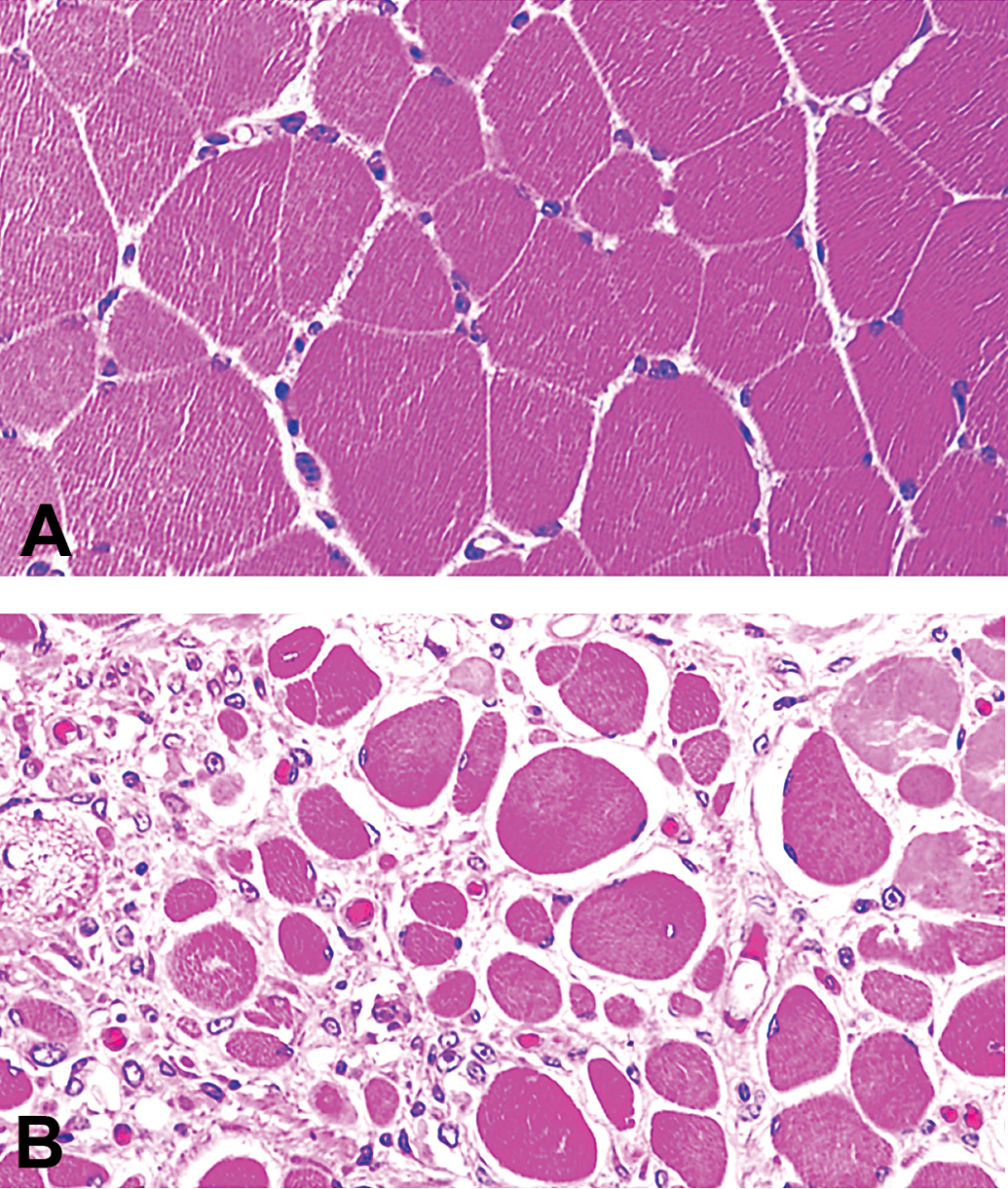
Severe muscular dystrophy in Duchenne muscular dystrophy (DMD) pigs. Cross sections of the biceps femoris muscle of a wild type (A) and a DMD pig (B) at 3 months of age. DMD pigs display variable muscle fiber diameters: large, rounded fibers with internalized central nuclei and necrosis of muscle fibers. Plastic (glycol methacrylate and methyl methacrylate) sections, 20× objective, H&E stains.
Transcriptome studies of skeletal muscle from young (2 days old) and older (around 3 months) DMD pigs and age-matched controls provided new insights into the hierarchy of physiological derangements of dystrophic muscle. The transcriptome changes in 3-month-old DMD pigs were similar to those of human DMD muscle, reflecting the processes of degeneration, regeneration, inflammation, fibrosis, and impaired metabolic activity. In contrast, the transcriptome profile of muscle samples from 2-day-old DMD pigs showed similarities with transcriptome changes induced by acute exercise muscle injury, suggesting mechanical stress on the muscle cell membranes as an early factor in the pathogenesis of DMD (Klymiuk et al. 2013).
