
Abstract
Understanding genetic causes of hearing loss can determine the pattern and course of a patient’s hearing loss and may also predict outcomes after cochlear implantation. Our goal in this study was to evaluate genetic causes of hearing loss in a large cohort of adults and children with cochlear implants. We performed comprehensive genetic testing on all patients undergoing cochlear implantation. Of the 459 patients included in the study, 128 (28%) had positive genetic testing. In total, 44 genes were identified as causative. The top 5 genes implicated were GJB2 (20, 16%), TMPRSS3 (13, 10%), SLC26A4 (10, 8%), MYO7A (9, 7%), and MT-RNR1 (7, 5%). Pediatric patients had a higher diagnostic rate. This study lays the groundwork for future studies evaluating the relationship between genetic variation and cochlear implant performance.
Cochlear implantation is an effective form of hearing restoration that improves quality of life and ameliorates the associated economic, social, emotional, and neurocognitive consequences of severe-to-profound hearing loss.1,2 Some patients, however, do not obtain the expected benefit from implantation.3,4 A growing body of literature has been dedicated to identifying factors that may affect cochlear implant (CI) postoperative speech outcomes, including social, clinical, and genetic factors.5,6 We have previously shown that CI recipients with genetic mutations affecting the spiral ganglion performed lower on speech perception tests than patients with mutations affecting the organ of Corti. 7
Additional research on CI genetics is needed to better understand this relationship. One knowledge gap includes the epidemiology of genetic mutations among CI recipients. While some studies have offered epidemiological data, these have been limited to smaller populations.6,8,9 The purpose of this study is to provide an epidemiological survey of causative genetic variants in a large cohort of adults and children with CIs to spur research focused on the relationship between CI outcomes and genetics.
Methods
Study approval was obtained from the University of Iowa Institutional Review Board. A prospective study was conducted whereby all patients undergoing implantation at our institution were evaluated between 2009 and 2018 for variants in genes associated with nonsyndromic hearing loss, as well as genes implicated in common syndromes using the OtoSCOPE panel, as previously described. 10 The current version of this panel sequences 158 deafness-associated genes, but previous versions included 66 to 133 genes. Clinical data included demographics, pattern of hearing loss, age at implant, and laterality. Epidemiological data were calculated for the overall population. Subgroup analyses were performed for children implanted age 18 years and younger, adults implanted over 18 years, and patients who underwent hearing-preservation implantation with short-electrode hybrid CIs for preservation of low-frequency hearing. Data on 155 patients were used in a previous study and are included here to provide a larger sample size. 7
Results
In total, 459 CI recipients (224 females; 100 children) underwent genetic testing ( Table 1 ). Ninety-eight percent were non-Hispanic white, and 128 tested positive for a diagnostic rate of 28% (see Suppl. Figure S1 in the online version of the article). The top genes implicated were GJB2 (20 patients, 16%), TMPRSS3 (13, 10%), SLC26A4 (10, 8%), MYO7A (9, 7%), and MT-RNR1 (7, 5%) ( Figure 1A ). Of 100 pediatric patients, causative mutations were identified in 48 (48%), affecting 17 genes. The most common genes were GJB2 (17 patients, 36%), SLC26A4 (6, 13%), MYO15A (4, 8%), and MYO7A (4, 8%) ( Figure 1B ). Of 359 adult patients, causative mutations were identified in 80 (22%), affecting 35 different genes. The most common genes were TMPRSS3 (13 patients, 16%), POU4F3 (6, 8%), MT-RNR1 (6, 8%), and MYO7A (5, 6%) ( Figure 1C ). Among 85 hearing-preservation patients (75 adults), causative mutations were identified in 26 (31%), affecting 15 genes. The most common mutations were TMPRSS3 (6 patients, 23%), TMC1 (3, 12%), POU4F3 (3, 12%), MYO15A (2, 8%), and MYO6 (2, 8%) ( Figure 1D ).
