
Abstract
Atherosclerosis has been recognized as an inflammatory/autoimmune disease. The long-standing low-grade inflammation which fuels its development is primarily focused on the components of the vessel wall. Originally, inflammation in atherogenesis was supposed to be driven by the pro-inflammatory Th1 cellular and cytokine immune response. On the basis of accumulating evidence, this view has been re-evaluated to include the Th17/Th1 axis which is shared by most diseases of sterile inflammation. The anti-inflammatory Th2 cellular and cytokine immune response is initiated concomitantly with the former two, the latter dampening their harmful reactions which culminate in full-blown atherosclerosis. Interleukin-33, a novel member of the IL-1 cytokine superfamily, was suggested to take part in the anti-atherogenic response by mediating the Th1-to-Th2 switch of the immune reactions. However, IL-33 is a multifaceted mediator with both pro- and anti-inflammatory activities, also called a “dual factor” or a “Janus face” interleukin. IL-33 occurs both in an extracellular (cytokine-like) and in a nuclear-bound (transcription factor-like) form, each of them performing distinct activities of their own. This review article presents the latest data relevant to IL-33’s role in atherosclerosis and cardiac diseases as perceived by a cardiologist and a cardiac surgeon.
Fundamental biology of IL-33
To begin with, IL-33’s occurrence – this name not yet existing at that time – was detected in the form of a transcriptional nuclear factor of high endothelial venules. That enigmatic substance was referred to as IL-1F11. Only in 2005 did Schmitz and coworkers report that this nuclear factor exhibited a surprising amino acid homology with the cytokine IL-18. 1 Thus, it was recognized as a new member of the IL-1 cytokine family and re-called by the proper name of interleukin-33. Right from the cradle, IL-33 has been shown to perform dual, apparently opposite activities. These two seemingly unrelated activities – one extracellular, the other intracellular – ultimately joined one another in the molecule of IL-33. The former was the paracrine/autocrine/juxtacrine/intracrine activity of an interleukin, principally identical with any other mediator acting on the target cell from the outside. The latter was that of a transcription factor which, under normal conditions, never gets out of the cell into the extracellular space. However, any time a cell succumbs to necrotic death, which is characterized by the rupture of its outer membrane, IL-33 is forced to leave the dying cell. Out of its parent cell, IL-33 then performs the function of an alarmin. Alarmins belong to a larger cluster of endogenous substances called the danger-associated molecular patterns or DAMPs. By analogy with the pathogen-associated molecular patterns or PAMPs, the DAMPs initiate sterile inflammation while the PAMPs elicit bacterial inflammation. Once the inflammation has been set off, further events, both on the molecular and on the clinical levels, occur in a strikingly similar manner. 2
IL-33 is being born: one cytokine and two receptors
Historically, the receptor ST2 was known well before its ligand, i.e., IL-33. Such receptors, whose partners have not yet been identified, are collectively known as “orphan” receptors. The ST2 gene – et tu, Brute! – encodes for at least two isoforms of the ST2 protein.
The membrane-bound ST2 isoform or ST2L is composed of extracellular IgG domains, one transmembrane domain and one intracellular TIR domain that is highly homologous to TLRs and other IL-1Rs. Accordingly, the IL-33 cytokine, by way of a network of intracellular proteins, ultimately activates nuclear factor NF-κB. Most importantly, the effects of NF-κB which has been activated by IL-33 are entirely different from those evoked by IL-1β or IL-18.