DMD pigs exhibit the functional and pathological hallmarks of the human disease but develop them in an accelerated manner. This offers improved opportunities for early and clear-cut readouts in efficacy studies of new treatments as compared to the currently available animal models. Since loss of exon 52 is a frequent mutation in human DMD, which can be treated by exon skipping (reviewed in Fairclough, Wood, and Davies 2013), this pig model has the potential to test and refine this therapeutic strategy.
A limitation of the published DMD pig model (Klymiuk et al. 2013) is that it cannot be propagated by breeding, since cloned male pigs do not survive until the age of sexual maturity. Therefore, we introduced the DMD Δexon52 mutation in female cells and generated female carrier pigs by SCNT. The first litter from a female carrier mated with a wild-type boar contained male DMD piglets, female DMD Δexon52 carriers as well as male and female wild-type piglets according to the expected Mendelian ratio (Figure 2A). Studies are in progress to characterize the phenotype of DMD pigs generated by breeding at the biochemical, clinical, and pathological level as done previously (Klymiuk et al. 2013). At the age of 1 week, the male DMD piglets showed already markedly elevated serum creatine kinase levels (Figure 2B), indicating degeneration of muscle fibers. Breeding of female DMD Δexon52 carriers will allow us to produce sufficient numbers of DMD piglets for systematic testing of targeted therapies as outlined subsequently.

Generation of Duchenne muscular dystrophy (DMD) pigs by breeding. (A) Heterozygous DMD mutant sow with her first litter produced by mating to a wild-type boar. The heterozygous DMD mutant sow was generated by somatic cell nuclear transfer from a female cell clone in which exon 52 of one DMD allele was deleted by bacterial artificial chromosome targeting as described by Klymiuk et al. (2013), while the other DMD allele was intact. Male DMD piglets (marked by an arrow), female carriers (marked by an asterisk), and wild-type pigs were obtained according to the expected Mendelian ratio. (B) Serum creatine kinase activities are elevated already in 8-day-old DMD pigs, indicating damage and decay of muscle fibers. Note that the heavily outlined circle in the wild-type group is from 2 animals (Nikolai Klymiuk, Barbara Keßler and Eckhard Wolf, unpublished).
DMD Δexon52 Yucatan minipigs have been developed by Exemplar Genetics, Inc., and a limited characterization is included in their patent application WO2014117045A2.
Pig Models of CF Provide Important Insights into Early Disease Mechanisms
CF is the most frequent inherited disease in Caucasians and affects ∼70,000 individuals worldwide (Cutting 2015). The causative gene CFTR encoding the CF transmembrane conductance regulator, an epithelial anion channel, was identified decades ago. Almost 2,000 mutations of CFTR have been reported, with deletion of phenylalanine at position 508 (F508del) being most common (Sosnay et al. 2013). The latter causes aberrant folding of CFTR and subsequent degradation of the majority of the synthesized protein. If F508del-CFTR is trafficked to the cell membrane, it has reduced membrane residency and aberrant chloride channel function (reviewed in Cutting 2015). CF is a multisystemic disease affecting the airways, the gastrointestinal tract including pancreas and hepatobiliary system, and the reproductive tract. Chronic bacterial infections and persistent inflammatory processes of the lung are the main cause of morbidity and mortality associated with CF (reviewed by Elston and Geddes 2007).
While defective transepithelial electrolyte transport plays a role, there is no comprehensive explanation of the disease pathogenesis in the affected organs. This is mainly due to the lack of translational animal models that reflect the human disease phenotype sufficiently well. Although numerous Cftr mutant mouse models have been established, they reproduce the disease processes going on in CF patients only partially (reviewed by Wilke et al. 2011). This is particularly true for the pathology of the respiratory tract, which is the most important cause of the declining patient’s quality of life leading to death. A CFTR-deficient rat model was reported to exhibit histological abnormalities in the ileum and increased intracellular mucus in the proximal nasal septa, reduced airway surface liquid and periciliary liquid depth, and abnormal submucosal gland size (Tuggle et al. 2014). However, although the CF rat recapitulates several aspects of human CF (aberrant chloride transport, intestinal obstruction, impaired growth, malformation of the trachea, and anomalous vas deferens), important hallmarks such as obstructive lung disease, dysfunction of liver and exocrine pancreas, and diabetes mellitus were not reported in this model (reviewed in Cutting 2015).
Only recently have nonrodent animal models of CF been established. CFTR-deficient pig models were generated by introducing a stop codon in exon 10 (Rogers, Stoltz, et al. 2008) or a STOP box that terminates both transcription and translation in exon 1 (Klymiuk, Mundhenk, et al. 2012). In a third CF pig model, the most relevant human CFTR mutation F508del in exon 10 was reproduced (Ostedgaard et al. 2011). In spite of the different CFTR mutations, the models revealed almost identical phenotypes. One of the major hallmarks is the (almost) 100% penetrance of meconium ileus (MI), a mechanical obstruction of the gut that occurs in human patients as well, albeit at a frequency of only 10–20% (Kelly and Buxbaum 2015). Neither ileostomy, that is, surgical removal of meconium, nor intensive enema, which resembles the standard treatment of MI in human patients, was sufficient to resolve this obstruction in CF pigs, which usually die at the age of several weeks. Stoltz et al. (2013) generated a “gut-corrected” CF pig expressing in the gut a CFTR transgene under the control of the rat fatty acid binding protein 2 promoter. This transgenic rescue can extend life to up to 12 months; however, in-depth evaluation of CF pigs has been performed only in the neonatal state and in a limited number of ileostomized pigs. Despite these limitations, the CF pig model has contributed tremendously to the understanding of CF pathogenesis. In particular, the availability of neonatal material that can be seen as a “native” tissue revealed novel insights into the very early steps of CF development.
