
Abstract
Cells grown in monocultures are widely used to model lung tissue. As a result of these culture conditions, these cells exhibit poor morphological similarity to those present in in vivo lung tissue. MucilAir™, a 3-D in vitro model comprising human basal, goblet and ciliated cells, represents a fully differentiated respiratory epithelium that can be used as an alternative and a more realistic system. The aim of our study was to compare the effects of short-term and long-term exposure to two polycyclic aromatic hydrocarbons (PAHs) — benzo[a]pyrene (B[a]P) and 3-nitrobenzanthrone (3-NBA) — using MucilAir as a model of human lung tissue. Two concentrations (0.1 μM and 1 μM) were tested at three time points (24 hours, 7 days and 28 days). Several aspects were assessed: cytotoxicity (lactate dehydrogenase (LDH) release), integrity of the cell layer (transepithelial electrical resistance (TEER)), induction of oxidative stress (reactive oxygen species production) and changes in the expression of selected genes involved in PAH metabolism (CYP1A1 and AKR1C2) and the antioxidant response (ALDH3A1, SOD1, SOD2, GPX1, CAT, HMOX1 and TXNRD1). The results showed that exposure to B[a]P caused a spike in LDH release at day 5. Exposure to 3-NBA caused a number of spikes in LDH release, starting at day 5, and a decrease in TEER after 11 days. CYP1A1 gene expression was upregulated after the 7-day and 28-day B[a]P exposures, as well as after the 24-hour and 7-day 3-NBA exposures. HMOX1 and SOD1 were downregulated after both 24-hour PAH treatments. HMOX1 was upregulated after a 1-week exposure to 3-NBA. There were no significant changes in the messenger RNA (mRNA) levels of AKR1C2, ALDH3A1, TXNRD1, SOD2, GPX1 or CAT. These results illustrate the potential use of this 3-D in vitro lung tissue model in studying the effects of chronic exposure to PAHs.
Keywords
Introduction
The toxicology of air pollutants has relied on in vivo and in vitro testing for decades. Although toxicological tests can be carried out in vitro on a wide range of cells, it is accepted that the complete response of the organism to a substance is only obtained when laboratory animals are used. As Flecknell 1 summarised, there is a high demand to find and implement an approach that encompasses the Three Rs principles (replacement, reduction and refinement). In inhalation toxicology, the need for non-animal alternatives gave rise to several types of complex epithelial models. The coculture models represent improved monocultures, grown under submerged conditions, in which different types of cells are used to simulate cell–cell interactions. Such models are usually a combination of target cells — for example, epithelial cells — and immune cells such as macrophages. 2,3 In view of the potential cellular interactions, coculture models can certainly provide more accurate information about the effects of chemical exposure, but the role of cell positioning and communication in the 3-D space is still not addressed in these models. In addition, the model lifespan is usually rather short, that is, days or weeks. Several companies have developed airway models that combine a 3-D tissue culture approach with the use of primary cells. 4 MucilAir™, developed by Epithelix Sárl (Geneva, Switzerland), is a 3-D in vitro model consisting of human basal, goblet and ciliated cells, cultured at an air–liquid interface (ALI). It represents a fully differentiated and functional respiratory epithelium and one which does not need trypsinisation as part of its preparation. 5,6 This model displays in vivo characteristics, including stratification, tight junctions, metabolic activity, mucus production and beating cilia. Each MucilAir insert consists of approximately 0.5 × 106 cells attached to the porous membrane and fed basolaterally. The lifespan of the inserts is up to 1 year after seeding, thus providing an opportunity to perform long-term exposures without the artificial selection and expansion of particular clones with certain properties that can incidentally occur as a result of successive passaging. 5
Although the use of 3-D cell models is promising, there is still minimal information available in the peer-reviewed literature comparing the short-term and long-term exposure of such models to air pollutants. As described previously, 7 –10 3-D cell models are generally more resistant to test compound exposure (e.g. can withstand higher doses and longer exposure) than are cell monolayers. Also, their biological response to air pollutants (e.g. cytotoxicity, production of cytokines, genotoxicity) is generally weaker when compared to widely used cell lines, such as A549 (human alveolar epithelial cells) or BEAS-2B (human bronchial epithelial cell line). One of the most common explanations given for the weaker response in the 3-D model is that the functional epithelium barrier protects lower layers of cells from apical exposure. 11
The current study addresses the lack of long-term studies (i.e. of several weeks’ duration) with 3-D cell models, comparing short-term (24-hour) and long-term (7-day and 28-day) exposures to polycyclic aromatic hydrocarbon (PAH) air pollutants. We chose two PAH representatives: benzo[a]pyrene (B[a]P), a human carcinogen 12 that is formed mainly by the incomplete combustion of fossil fuels, and 3-nitrobenzanthrone (3-NBA), a component of diesel exhaust and possible human carcinogen. 13 The PAH test concentrations used were based on the results of our previous study with standard monocultures. 14 The exposure time was extended to reflect differences between the cell models. Our aim was to characterise cytotoxicity, integrity of the cell layer, induction of oxidative stress (reactive oxygen species (ROS) production) and changes in the expression of selected genes involved in PAH metabolism (cytochrome P450 1A1 (CYP1A1) and aldo-keto reductase 1C2 (AKR1C2)) and the antioxidant response (aldehyde dehydrogenase 3A1 (ALDH3A1), superoxide dismutase (SOD1 and SOD2), glutathione peroxidase 1 (GPX1), catalase (CAT), haem oxygenase 1 (HMOX1) and thioredoxin reductase 1 (TXNRD1)) after exposure to the PAHs.
Materials and Methods
MucilAir primary cultures
MucilAir (Epithelix Sárl), a fully differentiated bronchial epithelial model reconstituted from primary human cells from healthy donors, was used for all the PAH exposures. The primary human cells used to construct the model were obtained from a 65-year-old Caucasian male, non-smoker and with no known pathology. A total of 45 inserts were divided into three main groups (for 24-hour, 7-day and 28-day exposure periods), with each group comprising 15 inserts. Two concentrations of B[a]P and 3-NBA (0.1 μM and 1 μM) were tested, as well as a negative control (0.1% v/v dimethyl sulphoxide (DMSO)), each in triplicate. The samples were maintained in MucilAir culture medium (Epithelix Sárl) in 24-well format Transwell® cell culture inserts (Sigma-Aldrich, St Louis, MO, USA) in a humidified incubator (37°C; 5% v/v CO2). Prior to the experiments, the samples were monitored in culture for 3 weeks to determine their stability. During this period, the culture medium was changed every 2–3 days, and an apical wash was performed every week to eliminate the accumulated mucus. Any changes in the morphology of the cells, specifically with regard to protrusions on the MucilAir surface or perforations in its structure, were assessed under a light microscope (CKX41; 200× magnification), as was the presence of beating cilia.
Exposure scheme
To compare the effects of short-term (24-hour) and long-term (7-day and 28-day) PAH exposures, commercially available B[a]P (Merck, St Louis, Missouri, USA) and 3-NBA (Chiron AS, Trondheim, Norway) were used. Based on our previous short-term tests on two commonly used cell lines (human adenocarcinoma alveolar basal epithelial cells, type II (A549) and human embryonic lung fibroblasts (HEL12469)), we decided to use two concentrations of B[a]P and 3-NBA (0.1 μM and 1 μM) and 0.1% DMSO as a negative control. 14 Prior to every experiment, an aliquot of PAH stock (10 mM in DMSO) was diluted in fresh culture medium for the MucilAir exposure.