The soluble ST2 isoform or sST2 is present in the extracellular space or in the circulation due to the lack of both the transmembrane and the intracellular domains. Its extracellular domains bind available IL-33 before it can reach its target cell and occupy its membrane-bound receptor ST2L. Accumulated evidence suggests that such an interaction (sST2+IL-33) inactivates the cytokine which cannot associate with the ST2L membrane receptor. Receptors which inactivate their ligands are collectively known as “decoy” receptors. Studies in ST2 gene-deficient animals or administering to laboratory rodents either ST2-specific antibodies or treating them with exogenous sST2 to prevent their endogenous IL-33 from activating its target cells have confirmed the role of IL-33 in supporting the Th2 cellular/cytokine responses. 3
The membrane-bound receptor ST2L belongs to the Toll-like receptor TLR/IL-1R superfamily of receptors. Along with the more largely distributed co-receptor IL-1R accessory protein, it makes up the immediately usable receptor for IL-33 to transmit its signal into the cell as far as the nucleus. 4 Cells devoid of the ST2L receptor do not respond to IL-33. ST2L is highly expressed on activated Th2 lymphocytes, mast cells and basophils. From our viewpoint, it is of paramount importance that ST2L is also expressed on endothelial cells. Binding of IL-33 to the ST2L receptor gives rise to the production of Th2-associated cytokines, mostly IL-4, IL-5 and IL-13. However, under strictly defined conditions, lymphocytes targeted by IL-33 via its membrane receptor ST2L may instead elaborate pro-inflammatory mediators, such as IL-6, IL-8 (CXCL8), TNF and IFN-γ which is recognized as the hallmark of the Th1 response. 5 Voilà! A typical Janus face of a cytokine. These effects illustrate, mostly, the duality of IL-33 receptors, which sets the stage for its dichotomous results: activation versus the inability to activate the target cell. The latter mechanism, depending on the sST2 receptor, proves useful in down-regulating exaggerated IL-33 activities, e.g., such as encountered in unbridled asthma bronchiale, which is a typical Th2-dependent inflammatory condition or in some overwhelming bacterial infections. On the other hand, inactivating IL-33 effects seems harmful in excluding the onset of or in minimizing the extent of IL-33-mediated benefit in cardiovascular diseases, most importantly in ischemia-reperfusion injury. 6
IL-33: don’t mix up apples with oranges; this is a pineapple
Recently, functional analysis of IL-33-deficient mice has suggested that, despite all evidence favoring its effects on the Th2 cells, IL-33 amplifies both Th1 and Th2 responses, particularly by targeting innate immune cells in mucosal tissues. IL-33 has been shown to augment inflammatory cytokine production, particularly (TNFα, IL-1β and IL-6) by mouse macrophages. During bacterial sepsis, the administration of exogenous IL-33 does not elicit a Th1-to-Th2 shift, but, instead, restrains systemic pro-inflammatory cytokine activities and impacts ST2L-expressing neutrophils to support their antibacterial responses. Furthermore, NK cells and NKT cells express ST2L on their outer membranes and respond to IL-33 by producing IFN-γ. The role of IL-33 as an alarmin released from necrotic epithelial and endothelial cells has been just mentioned. As it is, depending on environmental conditions and the cells about to be influenced by IL-33, this interleukin may act either as a potent Th2 type signal or as a generalized pro-inflammatory signal.
Bourgeois and coworkers 5 showed that the capacity to promote a Th1 phenotype is a general quality of IL-33. The authors found that it can be recapitulated both in C57Bl/6 and in BALB/c mice, each of the two groups being genetically biased either to a pro-Th1 or to a pro-Th2 immune response, respectively. The exact mechanism by which IL-33 up-regulates cytokine production by iNKT or NK cells remains, so far, unclear. The interaction by IL-33 with its respective ST2L receptor is not by itself sufficient to elicit the IFN-γ secretion. IFN-γ production does not take place without either TCR or IL-12 signaling if the cell cultures are prepared with naive iNKT (CD5+ NK1.1+) or NK cells. The same holds true for IL-4 secretion, implying that IL-33 acts rather as a cofactor than an inducer per se. Somewhat paradoxically to the former hypothesis, IL-33 administration by itself activated both iNKT and NK cells in vivo, which must have occurred indirectly by way of as yet unknown endogenous factors that are generated by other cells influenced by IL-33, the latter different from the IFN-γ or IL-4 producing cells. This idea is supported by the fact that IFN-γ secretion was detected in response to IL-33 alone, unaided by IL-12, in cultures set up with whole spleen cells. These data emphasize the concept that IL-33 contributes as a co-stimulatory factor in innate cellular immune responses. The ability of IL-33 to contribute to IFN-γ production and, hence, supporting the Th1-profiled inflammatory/immune responses, was complemented by additional studies. 5 Yang and colleagues reported that Th1 cytotoxic cells (Tc1), when cultured in vitro, produced unexpectedly high amounts of the ST2 receptor. This expression of ST2 was dependent on the presence of T-bet, the master Th1/Tc1 transcription factor which regulates the Th1-profiled inflammatory/immune response. IL-33, aided or not by IL-12, augments IFN-γ production by Tc1 cells. 7 These results expand a hitherto unknown role of IL-33, i.e., that of a promoter of type 1 adaptive immune responses on top of the innate immune responses.