Progressive obstruction of the respiratory tract is the most important cause of morbidity in CF patients. While histological examination of newborn CF pigs revealed apparently normal lung tissue (Rogers, Stoltz, et al. 2008), the trachea had a triangular rather than a circular shape and the cartilage appeared thicker and more discontinuous than in wild-type samples (Meyerholz et al. 2010; Klymiuk, Mundhenk, et al. 2012). This was confirmed in human CF infants (Meyerholz et al. 2010; Diwakar et al. 2015). In accordance to the findings in CF patients, sinus disease developed spontaneously in older CF pigs, whereas at birth sinuses were hypoplastic but did not show evidence of infection or inflammation (Chang et al. 2012). Although the lungs of the newborn CF piglets did not show signs of infection, defective bacterial eradication was observed (Stoltz et al. 2010) and attributed to the decreased pH on airway epithelia, which has been postulated to impair bacterial killing (Pezzulo et al. 2012). Furthermore, mucus detachment from submucosal glands of the airways has been shown to be impaired (Hoegger et al. 2014).
Analyses of epithelial tissue and cultivated cells from CF pigs revealed that the lack of CFTR caused reduced transcellular transport of Cl− and HCO3 −, but no alteration of Na+ transport or liquid absorption was reported (J. H. Chen et al. 2010). This was later confirmed in primary epithelial cells from human CF patients (Itani et al. 2011). This was a major paradigm shift driven by interrogation of the pig model, as the thickening of mucus in CF airways has been postulated to occur from a disturbance of osmolaric balance, whereas the pig model data suggested a Na+-independent mechanism. Consequently, these data challenged the relevance of mouse models overexpressing the epithelial Na+ channel, the only mouse model showing mucus thickening in the airways, for CF research (Collawn et al. 2012).
In conclusion, the established CF pig lines provide excellent models to study early mechanisms of lung disease and to evaluate therapeutic strategies in newborn animals.
A ferret model of CF was generated by Sun et al. (2010). The phenotypic changes in this model correspond to those of the pig models and have been reviewed recently (Yan et al. 2015).
The Potential Role of DMD and CF Pig Models for Evaluating Targeted Therapies
The recent developments of large animal models are benefitting treatment strategies for both DMD and CF.
Genetic approaches to cure DMD include replacing the defective DMD gene, readthrough of translation stop codons, exon skipping to restore the reading frame, and increased expression of the utrophin (UTRN) gene, which may compensate the loss of dystrophin (reviewed in Fairclough, Wood, and Davies 2013).
Challenges for gene therapy of DMD include the large size of the DMD mRNA (14 kb) and the need to target all muscles. DMD mini- and micro-genes have been developed to overcome the size problem of full-length DMD complementary DNA (reviewed in Davies 2013). The most commonly used viral vectors to transduce muscle cells are based on AAV; however, DMD gene delivery by using this vector has resulted in immune responses against mini-dystrophin (reviewed in Davies 2013).
Readthrough strategies for nonsense mutations use small molecule drugs such as aminoglycosides or ataluren (PTC124) that introduce a conformational change in the mRNA and allow the ribosome to insert an amino acid at a premature stop codon site during translation. This approach has been estimated to be applicable in ∼13% of patients with Duchenne/Becker muscular dystrophy (Finkel 2010). Clinical studies of ataluren demonstrated dystrophin expression (Finkel et al. 2013) and a positive effect on the outcome of a 6-min walk distance (6MWD) test (Bushby et al. 2014). However, this study observed an unexpectedly large standard deviation of the 6MWD scores over 48 weeks, and the levels of dystrophin in muscle biopsies were difficult to interpret due to the poor sample quality.
Exon skipping is another strategy that could work for more than 80% of all DMD mutations, including most out-of-frame deletions (reviewed in Fairclough, Wood, and Davies 2013). The aim of this strategy is to restore an intact reading frame of the transcript. Skipping of specific exons can be induced by intramuscular or systemic treatment with RNaseH-independent antisense oligonucleotides (AONs), which hybridize to complementary sequences in or adjacent to the target exon. 2′-O-methyl-phosphorothioate AONs and morpholino phosphorodiamidate oligonucleotides have been tested in preclinical studies and clinical trials (reviewed in Fairclough, Wood, and Davies 2013) but have failed to show clear clinical benefit. A new class of AONs made of tricyclo-DNA (tcDNA) rescued dystrophin expression in skeletal muscles and heart and to a lower level in the central nervous system of mdx mice (Goyenvalle et al. 2015). Improvement of several clinical parameters was reported in tcDNA AON-treated mdx mice and also in double mutant mice which lack both dystrophin and UTRN and show a more severe phenotype than the mdx mice.