First, we added 200 μl of pre-warmed MucilAir culture medium to the apical side of each MucilAir insert and measured the transepithelial electrical resistance (TEER). Then, the basal medium was changed, collected and stored at −20°C for further testing. The model was exposed to the B[a]P, 3-NBA or negative control via the apical side of the insert (Figure 1). Depending on the experiment, the inserts were harvested or incubated until the next time point.
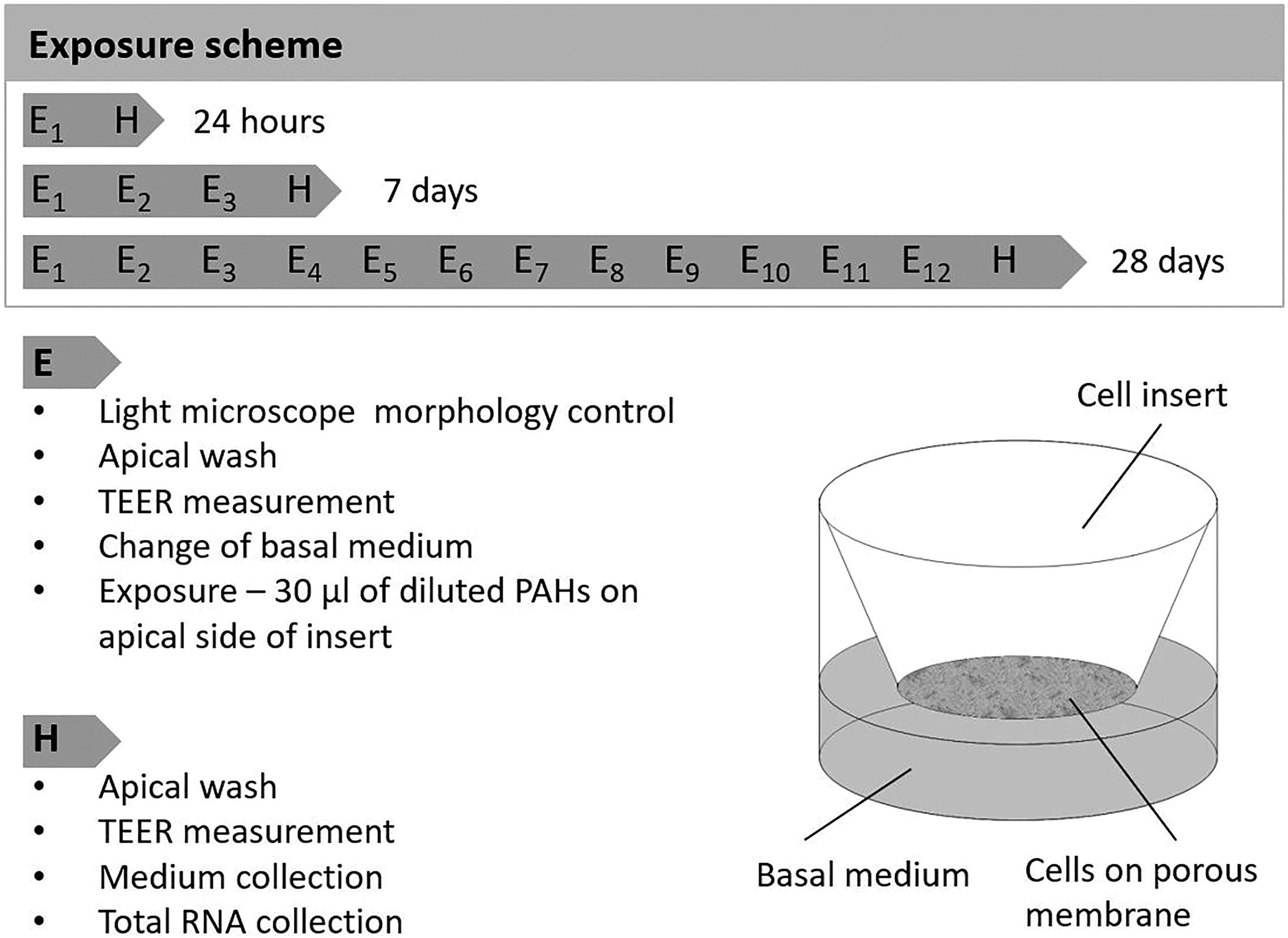
The MucilAir exposure scheme. Each MucilAir insert was washed with 200 μl of pre-warmed medium, the TEER was measured and the basal medium was changed. Exposure to B[a]P (0.1 μM and 1 μM), 3-NBA (0.1 μM and 1 μM), and the negative control (0.1% v/v DMSO), was carried out via the apical side of the inserts (E = exposure). The inserts with the test chemicals were either incubated until the next exposure or the cells were harvested (H = harvest). TEER: transepithelial electrical resistance; B[a]P: benzo[a]pyrene; 3-NBA: 3-nitrobenzanthrone.
Determination of TEER, cytotoxicity and ROS production
TEER measurements
TEER measurements were taken with an EVOM2 ohm meter (World Precision Instruments, Sarasota, FL, USA) in combination with an STX2 electrode. This method represents a quantitative non-destructive means of measuring the integrity of tight junction dynamics in cell culture models. In view of the ongoing culture of the cells, all TEER measurements were obtained under aseptic conditions. Prior to taking the measurements, 200 μl of preheated MucilAir medium was added to the apical side of the inserts, and the electrode was equilibrated by washing in sterile phosphate-buffered saline (PBS) and then in preheated medium before every use. Triplicate TEER measurements were taken for each insert, and the electrode was washed in sterile PBS and 70% v/v ethanol between each insert. TEER measurements were taken prior to every exposure to the test chemicals. Resistance values were calculated according to the formula: TEER (Ω*cm2) = (resistance of the test tissue (Ω) − resistance value of the untreated membrane (Ω)) × surface area of the epithelium (cm2), where the resistance value of the untreated membrane is 100 Ω and the surface of the epithelium is 0.33 cm2.
Cytotoxicity determination
Cytotoxicity was determined by means of the spectrophotometric quantification (SpectraMax® M5e; Molecular Devices, San Jose, CA, USA) of lactate dehydrogenase (LDH) activity in the harvested supernatants (the result of damaged cell leakage). The Roche Cytotoxicity Detection Kit (Roche, Basel, Switzerland) was used for this procedure, according to the manufacturer’s instructions. The results are presented as the percentage cytotoxicity relative to the value obtained from the positive (cytotoxic) control (a 1-hour exposure of the model to 1% v/v Triton X-100 in culture medium at 37°C).
ROS production
As a non-destructive method of measuring oxidative damage, thus allowing long-term observation of the same sample, extracellular ROS levels were measured in the harvested culture medium using Acridan Lumigen PS-3 Assay (Pierce™ ECL Plus Western Blotting Substrate; Pierce Biotechnology, Rockford, Illinois, USA), as described previously. 15 For the analysis, 50 μl of Acridan Lumigen PS-3 assay solution was mixed with 100 μl of the harvested medium and incubated in the dark for 5 minutes. Chemiluminescence was recorded with a SpectraMax® M5e spectrophotometer (integration time 500 ms). For the purposes of the ROS assay, MucilAir exposed to 250 mM hydrogen peroxide (H2O2) in the culture medium, for 1 hour at 37°C, was used as the positive control. As outlined in Uy et al. 15 all samples (including the positive control) were collected and stored at −20°C prior to subsequent analysis.