The happy life of IL-33
Both IL-33’s relatives, IL-1β and IL-18, are synthesized as biologically inactive precursors. In an inflammatory setting, both of them are activated by cleaving by way of caspase-1. By analogy, it was expected that IL-33 undergoes a comparable caspase-1-mediated processing to yield a functionally active molecule. Recent investigations even revealed that a proteolytic handling was not useful for IL-33-mediated signaling. On the contrary, any modification by caspases of the full-length IL-33 molecule results in its inactivation whenever apoptosis is the prevailing mechanism of cellular turnover or removal and, concomitantly, there are no signs to alert the inflammatory response. However, once homeostasis has suffered a blow which deviates the balance of the steady state, e.g., mechanical trauma or an injury inflicted by surgical operations, infection or, as it is, a cellular stress of any origin, no matter if local or systemic, cellular necrosis exceeds apoptosis as the principal way of removing damaged or otherwise useless cells. IL-33 is then set free out of the dying cells which have suffered a disruption of their plasma membranes. The extracellular appearance of IL-33, a formerly nuclear factor form of IL-33, i.e., a cytokine which normally does not associate with its receptor, converts IL-33 into an alarmin which now alerts the immune system of the impending danger, just the same as do penetrating bacteria. 8 Along with IL-33, the most important alarmins are high-mobility group box-1 (HMGB-1) and interleukin-1α (IL-1α). 8
To summarize briefly, we can state that, when bodily homeostasis is maintained, apoptosis takes care of tissue integrity by eliminating senescent and/or malfunctional cells. The free IL-33 molecule, if present in the extracellular milieu, is cleaved by caspases or sequestered by its decoy receptor sST2 to forestall its undue pro-inflammatory activities. When homeostasis is disturbed by any of the plethora of harmful stimuli, the full length, i.e., the biologically active form of IL-33 is extruded from necrotic cells in order to accomplish its mission of an endogenous danger signal or an alarmin which forewarns the immune system to get prepared.
The split “personality” of IL-33 is recapitulated in the ST2 receptor gene which, by alternative splicing, encodes for two isoforms of a protein. The membrane-bound ST2L isoform exerts ultimate transmission of the IL-33-borne signal via a cellular receptor complex which, itself, is a heterodimer consisting of ST2L, itself enforced by IL-1RAcP (IL-1R accessory protein). Il-1 RAcP is a receptor subunit shared by other members of the IL-1 cytokine family. The secreted or soluble ST2 (sST2) isoform neither associates with IL-1RAcP nor is equipped with membrane-inherent units of ST2; hence, it annihilates the IL-33’s signal transmission while, at the same time, acts as a decoy receptor, in that it switches off IL-33’s biological effects mainly in the circulation. On the outer membrane of IL-33-targeted cells, there is, at least, one more regulatory molecule, known as SIGIRR (single Ig IL-1R-related molecule). It joins the sST2 receptor isoform in abolishing IL-33 signal transmission into the cellular interior. In contrast to sST2, which is present exclusively in the extracellular space, SIGIRR stays attached to cellular membranes. 9