In spite of these promising results in dystrophic mouse models, it would be beneficial to test the efficacy of this new exon skipping strategy in a clinically severe large animal model before moving forward to clinical trials because a number of questions cannot be easily addressed in DMD patients. These include (1) the best timing to initiate AON therapy related to disease progression; (2) the amount of dystrophin required for near normal muscle function; (3) the optimal study duration, readouts, and outcome measures; (4) the best effective systemic administration route; and (5) the optimal dosage for a long-term therapy. Since our DMD exon 52-deficient pig model is amenable to correction by skipping of exon 51 or 53 and can now be provided by breeding in sufficient numbers for systematic studies, it appears to be ideally suited for testing this new promising approach of exon skipping. In addition, the DMD pig is useful to clarify efficacy and safety aspects of AAV-DMD mini-gene therapy, including potential immunological complications, and of readthrough treatment strategies or cellular therapies. In comparison with the existing canine DMD models, studies in DMD pigs may be ethically more acceptable.
The existing CF pig models are useful for optimizing gene therapy approaches, delivery, and safety. Gene therapy of CF using viral vectors (adenovirus [ADV], AAV2, and lentivirus [LV]) and nonviral vectors (reviewed in Griesenbach and Alton 2009; Prickett and Jain 2013) has not yet led to a clinically applicable therapy but have uncovered a number of problems limiting the efficacy of gene therapy for CF patients. These difficulties include challenges with the local delivery of gene therapy vectors into epithelial cells through a thickened mucus layer and immune reactions against the viral vectors. A recent clinical trial of repeated nebulization of nonviral CFTR gene therapy in CF patients revealed a significant, albeit modest, treatment effect with a stabilization of lung function (Alton et al. 2015). Large CF animal models with an airway and lung structure similar to CF patients will help improve vector design and delivery strategies. For example, Cao et al. (2013) demonstrated efficient transfer of LacZ reporter genes and human CFTR expression cassettes into airway epithelia and submucosal glands of normal pigs after intratracheal application of aerosolized helper-dependent ADV. In addition, intratracheal delivery of transfected airway epithelial cells has been suggested as treatment of CF, and proof of principle for efficient delivery of such cells has been shown in mice and wild-type pigs (Gui et al. 2015). It will be interesting to test these strategies in CF pigs, where gene or cell delivery may be more challenging because of the preexisting mucus and inflammation. CF pigs are also an interesting model for testing viral CFTR gene delivery via the celiac artery into the pancreas, a technique that has been recently established in wild-type piglets (Griffin et al. 2014).
The F508del-CFTR pig model can be used for evaluating combinations of CFTR correctors and potentiators. CFTR correctors, such as lumacaftor, reverse the folding defect of F508del-CFTR and increase its amount. CFTR potentiators, such as ivacaftor, increase the activity of the folding-corrected F508del-CFTR (reviewed in Cutting 2015).
Correction of Genetic Defects Using the CRISPR/Cas System
A recent major advancement is the use of genome editing tools like the CRISPR/Cas system. These systems are transforming the versatility, flexibility, and efficiency in the development of translatable animal models, including pigs (reviewed in Redel and Prather in press, this issue of Toxicologic Pathology), and offer great potential for treating diseases due to genetic defects. The concept of genome editing is based on the introduction by a programmable nuclease of a site-specific DNA double-strand break (DSB), which undergoes repair by different cellular repair mechanisms. One is nonhomologous end joining (NHEJ), which is induced by binding of KU heterodimers and associated repair proteins to the ends of DSB. The KU heterodimer is composed of the KU70 and KU80 subunits. Once the KU heterodimer is bound to the DSB ends, it recruits other NHEJ factors, including DNA-dependent protein kinase, catalytic subunit, X-ray cross-complementing protein 4 (XRCC4), DNA ligase IV, XRCC4-like factor, and aprataxin- and PNK-like factor, to process the DSB ends and facilitate their ligation (reviewed in Davis and Chen 2013). NHEJ is error prone, often leading to insertions or deletions, frameshift mutations, and gene knockouts. Alternatively, repair by HR can be directed by binding of RAD51 to DSB ends, followed by recruitment of accessory factors that direct HR with the matching sister chromatid or with homology regions of an exogenous repair template (Figure 3). The latter allows the introduction of precise genetic modifications or the correction of specific mutations (reviewed in Hsu, Lander, and Zhang 2014). The most widely used site-directed nucleases are ZFNs, TALENs, and the bacterial CRISPR/Cas system. The pros and cons of these systems have been subject to several recent reviews (Hsu, Lander, and Zhang 2014; Kim and Kim 2014).