Total RNA isolation and quality control
Total RNA from lysed MucilAir cells was isolated using TRIzol® (Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer’s protocol. Briefly, the inserts were washed with PBS containing Ca2+/Mg2+ and the well plate was placed on ice. A 150-μl aliquot of TRIzol was added to the apical side of each insert, the lysed cells scraped into 1.5 ml tubes and the tubes incubated on ice for 15 minutes. Then, 30 μl of chloroform was added to each tube and, after incubation on ice for 3 minutes, the homogenate was centrifuged (12,000 g, 15 minutes, 4°C). The upper aqueous phase containing total RNA was purified by extraction with phenol/chloroform/isoamyl alcohol (25:24:1), precipitated with 3 M sodium acetate/ethanol and then finally resuspended in 20 μl of RNase-free water.
RNA concentration was quantified with a Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific). The integrity of the RNA was checked with an Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, CA, USA). The threshold for RNA integrity was met by all samples (RNA Integrity Number > 8). Isolated RNA was stored at −80°C until further use.
mRNA expression analysis
For the reverse transcription, 1 μg RNA was used for complementary DNA (cDNA) synthesis using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche). The manufacturer’s protocol was modified to include the use of 2.5-μM oligo(dT) primers (18-mers) and 10-μM random hexamers. The RNA samples were mixed with the primers and incubated for 10 minutes at 65°C in a Mastercycler (Eppendorf, Hamburg, Germany). Then, they were placed on ice, and 1× reaction buffer, 20 U Protector RNase inhibitor, 1 mM dNTP mix and 5 mM THF reverse transcriptase were added. The total reaction volume was 20 μl. The reaction mixture was pre-incubated for 10 minutes at 25°C, followed by incubation for 30 minutes at 55°C and for 5 minutes at 85°C.
Real-time quantitative PCR (RT-qPCR) measurements were performed with a LightCycler 480 (Roche). Each RT-qPCR reaction was carried out in a final volume of 14 μl (0.7 μl primers, 7 μl master mix, 3.8 μl sterile water, 25 ng cDNA diluted in 2.5 μl sterile water). A custom-designed RT-qPCR assay with Double-Dye probe (PrimerDesign, Eastleigh, UK) was used. The cycling conditions were 2 minutes at 95°C, followed by 40 cycles of amplification (10 seconds at 95°C and 60 seconds at 60°C). Cycle threshold (Ct) values were generated by GenEx software (MultiD AB, Goteborg, Sweden). The expression levels of the target genes were normalised to the reference genes tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta (YWHAZ) and eukaryotic translation initiation factor 4A2 (EIF4A2). Relative changes in normalised gene levels were calculated using the 2−▵▵Ct method. 16 The sequences of the primers used in the RT-qPCR are presented in Table 1.
Sequences of the primers used for the RT-qPCR.
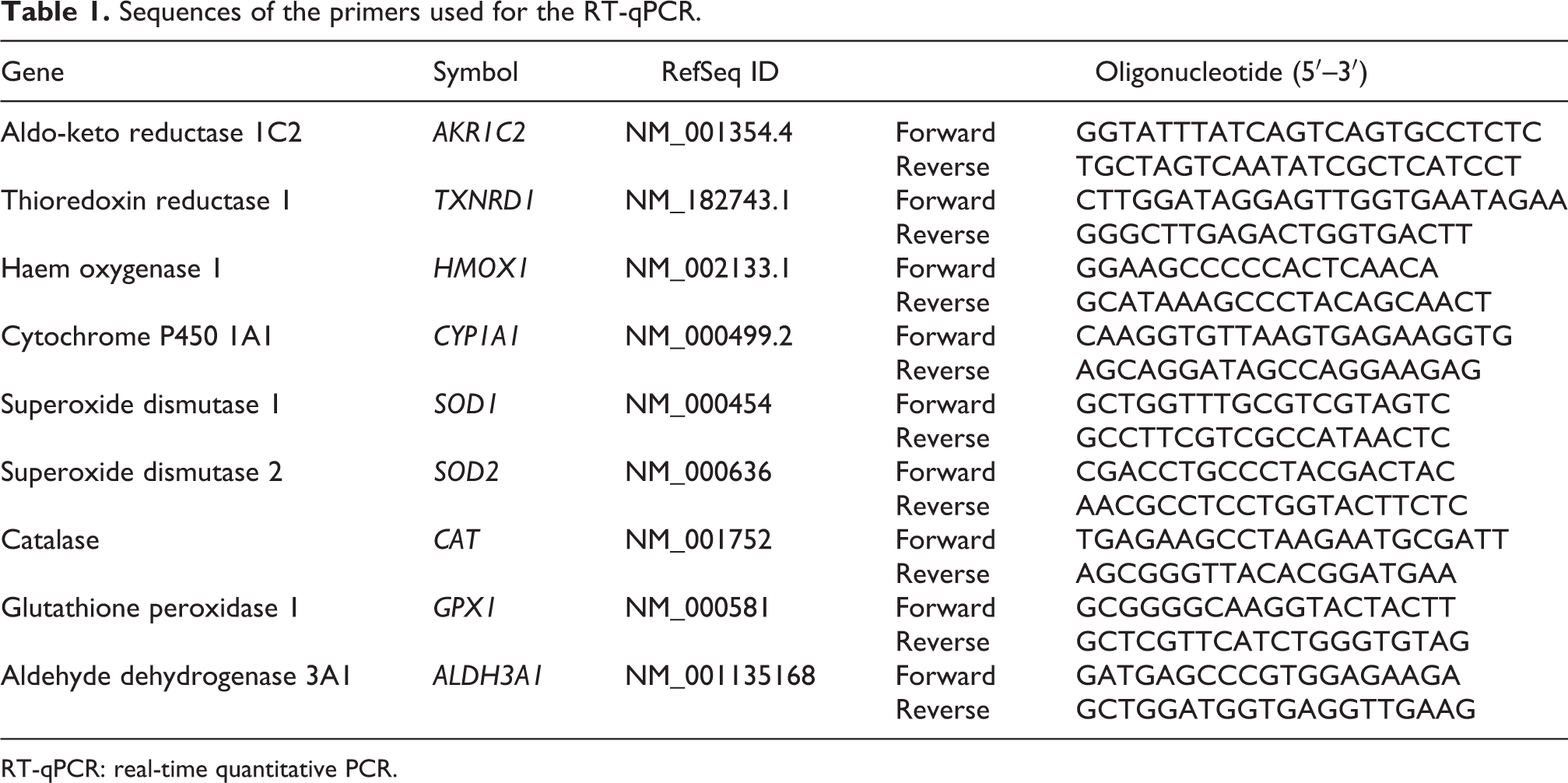
RT-qPCR: real-time quantitative PCR.
Statistical analysis
Statistical analysis of the data obtained in the study was carried out using a two-way analysis of variance. Differences with a p value of 0.05 or less were considered significant.
Results
Morphology and TEER measurements
The stability of the MucilAir tissue was monitored for 3 weeks prior to PAH exposure. Both before and during the experiments, no visible changes in the morphology of the cells were observed, specifically with regard to any perforations or surface protrusions. Beating cilia were readily visible under the light microscope.