Atherosclerosis is a stage and all the biomarkers are mere players: enter IL-33
Inflammatory cell penetration of the still healthy, but atherogenic noxi-susceptible vascular segments begins with the entry of CD4+ T lymphocytes. These are closely followed by monocytes and other populations from the bloodstream whereas, from the opposite direction, i.e., from the media, smooth muscle cells start to move to join the cellular infiltrate in the intima. This cellular accumulation is supplemented by mast cells and by dendritic cells traveling to the cellular assembly point in the arterial intima from the adventitia or even from the perivascular space. Thus, a favorable microenvironment is instituted in the arterial wall for a local immune reaction to take place, starting in and disseminating from the intima. 10 The inflammatory nature of these events is beyond any doubt since macrophages resident in healthy vessels do not display the appearance of foamy cells, just the same as no lipid deposits are detectable in atherosclerosis-resistant vessels. As atherosclerosis goes on, the continually increasing number of foam cells convert into so-called fatty streaks, which make up a morphological gangway for the development of atherosclerosis. Up to this stage, at least, the disease is supposed to be accessible to substantial retarding in its progression or even to a reversal by natural, endogenous anti-inflammatory reactions or, lately, by anti-inflammatory and/or hypolipidemic drugs. 11 Both sets of protective means are operative in full-blown stages of atherosclerosis as well when they ward off atherosclerosis-associated complications, such as myocardial infarction or cerebral stroke. The endogenous, body-own anti-inflammatory resources are represented, among others, by the IL-33/ST2 axis.
To get an insight into early atherogenetic events, the disease development has been able to be studied, both extensively and in depth, ever since the availability of genetically modified mice. Among these animal models, ApoE–/– mice, which do not express any traces of apolipoprotein E, recapitulate faithfully the course of human atherosclerosis from the very beginning up to fatal complications. When exposed to the impact of disease promoting or, on the other hand, of disease-protective external influences, these animals experience the imprint on atherosclerosis development of both kinds, harmful or beneficial, every day, according to their genetic background and the external events to which they are exposed. Recently, ST2–/– mice have been generated which lack the membrane receptor ST2L, which is strictly required for IL-33-borne intracellular signal transduction. These living beings, although only rodents, can furnish additional new information regarding the atherosclerosis-modifying effects of the IL-33/ST2 axis which, otherwise, would be available only in a fragmented mode from in vitro or ex vivo experiments. 12 Continued research has shown that the IL-33/ST2 axis, originally assumed to produce exclusively anti-inflammatory and anti-atherogenetic effects, is basically capable of acting in both ways, i.e., both in the pro- and in the anti-inflammatory fashion, depending on environmental conditions. This conclusion supplies yet another piece of evidence implying that atherosclerosis is not predestined to an unrestrained, helpless progression, but a regression of varying extent and varying durability is feasible under favorable conditions. 13