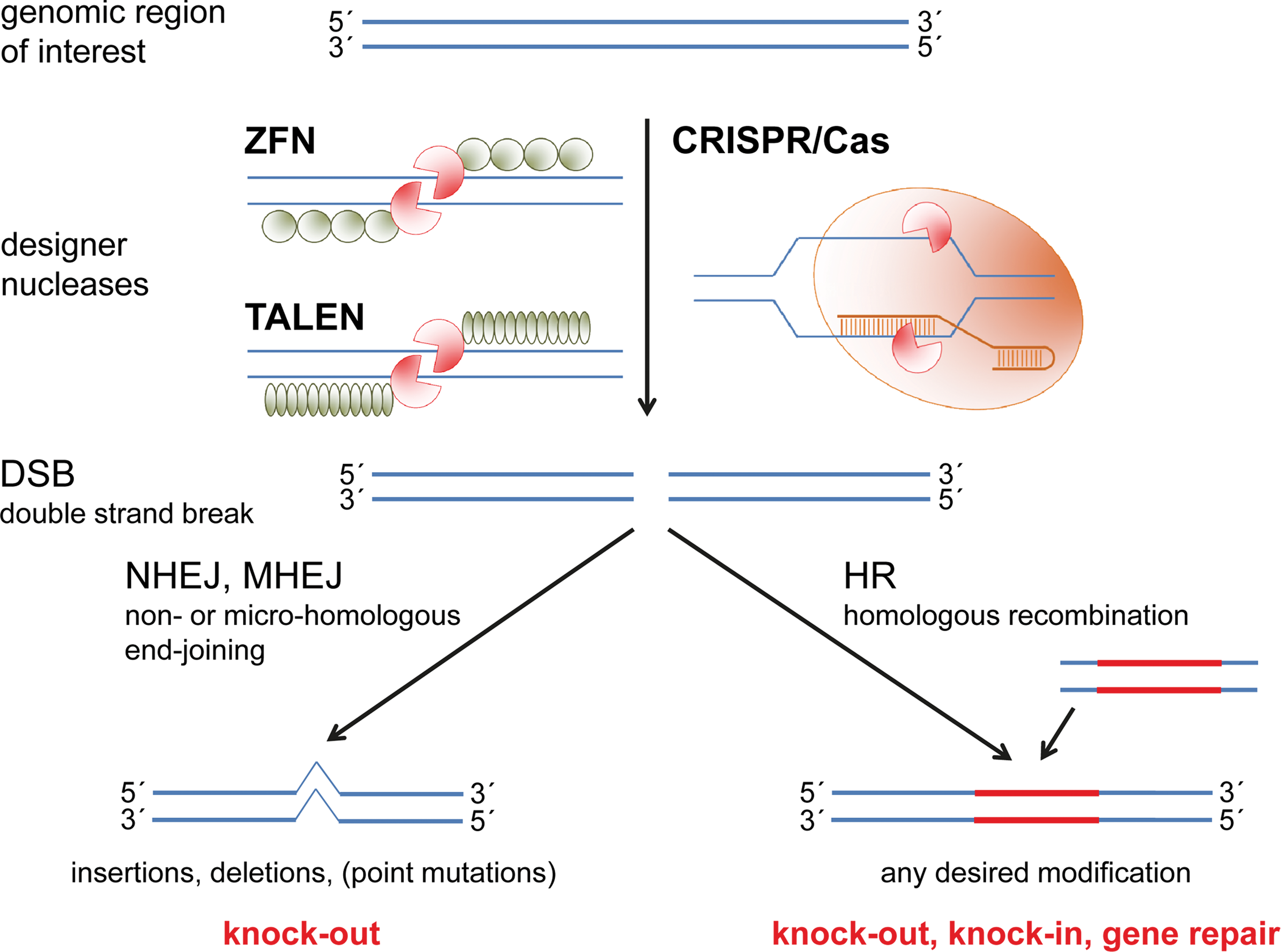
Principles of genome editing. Designer nucleases comprise a DNA-cutting domain as well as a unit that directs the DNA-cutting domain to a desired site in the genome. Three types of nucleases are regularly used: zinc finger nucleases and transcription activator-like effector nucleases are synthetic enzymes that both use FokI as a DNA-cutting domain, whereas the DNA-binding unit is either constituted of serially assembled zinc finger motifs or of DNA-binding domains of transcription activator-like effectors. As FokI induces only single-strand breaks, a pair of inverted nucleases is necessary to induce a double-strand break (DSB). In contrast, CRISPR/Cas9 and other RNA-guided endonucleases bind to their targets via a specific RNA element that is linked to a protein that directly introduces a DSB via its RuvC and HNH domains. Genomic modifications are introduced during DSB repair by nonhomologous end joining (NHEJ), micro-homologous end joining (MHEJ), or homologous recombination (HR). NHEJ fixes the DSB via blunt end ligation, whereas MHEJ uses micro-homologies of a few base pairs in size located around the DSB. In both processes, the exact kind of modification cannot be predicted but often insertions or deletions result in the knockout of the targeted gene. For introducing a defined mutation, HR with exogenous DNA that carries the desired modification as well as regions of homology to the target site has to occur. Exogenous DNA has to be cotransfected with the DSB-introducing nuclease and can be used for the generation of a knockout, a knockin, or the repair of a gene.
The CRISPR/Cas9 system was discovered when investigating the natural innate immune system of bacteria against virus infections (reviewed in Sander and Joung 2014). The breakthrough was in the recognition that the CRISPR/Cas9 system from Streptococcus pyogenes could be adapted for targeting and editing the genome precisely in any in vivo or ex vivo model (Deltcheva et al. 2011). The CRISPR/Cas system has attracted major attention and is now widely used since the site specificity of the Cas9 nuclease is provided by a single-guide RNA (sgRNA), which is easier and faster to engineer than ZFNs or TALENs where FokI endonuclease needs to be fused to specific DNA-binding proteins to generate programmable site-specific nucleases.