TEER values, and therefore the integrity of the MucilAir model with regard to its tight junction function, were negatively affected after a 7-day exposure to 0.1 μM and 1 μM B[a]P (Figure 2(a)). After that time point, TEER values slightly increased above the level of the control for both B[a]P concentrations — reaching a statistically significant high level on day 18 in the case of 0.1 μM — and decreased again on days 21 and 28.
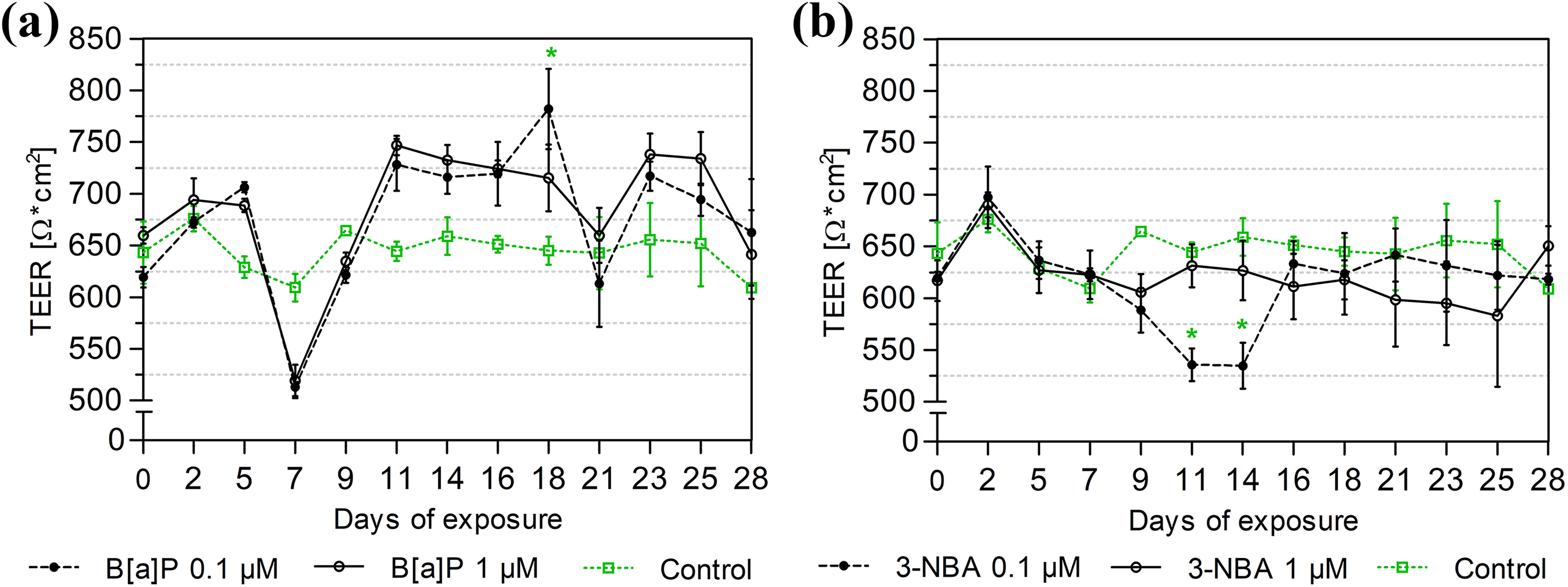
Changes in TEER after exposure to B[a]P and 3-NBA for 28 days. MucilAir inserts were exposed for a total of 28 days to the 0.1% DMSO control: (a) B[a]P (0.1 μM, 1 μM) and (b) 3-NBA (0.1 μM, 1 μM). TEER was measured every 2–3 days. The results are presented as average values (± SD) from three independent measurements. Significant differences (p < 0.05), as compared to the control values, are denoted by asterisks. TEER: transepithelial electrical resistance; B[a]P: benzo[a]pyrene; 3-NBA: 3-nitrobenzanthrone; SD: standard deviation.
In contrast, the detrimental effect of 3-NBA exposure on TEER values was evident after 11 days of exposure to a concentration of 0.1 μM (Figure 2(b)). Exposure to 1 μM 3-NBA led to a continuous slight decrease of TEER from day 9.
Cytotoxicity and ROS production
A spike in LDH activity in the harvested supernatants (the result of damaged cell leakage) was apparent after 5 days of exposure to the test compounds at both concentrations (0.1 μM and 1 μM) (p < 0.05; Figure 3). By day 7, LDH activity had fallen back to values similar to those of the control sample for both compounds. A longer exposure to B[a]P had no further significant effect on cytotoxicity (Figure 3(a)). In contrast, continued exposure to 0.1 μM 3-NBA led to a further spike in LDH activity on day 9, with the values remaining elevated (p < 0.05) until day 14. Treatment with 1 μM 3-NBA led to a further spike in LDH activity from day 21 (p < 0.05; Figure 3(b)).
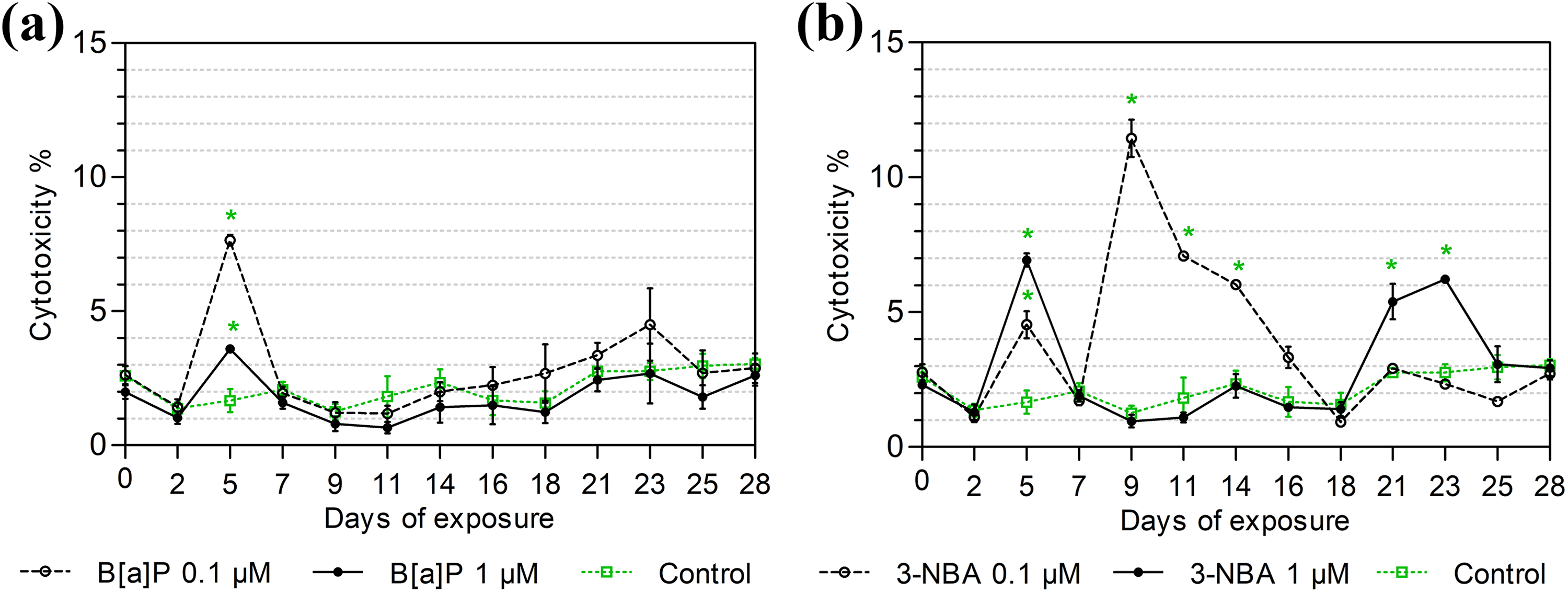
LDH release after exposure to B[a]P and 3-NBA for 28 days. MucilAir inserts were exposed for a total of 28 days to the 0.1% DMSO control: (a) B[a]P (0.1 μM, 1 μM); and (b) 3-NBA (0.1 μM, 1 μM). LDH activity in the culture medium was measured every 2–3 days. The results are shown as the % cytotoxicity relative to the control and represent average values (± SD) from three independent measurements. Significant differences (p < 0.05), as compared to the control values, are denoted by asterisks. LDH: lactate dehydrogenase; B[a]P: benzo[a]pyrene; 3-NBA: 3-nitrobenzanthrone; SD: standard deviation.