IL-33 and natural IgM antibodies: a way to combat in atherosclerosis
In vitro, IL-33 has been shown to attenuate macrophage foam cell formation by decreasing modified, i.e., acetylated or oxidized, AcLDL or OxLDL, low-density lipoprotein uptake. Both total and esterified macrophage cholesterol volume is diminished whereas cholesterol efflux out of the cell is increased to be evacuated by the reverse transport from inside the vascular wall into the liver and out of the body via the feces. These IL-33-mediated beneficial effects originate, for the most part, from a genetic reprogramming of its target cell, i.e., the macrophage. This process results in the down-regulation of genes and their products which are responsible for the LDL uptake, such as the CD36 receptor. In parallel, genes implicated in cholesterol efflux, such as Apo E, are upregulated. 14 The in vitro atheroprotective capacity of IL-33 has been confirmed in vivo in Apo E–/– mice which were fed lipid-rich “Western” diet along with the concomitant application of IL-33 peptide by the intraperitoneal route. In these genetically modified animals, IL-33 decreased the absolute number of vessel wall-associated F4/80+ macrophages, i.e., those cells which would otherwise have converted into foam cells. This IL-33-mediated effect is highly reminiscent of the clearance by apoptosis of lipid-accumulating macrophages, a process that occurs naturally in the early stages of atherosclerosis. Moreover, ApoE–/– mice treated with IL-33 produce higher-than-normal titers of anti-ox-LDL IgM antibodies, which are expected to play important atheroprotective roles. Natural IgM antibodies (Abs), i.e., those available without prior encounter with an antigen, are elaborated by subset B1 of B cells. IL-33 not only impacts positively on B1 B cell activity, but, maybe more importantly, in supporting the Th2 immune response, IL-33 also supports the activities of IL-5, a cytokine which is supposed to be the most important regulator of auto- or atheroprotective antibodies. Their nearly immediate availability in an ample quantity makes it possible for natural IgM Abs to exert a non-redundant contribution to the first-line defense against any invading pathogens. Natural IgM Abs are also highly reactive with neo-self or modified self-structures that stem from the debris of their own tissues, which is an inevitable by-product of all defense reactions. These housekeeping functions promote the clearance of damaged cells and their extruded intracellular components which, under normal conditions, are out of reach of the immune system.15,16 On the other hand, whenever necrotic death of its own cells occurs, the now free contents of the cellular interior are recognized by the immune system as inappropriate, potentially harmful particles whose unbridled accumulation might further boost defense responses against the self-constituents, which are now perceived as the alarmins or DAMPs. Natural IgM Abs do their best to diminish the potentially reactive volume of these alarmins. Accordingly, by way of comparable activities, natural IgM Abs are supposed to play protective roles in atherosclerosis. Nowadays, it can be taken for granted that natural IgM Ab of the T15/EO6 clonotype exerts protection from the development of chronic inflammatory plaques through the recognition of atherogenic noxious particles, primordially OxLDL. T15/EO6 IgM displays the capacity to exclude the ingestion of the OxLDL particles via macrophage scavenger receptors CD36 and SRB-1. 