Several modifications of the CRISPR/Cas system have been developed, such as a new generation of Cas9, which increases the direction of DNA repair to HR and at the same time reduces the risk of off-target effects. A mutated variant of the enzyme, called nickase, is only capable of cutting 1 single DNA strand at a time (nick). By inducing 2 neighboring single-strand breaks, a DSB-like cut can be achieved and direct the concluding repair event to the HR pathway (reviewed by Sander and Joung 2014). In addition, the sgRNA-directed catalytically inactive or “dead” Cas9 can be fused to activation domains to mediate upregulation of specific endogenous genes (reviewed by Sander and Joung 2014). A potential application in DMD would be the targeted upregulation of UTRN to compensate for the loss of dystrophin.
Recently the CRISPR/Cas9 system has been used to correct the Dmd mutation in exon 23 of the mdx mouse (Long et al. 2014). The authors injected an sgRNA targeting Dmd exon 23, Cas9 mRNA, and a single-stranded oligodeoxynucleotide as template for HR-mediated gene repair into zygotes from mdx mice and transferred the embryos to pseudopregnant recipients. The resulting offspring turned out to be genetic mosaics with 2–100% Dmd alleles corrected by HR. In addition, restoration of an intact reading frame by NHEJ was observed. Strikingly, correction of less than 20% of the mutant Dmd alleles was sufficient to restore dystrophin expression in a majority of myofibers with a level comparable to that of wild-type mice. This was explained by the multinucleated structure of myofibers, where correction of the Dmd gene in a subset of nuclei may be sufficient to restore dystrophin in the entire myofiber. Fusion of corrected satellite cells with dystrophic fibers might also contribute to the restoration of dystrophin expression and to the regeneration of dystrophic muscle (Long et al. 2014).
Another study used CRISPR/Cas9 with single or multiplexed sgRNAs to restore the DMD reading frame by targeting the mutational hot spot at exons 45–55 and introducing shifts within exons or deleting one or more exons in cultured myoblasts from DMD patients. Dystrophin expression could be restored in vitro and remained stable after transplantation of the cells into tibialis anterior muscles of immunodeficient mice (Ousterout et al. 2015).
The promising results of these studies clearly open the perspective for CRISPR/Cas9-mediated correction of DMD mutations in postnatal muscle cells of Duchenne patients, if appropriate delivery systems for the components of the CRISPR/Cas9 system can be developed. AAV, in particular AAV9 serotype providing robust expression in skeletal muscle, heart, and brain, as well as injection of naked plasmid DNA, chemically modified mRNA, and nanoparticles containing nucleic acid have been discussed as potential solutions to meet this challenge (Long et al. 2014).
For CF, proof of concept for a functional repair of mutant CFTR by the CRISPR/Cas9 system was first demonstrated in intestinal stem cell organoids of CF patients (Schwank et al. 2013), and efficient vectors based on helper-dependent ADV (Cao et al. 2013) are available for testing this strategy in vivo. Due to the small size of rodents, concepts proven in these models may be difficult to scale up to a level where clinical feasibility can be demonstrated. In this respect, large animal models such as the DMD pig (Klymiuk et al. 2013) and CFTR mutant pigs will be critically important to evaluate efficacy and safety issues of the CRISPR/Cas9 system in vivo, especially for long-term use.
Risk Assessment for Genome Editing Technologies
The therapeutic use of genome editing technologies in vivo or ex vivo requires very specific safety considerations with regard to the used technology, the choice of the gene delivery system, and the most relevant preclinical species. For all areas, the toxicological pathologist can contribute to a successful design and development of a gene editing strategy and therapy.
Briefly, for all genome editing platforms, the specificity of targeting a gene is of major importance and off-target effects, that is, mutagenesis of nontarget sequences, must be reduced to a minimum. Compared to other gene therapeutic approaches, the introduction of the CRISPR/Cas9 system promises a huge step ahead toward a safer and more specific gene editing technology (Sander and Joung 2014). Recent progress in the design of the sgRNA, the engineering of the Cas9 enzyme, and a better understanding of the correlation between the concentration of the Cas9 protein and its on- or off-target activity leads further toward these goals (Corrigan-Curay et al. 2015). Off-target sites can be predicted using sophisticated bioinformatic tools such as PROGNOS (http://baolab.bme.gatech.edu/Research/BioinformaticTools/prognos.html) and off-target DSB can be identified in a genome-wide unbiased manner by sequencing-based methods, such as GUIDE-seq (Tsai et al. 2015). Despite these technical advances, evaluating off-target activity of any of these gene editing systems is a challenge in animal models. The differences between the animal and human genomes will result in off-target activity that likely will not accurately predict effects in man. Human in vitro systems using patient or volunteer-derived cells (e.g., induced pluripotent stem cells) are likely necessary in addition to animal models for gaining a full understanding of the potential for off-target gene editing effects preclinically.