ROS levels in the harvested supernatants after PAH exposure were unchanged relative to the negative control (0.1% DMSO exposure). However, the assay data from the positive control sample (250 mM H2O2 exposure for 1 hour) were at least 28-fold higher than the corresponding 0.1% DMSO control (Table 2).
ROS production by MucilAir during a 28-day exposure to B[a]P or 3-NBA.a
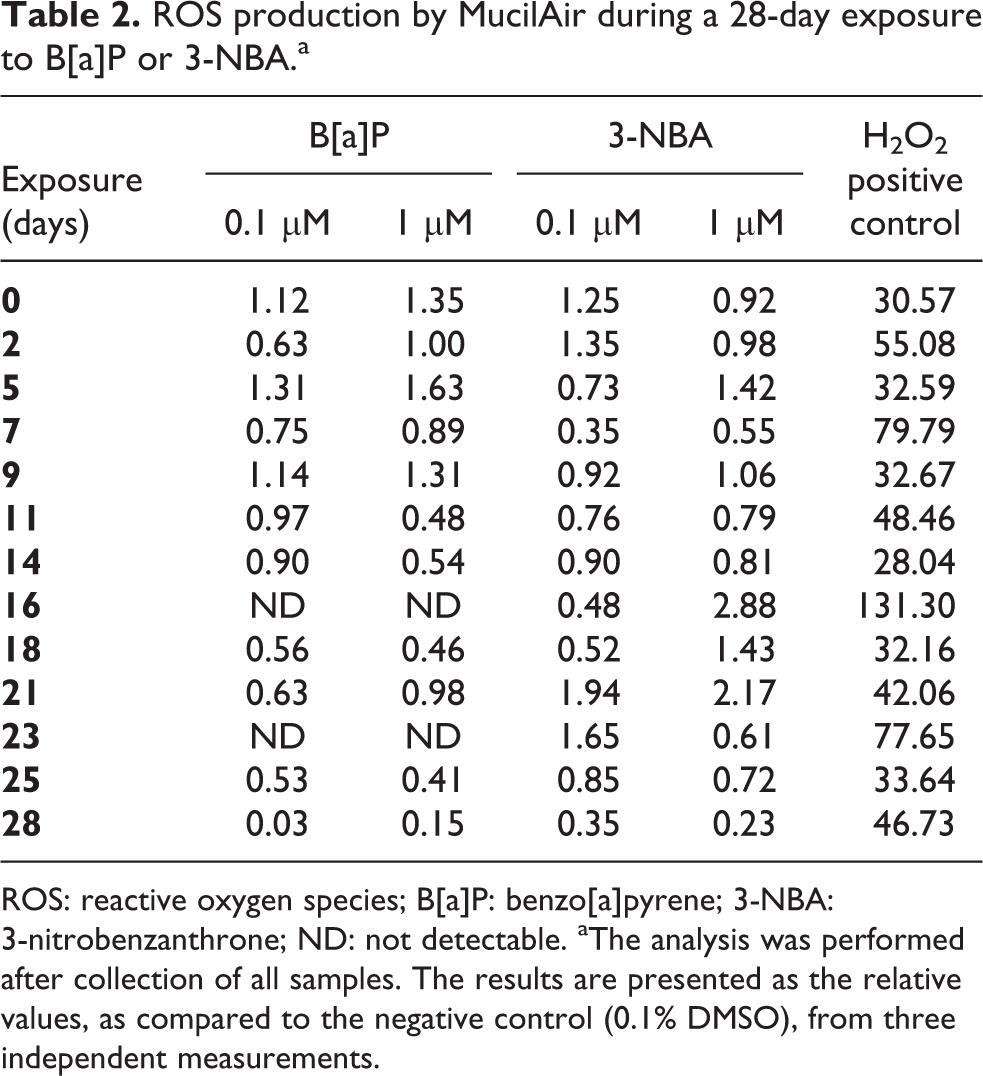
ROS: reactive oxygen species; B[a]P: benzo[a]pyrene; 3-NBA: 3-nitrobenzanthrone; ND: not detectable. aThe analysis was performed after collection of all samples. The results are presented as the relative values, as compared to the negative control (0.1% DMSO), from three independent measurements.
mRNA expression analysis
After the 24-hour, 7-day and 28-day exposure time points, the expression levels of selected genes (CYP1A1, AKR1C2, ALDH3A1, SOD1, SOD2, GPX1, CAT, HMOX1, TXNRD1) were assessed (see Figures 4 to 6 and summary in Table 3). Expression of CYP1A1, a gene encoding the PAH-metabolising cytochrome P450 1A1, significantly decreased following a 24-hour exposure to 0.1 μM B[a]P (p < 0.05; Figure 4(a)). However, after 7-day and 28-day exposures to B[a]P at this same concentration, CYP1A1 gene expression was significantly upregulated (p < 0.05; Figure 4(a)). After 24-hour PAH exposure, the expression of HMOX1, a gene which plays a protective role against oxidative damage, was significantly downregulated (p < 0.05) for both concentrations of the tested compounds (Figures 4(a) and (b)).