17 Consequently, the production of foam cells, the rate-limiting step in early atherogenesis development, is substantially diminished. Into the bargain, apoptotic cells, which originate spontaneously due to accelerated cellular turnover in continuing atherogenesis, exhibit prominent oxidation-specific epitopes on their surfaces. Thanks to an abundance of oxidation epitope-specific IgM Abs in the sera of B1 cell reconstituted mice, it could be shown that the cluster of B1 cell-produced IgM Abs associated avidly with the surface of these apoptotic cells. The clearance of these oxidation-epitope(s)-endowed apoptotic cells is robustly aided by IL-5. In addition, natural IgM Abs possess another protective role which wards off the inflammatory reactions that would be otherwise elicited by oxidized lipoproteins. 18 Apoptotic cells carrying oxidation-specific epitopes dispose of the capacity to activate quiescent endothelial cells so as to increase the adhesion of circulating monocytes on their surfaces. Natural T15/EO6 IgM Abs can inhibit this inflammatory reaction, which likely occurs in vivo, thus, supporting the development of atherosclerosis. It has been reported that IL-33 activates B1 cells which express its receptor ST2. IL-33 can support B1 cell proliferation and, thereby, increase the production of anti-inflammatory and anti-atherogenic substances, such as natural IgM, IL-5 and IL-13 in vitro and in vivo. 19
IL-5 and IL-13, in turn, promote the production of antigen-specific atheroprotective antibodies. By contrast, recent in vitro studies have shown that, in isolated human endothelial cells, IL-33 addition led to an inflammatory activation of the former, with increased expression of adhesion molecules and pro-inflammatory cytokines by way of the ST2 receptor. This finding seems to be in line with IL-33’s membership of the IL-1 family, notably, with the established atherogenic effects of IL-1β and IL-18. 20 On the other hand, these findings omit the plethora of co-regulatory effects which are operative in any intact organism. Most interestingly, IL-33 has been shown to produce virtual anti-inflammatory effects in ischemia-reperfusion injury as well as in atherosclerosis via its ST2 receptor while deteriorating inflammatory endothelial damage in acute renal injury when administered in excess without its receptor. These findings will be touched upon later, after finishing the molecular-based part of IL-33’s effects. (Figure 1)
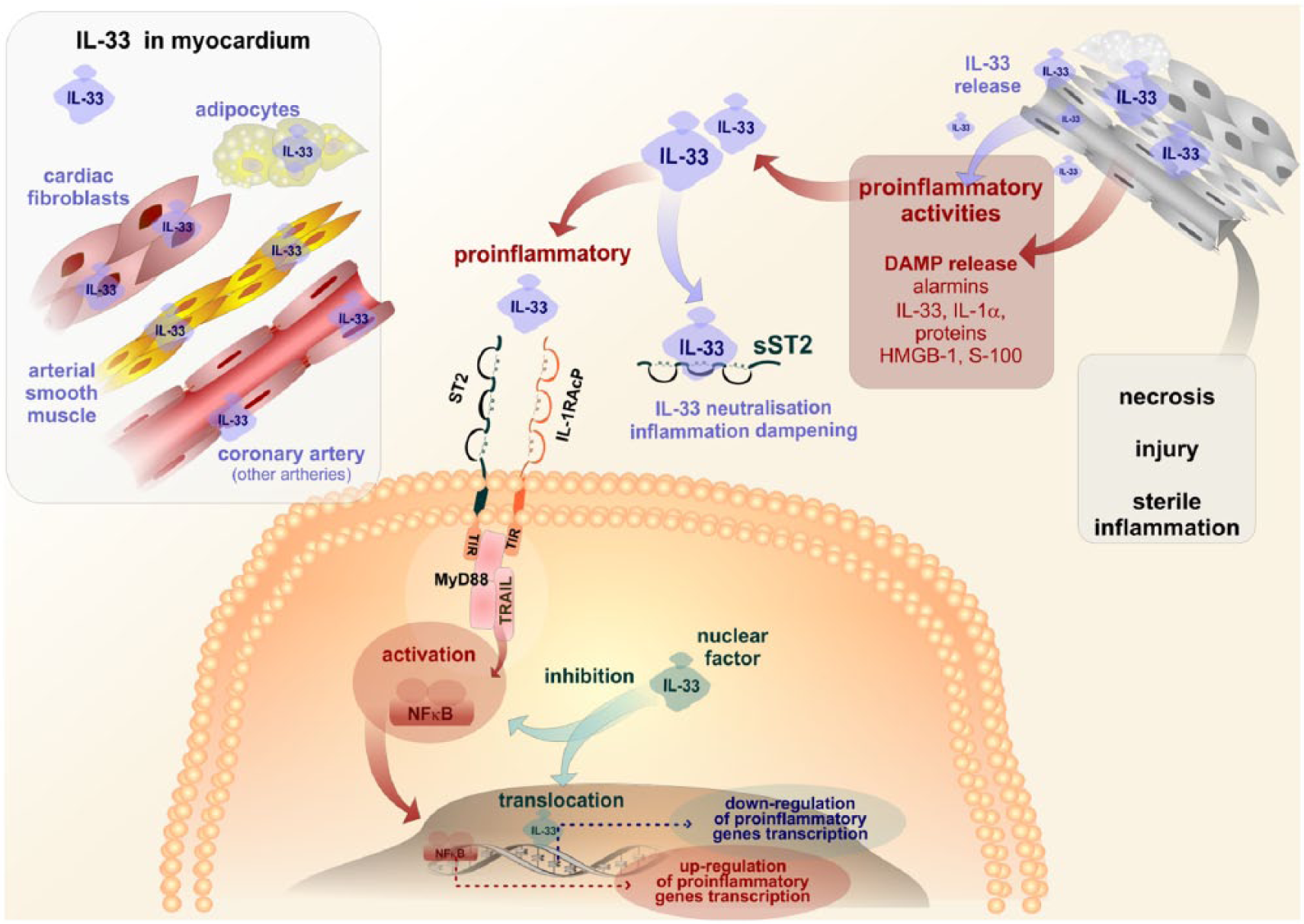
Interleukin-33 is produced by cellular components of the myocardium. Damage of myocardial tissues caused by either necrosis, injury or sterile inflammation is followed by the release of alarmins among which an important role is due to IL-33. Binding of IL-33 to its specific receptor is followed by the activation of intracellular signaling, terminating in NFκB activation and the stimulation of pro-inflammatory genes transcription. In contrast, IL-33 present in cell cytoplasma can serve by both inhibiting NFκB activation and/or stimulating anti-inflammatory gene transcription, thus, displaying its anti-inflammatory effects. In addition, the soluble form of sST2 receptor for interleukin-33 is neutralizing pro-inflammatory activities of IL-33.