Study Population Characteristics for Overall Population, Pediatric Patients, and Adult Patients. a
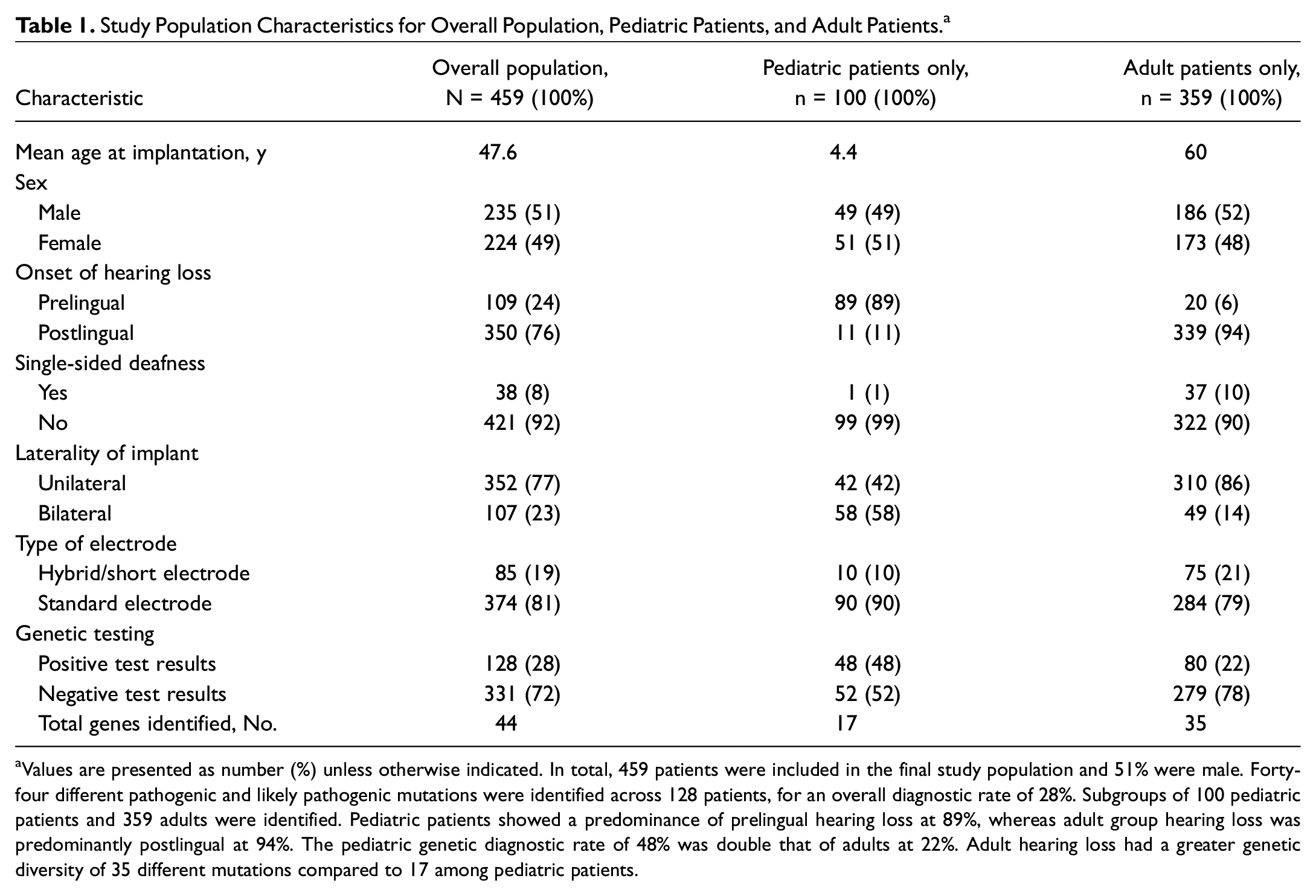
Values are presented as number (%) unless otherwise indicated. In total, 459 patients were included in the final study population and 51% were male. Forty-four different pathogenic and likely pathogenic mutations were identified across 128 patients, for an overall diagnostic rate of 28%. Subgroups of 100 pediatric patients and 359 adults were identified. Pediatric patients showed a predominance of prelingual hearing loss at 89%, whereas adult group hearing loss was predominantly postlingual at 94%. The pediatric genetic diagnostic rate of 48% was double that of adults at 22%. Adult hearing loss had a greater genetic diversity of 35 different mutations compared to 17 among pediatric patients.