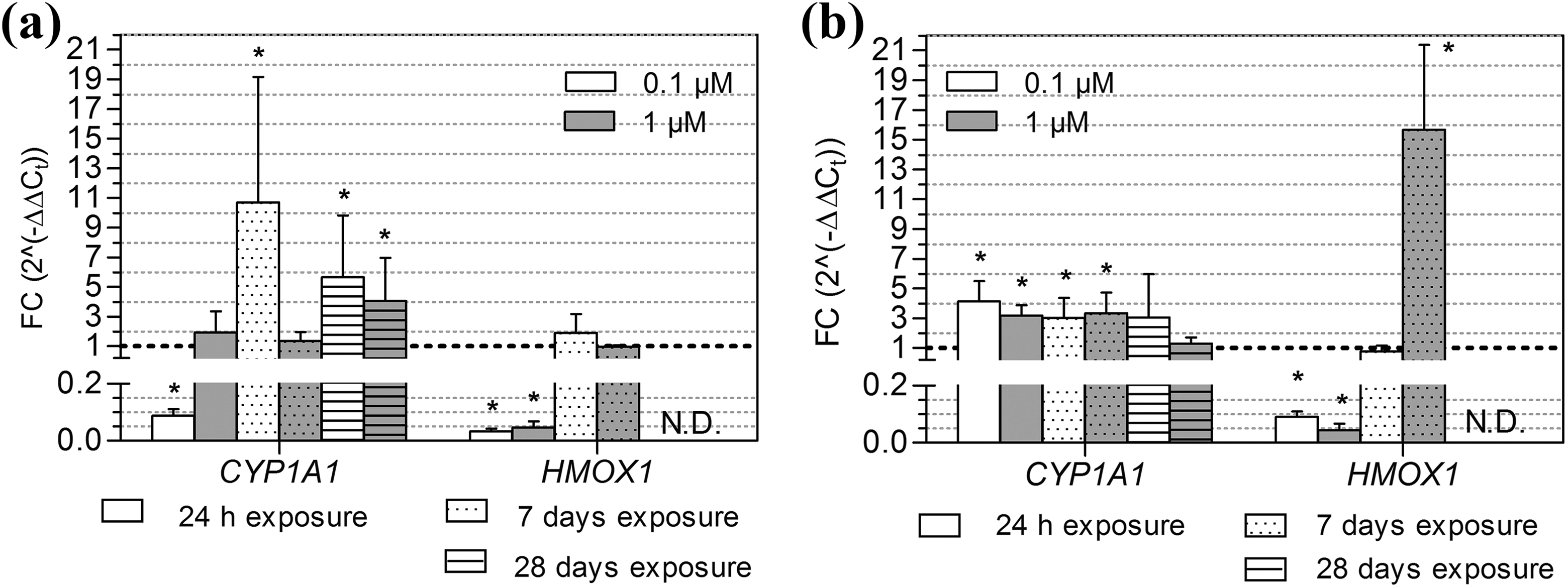
Gene expression of CYP1A1 and HMOX1 after exposure to B[a]P and 3-NBA. The average expression (± SD) of CYP1A1 and HMOX1 relative to the 0.1% DMSO control after a 24-hour, 7-day and 28-day exposure of MucilAir to (a) B[a]P (0.1 μM, 1 μM) and (b) 3-NBA (0.1 μM, 1 μM). The dashed horizontal line indicates the expression level after exposure to the DMSO control. Significant differences (p < 0.05), as compared to the control values, are denoted by asterisks. CYP1A1: cytochrome P450 1A1; HMOX1: haem oxygenase 1; ND: not determined; B[a]P: benzo[a]pyrene; 3-NBA: 3-nitrobenzanthrone; SD: standard deviation.
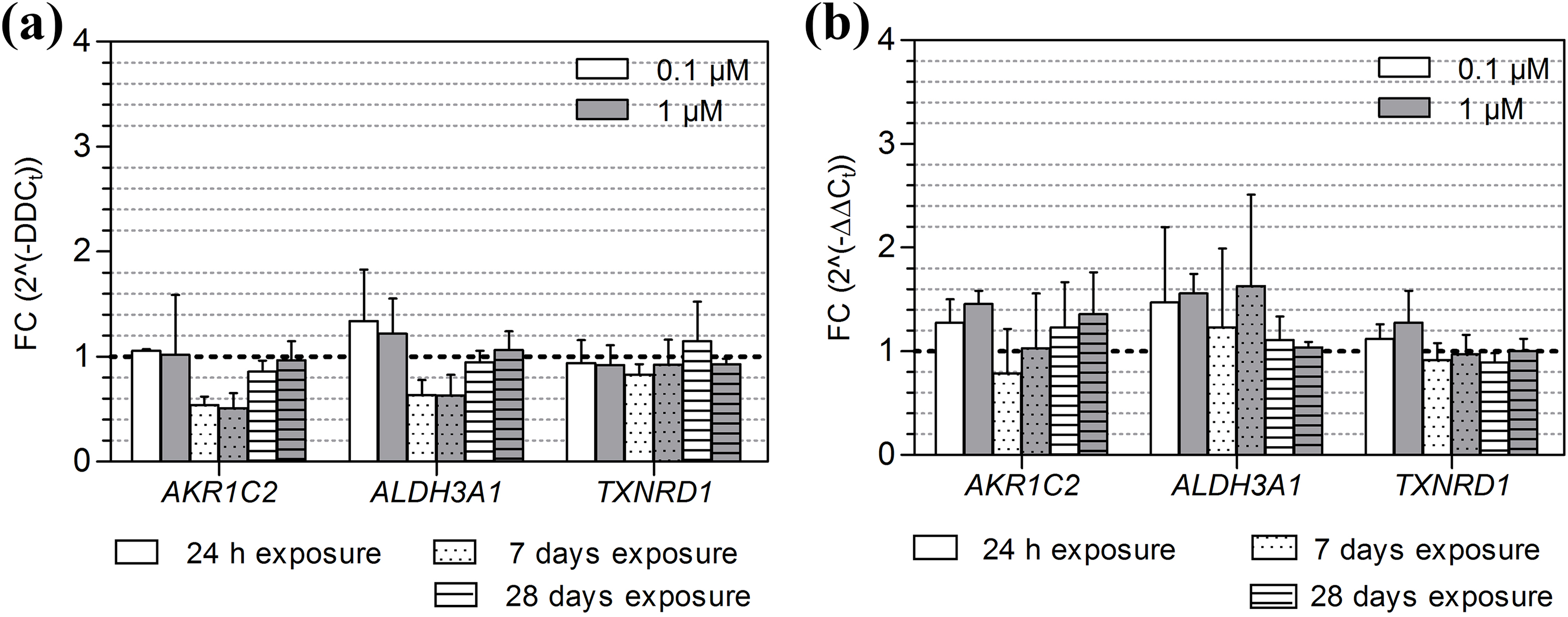
Gene expression of AKR1C2, ALDH3A1 and TXNRD1 after exposure to B[a]P and 3-NBA. The average expression (±SD) of AKR1C2, ALDH3A1 and TXNRD1, relative to the 0.1% DMSO control, after a 24-hour, 7-day and 28-day exposure of MucilAir to (a) B[a]P (0.1 μM, 1 μM) and (b) 3-NBA (0.1 μM, 1 μM). The dashed horizontal line indicates the expression level after exposure to the DMSO control. AKR1C2: aldo-keto reductase 1C2; ALDH3A1: aldehyde dehydrogenase 3A1; TXNRD1: thioredoxin reductase 1; B[a]P: benzo[a]pyrene; 3-NBA: 3-nitrobenzanthrone; SD: standard deviation.