Choice of the weapons: IL-33 and gene expression in atherosclerosis
The particular mechanism accounting for IL-33’s protective effects in the arterial wall can be found primordially in the altered expression of genes that are implicated in cholesterol trafficking. The excellent study by McLaren and colleagues has supplied ample material attesting to this idea. 12 To summarize basic principles, internalization of LDL particles by macrophages is carried out via scavenger receptors SR-A, SR-B1 and CD36. 21 Cholesterol efflux out of macrophages is effectuated by so-called reverse cholesterol transport. This is an active process which is brought about by members of the ABC transporter family, namely ABCA-1 and ABCG-1. 22 At the same time, it is dependent on the availability of cholesterol acceptors ApoE and ApoA-I. 23 In these complex events, as McLaren and colleagues have shown, IL-33 is able to down-regulate both mRNA and protein expression in human monocyte-derived macrophages of SR-A, SR-B1 and CD36 and, in parallel, to up-regulate mRNA and protein expression of ApoE, ABCA-1 and ABCG-1. Importantly, the IL-33-mediated increase in ApoE, ABCA-1 and ABCG-1 availability has been documented even in LDL-containing ST2+/+ foam cells.
Intracellular lipid accumulation, which results in underlies macrophage/foam cell conversion, involves not only a disturbance in uptake/efflux ratio of atherogenic lipoprotein particles, 24 but distinct processes regulating the intracellular storage of cholesteryl esters and triglyceride-rich lipid droplets are required as well. 25 Our current knowledge suggests that IL-33 is operative also on the cytoplasmic level. IL-33 decreases the total and esterified cholesterol content in human monocyte-derived macrophages and, even more importantly, in macrophage- or smooth muscle cell-derived foam cells. The mechanisms that are in charge of the respective cytoplasmic volumes of cholesteryl esters and triglyceride-rich droplets are continuously watched over by a network of proteins, the activity of which is, in turn, cytokine-sensitive and responsive to various cytokines, including IL-33. 26
Among many others, the following compounds of particular importance in the above-mentioned process are Niemann Pick type C protein (NPC-1 and NPC-2), carnitine palmitoyltransferase-1 (CPT-1), adipocyte differentiation-related protein (ADRP), acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) and neutral cholesteryl ester hydrolase (NCEH).
Their respective functions can be summarized as follows. NPC-1 and NPC-2 superintend cholesterol trafficking from the late endosome/lysosome compartment to the plasma membrane, thus, up- or down-regulating cholesterol efflux, respectively, in keeping with the demands of the particular tissue(s). 27 CPT-1 limits the accessibility of fatty acids for cholesterol esterification. 28 ADRP heightens triglyceride storage. 29 ACAT-1, in co-operation with the foam cells, esterifies cholesterol within the endoplasmic reticulum, thus, decreasing its cytotoxicity. 30 NCEH steers cholesteryl ester accumulation by regulating its hydroylsis. 31
As to the contribution of IL-33 itself, it can significantly increase NPC-1 and NPC-2 mRNA expression, which results in higher amounts of cholesterol efflux and higher mRNA/protein expression of ApoE, ABCA-1 and ABCG-1 in human monocyte-derived macrophages. In this respect, IL-33 activities resemble the therapeutic impact of the compound ezetimibe which acts in conjunction with the statins or, even, all by itself.