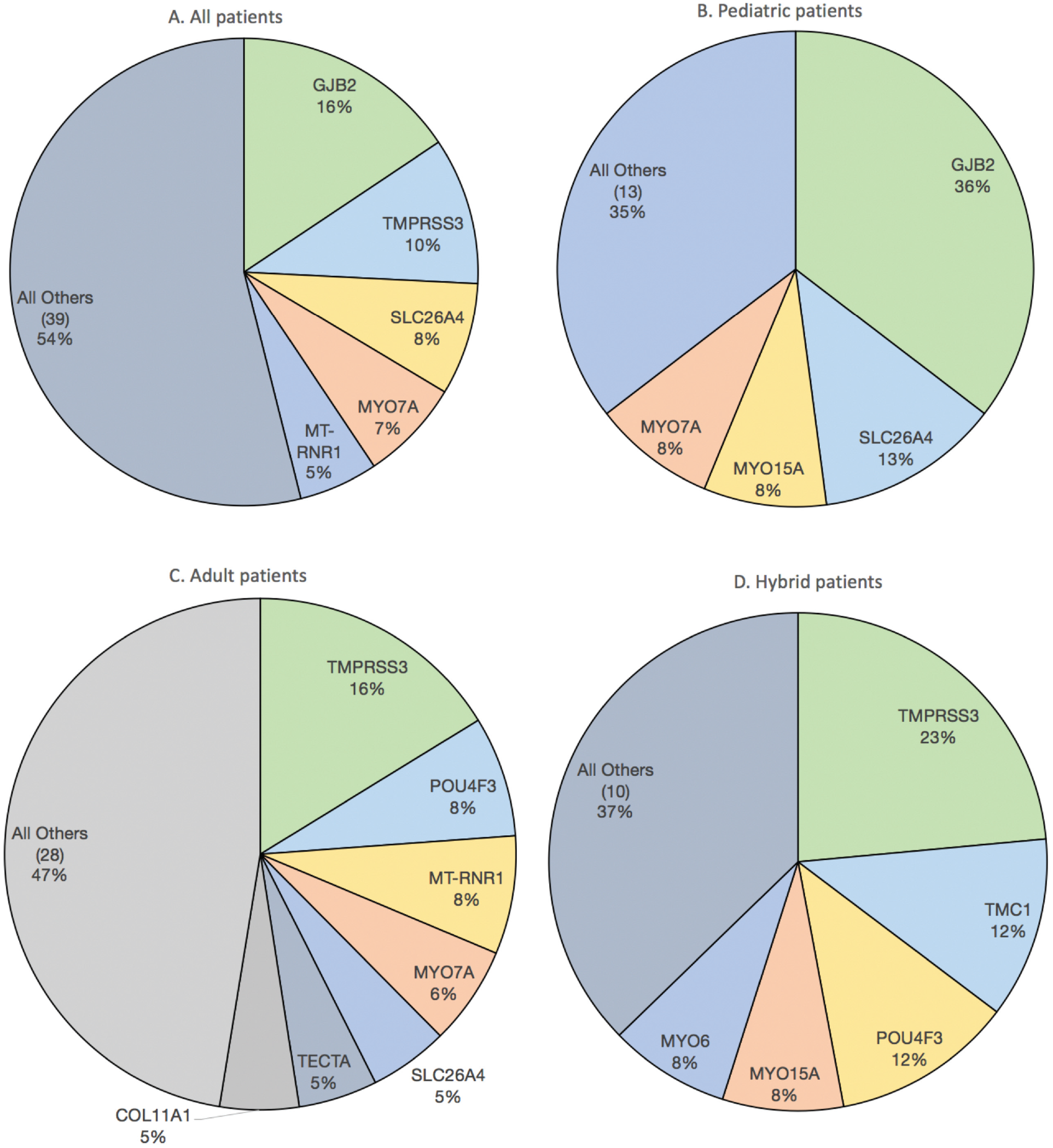
Distribution of mutations among (A) all patients with positive genetic testing (n = 128), (B) children with positive testing (n = 48), (C) adults with positive testing (n = 80), and (D) hybrid cochlear implant patients with positive testing (n = 26).