The choice of the right delivery system for gene editing tools depends on the capacity, efficacy of delivery, targeting of specific tissues and cell types, and the possibility of species-specific adverse reactions. The 2 major classes of delivery systems, which have been entered into clinic trials, are based on viral or nonviral vector platforms (reviewed by Sheridan 2011). Common viral delivery systems are genome integrated retroviruses (RV) and LV as well as genome nonintegrated ADV, AAV, or modified herpes simplex virus (HSV; Hareendran et al. 2013; Thomas, Ehrhardt, and Kay 2003), and for most, except the HSV, the use of CRISPR/Cas9 has been successfully demonstrated (Schmidt and Grimm 2015). All systems need to overcome the host immune response that occurs at different levels. In principle, all viral vectors have the potential to be recognized as an infection and therefore stimulate the immunological defense system by recognition of viral gene products, capsid proteins, foreign components of the gene editing tool package, like the Cas9, or foreign transgene products (Thomas, Ehrhardt, and Kay 2003).
The toxicological pathologist should be prepared to face a spectrum of morphological changes, which can range from local, tissue-limited, inflammatory reactions to massive systemic, cytokine-driven inflammation, eventually resulting in disseminated intravascular coagulation, multiorgan failure, and death. Furthermore, for ADV, a dose relationship has been described between virus dose and cell toxicity and is characterized by a threshold-like immune response (Thomas, Ehrhardt, and Kay 2003).
LV and AAV vectors have become more attractive since they seem to avoid initial immune responses, but still the transgene product can elicit such an effect (Hareendran et al. 2013). To limit the drawbacks of other virus vehicle systems, engineered AAV vectors like recombinant AAV serotype variants have become a focus of intensive research due to their nonpathogenic character, lower immunogenicity profile, lower oncogenic risk for insertional mutagenesis, and specific tissue tropism compared to other vectors (Hareendran et al. 2013; Masat, Pavani, and Mingozzi 2013). In preclinical studies, AAV vectors demonstrated favorable characteristics, targeting a broad variety of tissues and providing a stable transgene expression and a low immunogenicity (Asokan, Schaffer, and Samulski 2012). The integration of virus genome, which is the case for LV and RV transduction, may result in an increased risk for genotoxicity and insertional mutagenesis by stimulation of oncogenes or inhibition of tumor suppressor genes with an increased risk for tumorigenesis (Schmidt and Grimm 2015).
A significant spectrum of nonviral gene delivery methods including naked plasmids, polymers, liposomes, peptides, and inorganic particles have been explored both preclinically and clinically (reviewed by Yin et al. 2014). Lipid nanoparticles (LNPs) have been investigated for delivery of different DNA and RNA species (microRNA and small interfering RNA) as well as plasmid vectors. LNPs have a lower rate of efficacy compared to viral vectors and an increased risk of stimulation of the immune system and inducing inflammation, limiting the spectrum of recommended tissue for genome targeting approaches (Yin et al. 2014). As discussed subsequently, for evaluating the safety of these delivery methods, the toxicologic pathologist must consider the spectrum of safety relevant end points appropriate for the gene delivery method and preclinical safety plans have to be adapted.
In general, the therapeutic use of genome editing technologies in vivo or ex vivo requires very specific considerations with regard to preclinical risk assessment. Regulatory guidance is available (U.S. Department of Health and Human Services, Food and Drug Administration 2013), but this field is developing rapidly and close communication with regulators will enable a fit-for-purpose perspective and approach, both critical for effective and efficient clinical development. The industry guidance was developed by the Center for Biologics Evaluation and Research and supports preclinical design for investigational cellular therapies, gene therapies, therapeutic vaccines, xenotransplantation, and biologic-device combination products. For the purpose of this summary, we will focus on discussions most germane to the toxicologic pathologist as a principal investigator.
One of the most important considerations for the preclinical development program is the selection of the animal model for safety assessment. When evaluating safety it is critical that the animal demonstrates a biological response to the test product and that there is sufficient comparability of anatomy and physiology to humans. Compared to the rodent, pig anatomy and physiology are more similar to humans in regard to the cardiovascular, immune, respiratory, skeletal muscle, and metabolic systems. The same is true for the organization and sequence homology of the genome. Gene editing methodologies allow the design of pig models that may reflect the clinical disease better than rodents, as exemplified by the DMD and CF pig models discussed in this review. The pig also affords flexibility in the development of delivery systems/procedures that closely imitate the approach for the clinic. Therefore, innovative models based on genetically engineered pigs are an area of rapid growth in the safety assessment of gene therapy products. The toxicologic pathologist should be a key partner for summarizing the rationale of a specific animal model in the preclinical section of the investigative new drug (IND) application or similar regulatory document.
As implied earlier in this review, the use of animal models that approximate the human disease may better define the risk–benefit ratio associated with a gene therapy product. Challenges with these disease models include technical limitations and inherent variability, further complicated by the limited historical safety data and the fact that these models are often not performed in good laboratory practices (GLP)-compliant facilities. The regulators afford some flexibility for GLP in these situations; therefore, the lack of a GLP-compliant facility is not an insurmountable hurdle. While historical safety data may not be available for the disease model, the use of pretest data for clinical pathology parameters and the information available for the founder animals can overcome some of this risk. The toxicologic pathologist should take a leadership role in guiding the team on the inherent limitations of any model and put forward a tiered, balanced recommendation and risk mitigation plan for the project.