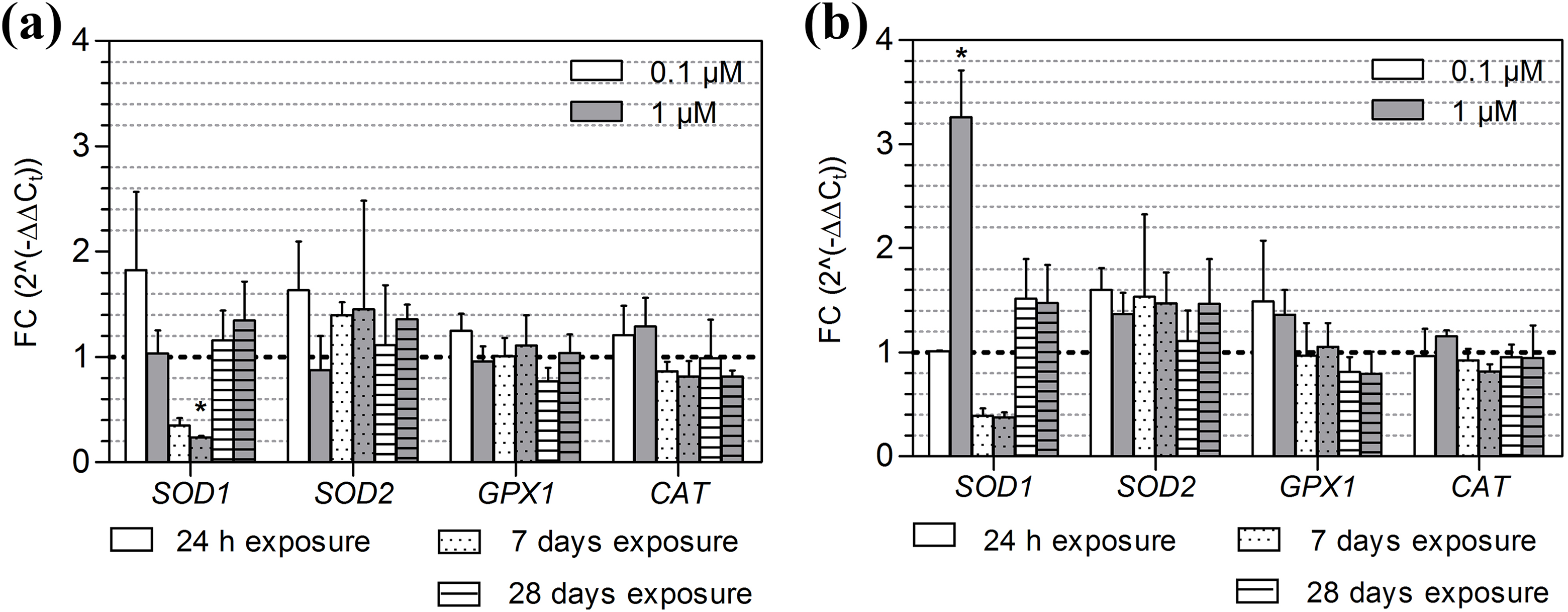
Gene expression of SOD1, SOD2, GPX1 and CAT after exposure to B[a]P and 3-NBA. The average expression (±SD) of SOD1, SOD2, GPX1 and CAT, relative to the 0.1% DMSO control, after a 24-hour, 7-day and 28-day exposure of MucilAir to (a) B[a]P (0.1 μM, 1 μM) and b) 3-NBA (0.1 μM, 1 μM). The dashed horizontal line indicates the expression level after exposure to the DMSO control. Significant differences (p < 0.05), as compared to the control values, are denoted by asterisks. SOD1: superoxide dismutase 1; SOD2: superoxide dismutase 2; GPX1: glutathione peroxidase 1; CAT: catalase; B[a]P: benzo[a]pyrene; 3-NBA: 3-nitrobenzanthrone; SD: standard deviation.
Gene expression results summary.a
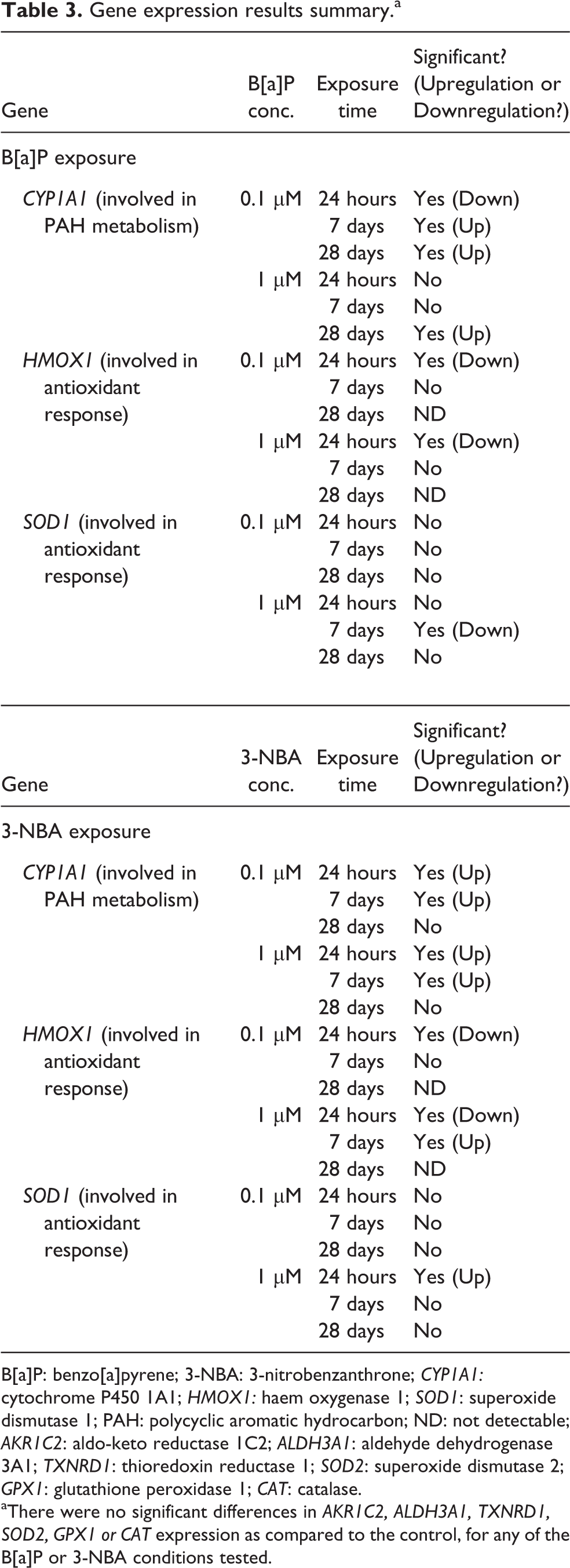
B[a]P: benzo[a]pyrene; 3-NBA: 3-nitrobenzanthrone; CYP1A1: cytochrome P450 1A1; HMOX1: haem oxygenase 1; SOD1: superoxide dismutase 1; PAH: polycyclic aromatic hydrocarbon; ND: not detectable; AKR1C2: aldo-keto reductase 1C2; ALDH3A1: aldehyde dehydrogenase 3A1; TXNRD1: thioredoxin reductase 1; SOD2: superoxide dismutase 2; GPX1: glutathione peroxidase 1; CAT: catalase.
aThere were no significant differences in AKR1C2, ALDH3A1, TXNRD1, SOD2, GPX1 or CAT expression as compared to the control, for any of the B[a]P or 3-NBA conditions tested.
The expression of SOD1 was downregulated after a 7-day exposure to 1 μM B[a]P (p < 0.05; Figure 6(a)) but was significantly induced after exposure to 1 μM 3-NBA (Figure 6(b)). 3-NBA significantly induced the expression of CYP1A1 after 24-hour and 7-day exposures (both concentrations) and also induced HMOX1 gene expression at the higher concentration of 1 μM, after a 7-day exposure. For both test compounds, the expression of HMOX1 was not detectable after a 28-day exposure (Figures 4(a) and (b)). There were no significant differences in AKR1C2, ALDH3A1, TXNRD1, SOD2, GPX1 or CAT expression as compared to the control, for any of the B[a]P or 3-NBA conditions tested.