Discussion
Several studies have published epidemiological data for CI patients, but these have been limited in scope or population size.5,6,9,11 To our knowledge, our data set of 459 CI recipients is the largest to include comprehensive genetic testing. The difference in diagnostic rate between adults and children is expected and reflects our greater understanding of genetic contributors to congenital and childhood hearing loss. Further research is needed to elucidate the genetic contributors to later-onset hearing loss.
Our study population is most similar to Miyagawa et al, 8 who performed comprehensive testing on 173 consecutive CI recipients. In total, 59.8% of prelingual hearing loss patients and 35.8% of postlingual patients tested positive. Although their prelingual diagnostic rate is higher than our rate of 48% among our predominantly prelingual pediatric group, this difference may be attributed to a variety of factors, including increased genetic testing among members of the same family in the Miyagawa et al 8 study as well as differing ethnicities and limited sample sizes in both study populations. We similarly found a lower rate of 22% among our predominantly postlingual adult group.
The primary weakness of this study is its limitation to a single tertiary institution with a predominantly non-Hispanic white population. It is clear that genetic contribution to hearing loss varies significantly based on ethnicity. 12 Evaluation of genetics in CI patients from varying ethnicities is an area of future research. In addition, this study is epidemiological and does not address postimplantation speech perception outcomes, which is of interest to CI surgeons and the greater scientific community. 13 CI performance is influenced by a complicated array of factors, such as residual hearing, duration of hearing loss, age at onset of hearing loss, type of implant, and socioeconomic status.3,14 Further study of CI outcomes will be required to clarify the relationship between genetics and CI speech perception outcomes. Finally, 96 patients underwent testing with the most current OtoSCOPE panel of 158 genes; the remainder underwent testing with earlier panels. We have found only a small increase in diagnostic rate as more genes are added to the panel (unpublished data).
As we have shown, classification of genetic variants is difficult and nuanced, and reclassifying variants of uncertain significance as benign or pathogenic requires complex decision making based on clinical information. 15 However, comprehensive genetic testing for hearing loss provides valuable information to patients and clinicians and has become standard of care. 16 The data presented here provide insights into the genetic contribution to hearing loss in a large population of CI users. These data lay the groundwork for future research into the complex interaction between genes and environment that underlies CI outcomes.
Supplemental Material
sj-pdf-1-oto-10.1177_01945998211021308 – Supplemental material for Genetic Causes of Hearing Loss in a Large Cohort of Cochlear Implant Recipients
Supplemental material, sj-pdf-1-oto-10.1177_01945998211021308 for Genetic Causes of Hearing Loss in a Large Cohort of Cochlear Implant Recipients by Kristen L. Seligman, A. Eliot Shearer, Kathy Frees, Carla Nishimura, Diana Kolbe, Camille Dunn, Marlan R. Hansen, Bruce J. Gantz and Richard J. H. Smith in Otolaryngology–Head and Neck Surgery
Footnotes
This article was presented virtually at the AAO-HNSF 2020 Virtual Annual Meeting & OTO Experience, September 13 to October 25, 2020.
Author Contributions
Disclosures
Supplemental Material
Additional supporting information is available in the online version of the article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.