Once the animal model(s) is/are selected, the design of the studies must develop a data set that allows for the safe conduct of clinical proof-of-concept studies. Ideally from the animal model(s), one has a rigorous data set that highlights the projected pharmacologically effective dose range and optimal dosing schedule as well as a thorough understanding of the tissue distribution of the gene therapy product and the potential biological activities, both wanted and unwanted. Taken together, these data are used for the IND filing or similar and will inform the clinical investigator and patient on the projected risks and benefits of the therapy. Safety end points are included for characterization of the potential toxicities associated with the pharmacological activity associated with the gene therapy product and potential effects of the delivery vehicle, transporter system, or the gene editing tool and its constituents. For the toxicologic pathologist, the parameters evaluated are very similar to those for a program evaluating the safety of a small molecule or biologic and include clinical pathology, organ weights, and gross and histopathology. Specialized histopathology such as immunohistochemistry and tissue microarrays in addition to molecular tools like deep sequencing may be a useful adjunct for the quantification of gene product expression in different tissues (both target and nontarget). Also because of the inherent risk for immunologic responses to the transporter system or key constituents of the gene editing tool, advanced methods for examination of immunopathologic effects may be warranted. These could include cytokine analysis, flow cytometry, or other immunotoxicologic measures of innate and adaptive immunity.
There are a few unique characteristics for the design of a preclinical safety package for gene therapy products. First, the normal paradigm of studies progressing from shorter term (2 weeks–1 month) to longer (up to 6 month–1 year) in a rodent and a nonrodent model may not be appropriate. However, the understanding of the chronic effects of the gene engineering product (or delivery system/transporter system/gene editing tool) may require longer-term studies. In fact, because integrating viral vectors or stem cells may be key constituents of a gene therapy product, studies assessing the carcinogenicity potential often occur earlier in a development program than would be the case for a small molecule or biologic. Also it is possible that the species specificity of the gene therapy product would justify a rationale for only using 1 species (rodent or nonrodent) or alternatively a translatable animal disease model for preclinical safety studies. In any case, close interaction between the toxicologic pathologist and the regulatory agencies is key in the planning of these studies. Generally, dosing (number and frequency) in animal models mimics the clinical approach. Therefore, the number of doses for a gene therapy product may be limited no matter what the observation period is. Notably, the limited number of doses and the lower milligram per kilogram requirements for gene therapy products may result in less of a preclinical pharmaceutical development hurdle for these studies as compared to preclinical studies for a small molecule or biologic in larger species like the pig.
Conclusions and Perspectives
Tailored large animal models, especially pigs, have the potential to bridge the gap between proof-of-concept efficacy and safety studies in rodent models and clinical trials. Due to major advancements in technologies for genetic engineering of pigs, there are models for numerous disease areas, including cancer (reviewed by Flisikowska, Kind, and Schnieke 2013), metabolic diseases (reviewed by Wolf et al. 2014), and neurodegenerative diseases (reviewed by Dolezalova et al. 2014). As a next step, standardized phenotyping protocols such as those described by Albl et al. (in press) in this issue of Toxicologic Pathology will be required to fully exploit the translational potential of these novel models. These should be developed in close collaboration between academic partners and experts from the pharmaceutical industry including toxicologic pathologists and clinicians to ensure the translational value of the acquired data. Because the pig is accepted as an appropriate large animal species for safety assessment, combined safety/efficacy studies can be designed for the support of clinical trials. The growing body of knowledge and experience with genetically engineered large animal models will increase their acceptance by funding agencies, industry, and regulatory authorities as an important new element in drug development.
Footnotes
Authors’ Contributions
NK, FS, DR, and EW wrote the manuscript with contributions from all other authors. AB, NK, FS, DR, and EW contributed illustrations. MB and DR reviewed and edited the manuscript. All authors read and approved the final manuscript.
Authors’ Note
Nikolai Klymiuk and Frank Seeliger contributed equally to this work. Daniel G. Rudmann and Eckhard Wolf are senior authors who contributed equally to this study.
Declaration of Conflicting Interests
The author(s) declared no conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Projects at LMU Munich involving the generation of genetically engineered pigs are/were financially supported by the German Research Council (FOR 535 “Xenotransplantation,” FOR 793 “Mechanisms of Fracture Healing in Osteoporosis,” and Transregio-CRC 127 “Biology of xenogeneic cell, tissue and organ transplantation—from bench to bedside”), by the Federal Ministry for Education and Research (Leading-Edge Cluster “m4—Personalised Medicine and Targeted Therapies”), the Bavarian Research Council (FORZebRA, Az. 802-08), and by the Mukoviszidose Institut gemeinnützige Gesellschaft für Forschung und Therapieentwicklung mbH. The authors are members of COST Action BM1308 “Sharing Advances on Large Animal Models—SALAAM.”