Discussion
In this study, we aimed to assess the suitability of the MucilAir model to compare the effects of short-term and long-term exposure to two PAHs (B[a]P and 3-NBA). The toxic effects of PAHs, which are products of the incomplete combustion of organic material and are ubiquitous air pollutants, have been analysed in numerous studies, including those based on monocultures of human lung cells. 17 –21 However, comparable studies based on the use of 3-D models are scarce. To the best of our knowledge, fewer than 20 peer-reviewed articles have been published on studies involving the MucilAir model, and less than half of these address the effects of air pollutants. 22 –25 Furthermore, none of these studies (including those based on other ALI cell models 26 –28 ) have focused on the effects of long-term exposure (i.e. lasting several weeks). Our study is also the first to look at the exposure of MucilAir to these two particular PAHs.
To characterise the response of MucilAir to PAH exposure, we focused on the basic parameters of cytotoxicity and tissue damage (LDH release and TEER measurement), as well as on effects related to the specific chemical properties of the compounds (the induction of ROS production and changes in expression of genes encoding metabolic activation and antioxidant enzymes). Generally, cytotoxic effects were mostly limited to those exposures lasting around 5–7 days.
Significant LDH leakage, which indicates damage to the cell membrane, was observed after 5 days of exposure to PAHs at both concentrations. After that time point, the LDH values mostly fell back to the level of the control values. Exceptions were the exposure to 0.1 μM 3-NBA, which led to a second spike in LDH leakage after the 9-day exposure, while concentration of 1 μM 3-NBA led to a similar spike at day 23. These data suggest that, after a certain time period, the model is able to recover from and/or adapt to the damage. 5 This capacity for cell damage recovery has been shown in well-differentiated human airway epithelial cell (HAEC) 2-D cultures by Crespin et al. 29
The TEER data, which are used to check tissue integrity and tight junction function, followed a similar trend to the LDH leakage (cellular damage) data, suggesting a link between the two effects. In contrast, when ROS production was assessed, no significant changes were apparent for any of the B[a]P or 3-NBA conditions tested. This suggests that these compounds have a limited capacity to induce oxidative damage in the MucilAir system. This finding is supported by the gene expression results, which showed no induction of genes involved in the antioxidant response.
As cytochrome P450 enzymes are primarily responsible for the metabolic activation of PAHs, we analysed the modulation of CYP1A1 gene expression. We found that the gene was upregulated after B[a]P treatment, but only after longer exposures (at least 1 week). In contrast, the results obtained with cell monocultures showed that even a short 4-hour incubation with B[a]P resulted in a significant increase in CYP1A1 expression. 20 This observation is in line with the conclusions of recent studies, 7 –9 which report that 3-D models like MucilAir are more resistant to harmful substance exposure. Another study focusing on gasoline exhaust effects in various types of ALI cell models showed that MucilAir is a suitable model for long-term (3-day) exposure at the ALI, in view of the lack of damage sustained by the cells in the short term. 24 Although it is not clear why 3-D models are more resistant to the effects of toxic compounds, it might be that the mucus layer and functioning tight junctions of MucilAir limit the penetration of the PAHs into the lower layers of the tissue. 11 This might explain why the gene expression analysed in our study was mostly unchanged.
We did not compare 3-D models with cell monocultures, and it is generally accepted that the biological responses of 3-D cultures differ from those observed for 2-D cell monocultures, which have been used for many years and are considered the ‘gold standard’ in toxicology testing. Cell monocultures have numerous advantages, particularly if the cells used are the ones recommended by the Organisation for the Economic Co-operation and Development guidelines. Ideally, these cell lines should be genetically stable, with low DNA damage background and functional p53 activity, 30 as the use of p53-deficient cells may lead to false-positive results. 31 However, these recommendations are often ignored, and immortalised cells or cells of neoplastic origin are used by many researchers. Although these types of cell line are suitable for use in basic genotoxicity tests, they may lack (or have gained) some important properties (e.g. the activity of metabolic enzymes, cell cycle regulation, DNA damage response), their genetic background might have changed or new clones might have been unknowingly selected due to repeated passages. Moreover, cells maintained in 2-D monoculture (i.e. cells of a single type growing in one layer) lack an extracellular matrix and the intercellular and intracellular communication that is typical of in vivo tissues and organs. This affects numerous cellular functions — including gene expression modulation, cell cycle control or apoptotic responses following exposure to chemicals — which might lead to potentially incorrect conclusions from the data generated. Therefore, the use of 3-D tissue cultures is advocated as a better alternative for toxicity testing, 30,32,33 as they represent a more relevant system. However, some limitations need to be addressed before 3-D cultures can be regarded as suitable replacements for cell monocultures. Most importantly, the tests with 3-D models need to be standardised and more results from independent laboratories should be made available. Thus, although the use of MucilAir inserts as an ALI cell model for long-term air pollution toxicity studies is promising, future studies should aim to design dose–response experiments for various air pollutants, as has been previously carried out for the ‘gold standard’ 2-D cell monocultures.
An issue specific to this type of 3-D model is the potential for variability in the data obtained, due to the use of different tissue donors. This was addressed by Kooter et al. 23 in a study that focused on the effects of exposure to aerosols of cupric oxide (CuO) nanoparticles. According to their results, the main source of variation was the concentration of CuO, but there were also substantial differences apparent among donors and between the test sessions. Since the aim of our study was to compare short-term (24-hour) and long-term (7-day and 28-day) exposure to PAHs, donor variability was not a test parameter. One of the biggest advantages of 3-D models is the possibility to purchase ready-to-use cells with a relatively long shelf life. These commercially available models permit studies of repeated exposure to pollutants that would not be possible, or would be less informative, with immortalised or transformed cell lines.
Our experiments with MucilAir revealed some practical limitations to the system. In the current study, a standard 24-well plate with approximately 0.5 × 106 cells per insert was used. If a long-term study protocol involves the destruction of a sample for analysis, then multiple inserts would be required, thus increasing the material cost and staff labour time involved. This was the main reason why our study did not include the microscopic analysis of histological slices or other imaging technique that would involve tissue destruction. However, the stability of the MucilAir inserts was visually monitored for 3 weeks prior to PAH exposure and during the experiments, and the tissue surface checked for protrusions or gaps. In addition, beating cilia were visualised under a light microscope.
Although the ALI exposure system is not the standard laboratory equipment, it is possible to expose the cells via the manual addition of droplets (10–30 μl) on the apical side of the insert, which quickly evaporate and allow further maintenance of the ALI conditions. 7 Finally, as the MucilAir models can be generated from various donors, they can be used to study the effects of exposure to air pollutants in diseased populations (e.g. chronic obstructive pulmonary disease patients, asthmatics, etc). In summary, MucilAir has the potential to be a useful model for long-term exposure to air pollutants, although further studies, validation of the results and comparisons with 2-D cell monoculture systems are needed.
Conclusions
The MucilAir model is a robust and long-lasting tool for conducting air pollution research, which is amenable to a wide variety of testing approaches. It displays in vivo characteristics, including stratification, tight junctions, metabolic activity, mucus production and beating cilia. In the current study, the adverse effects of 0.1 μM and 1 μM B[a]P and 3-NBA were mostly limited to long-term exposures, illustrating the potential use of this 3-D in vitro lung tissue model in studying the effects of chronic exposure to PAHs.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Czech Science Foundation [grant number 16-14631S].
Ethics approval
Ethics approval was not required for this research article.
Informed consent
Informed consent was not required for this research article.