IL-33 can also support CPT-1 mRNA expression, which results in less cholesterol volume within the cell. Furthermore, IL-33 is able to decrease ADRP and ACAT-1 mRNA expression. Down-regulation of these two genes would bring about less cholesteryl ester and less triglyceride cellular accumulation. IL-33 is also able to reduce NCEH mRNA expression, much as the impact of this last effect remains unclear. Collectively, these gene-modifying data underscore IL-33’s effect on restraining the total and esterified cholesterol levels in human macrophages. These achievements of IL-33 are mediated by its membrane receptor ST2L on macrophages or smooth muscle cells, while they are annihilated whenever the soluble isoform sST2 prevails. Such may be the case in atherogenesis.
Choice of the battle-field: IL-33 in the adipose tissue
The lipid-modulating effects of IL-33 extend from subendothelial macrophages transforming (or not) into foam cells to a relatively compact tissue, predominantly the white adipose tissue (WAT). The compactness of WAT is dubious since many of its components are disseminated throughout the body. From the phylogenetic point of view, adipose tissue is a predecessor of today’s immune system which is anatomically composed of the thymus, the spleen and the lymph nodes. Even today, WAT is endowed with the capacity to perform some less sophisticated immune reactions. 32 Actually, adipocytes and macrophages stem from the same precursor cell. Persons affected by obesity exhibit a low-grade inflammation which is reminiscent of, if not identically merges with, that underlying atherogenesis. In obesity, just the same as in atherosclerosis-afflicted vessels, WAT is infiltrated by inflammatory/immune cells; in the first place by CD4+ T-lymphocytes and macrophages, tightly followed by neutrophils, CD8+ T lymphocytes and mast cells. 33 The two inflamed anatomic regions, i.e., WAT and atherosclerotic arteries, reciprocally support each other’s inflammatory conditions. 34 This is an outstanding effect since both WAT’s components and the arterial tree run throughout the human body. WAT inflammation results in tissue insulin resistance despite compensatorily elevated insulin levels. Thus, an increase in circulating insulin which exceeds the requirements of an as yet healthy body to metabolize actual glucose levels heralds the impending development of type 2 diabetes and the metabolic syndrome. 35 As often as not, these two comorbidities accompany atherosclerosis. By contrast, a normal lean WAT is occupied by eosinophils, alternatively activated macrophages, invariant natural killer T lymphocytes (iNKTs) and regulatory T cells (Tregs). Eosinophils appear to be the most important cell population in lean WAT since they produce IL-5. This interleukin, in its turn, contributes to the elaboration of eosinophils in the bone marrow and their subsequent tissue dissemination throughout manifold tissues. Moreover, IL-5, along with appropriate numbers of eosinophils and with fully competent IL-33/ST2 axis, is required for intestinal helminth expulsion. On top of IL-5 and IL-33, chemokines known as eotaxins, namely CCL11 and CCL24, also contribute to eosinophil recruitment to the gastro-intestinal tract. 36 Eosinophils, themselves, produce IL-4 and IL-13, both potent anti-inflammatory Th2 cytokines. By an effective feedback loop, eosinophils produce additional amounts of IL-5 and IL-33. Contrary to lean WAT, the inflamed adipose tissue is pervaded by T-lymphocytes which exhibit a Th1 pattern of activation, elaborating predominantly IFN-γ. The prevalent Th1 immune response in inflamed WAT is active, of course, at the expense of anti-inflammatory Treg and Th2 responses. 37
Importantly, WAT is a rich source of so-called innate lymphoid type 2 cells (ILC2s). 38 These cells may be also called innate helper type 2 cells or nuocytes or natural helper cells. 39 All of these synonyms are, so far, interchangeable. ILC2s are broadly distributed within different body tissues and they are fully active without prior antigenic stimulation. ILC2s display features that are common to other populations of innate lymphocytes, such as NK cells ILC1 and ILC3 and the RORγt-dependent ILC, 40 lymphoid tissue-inducer cells (LTic), innate IL-22 producing cells (NK22, ILC22, NCR22 or NKR+LTic) and innate IL-17-producing cells.
Forewarned is forearmed: IL-33 and gene expression in adipose tissue
All ILCs share the dependence on the transcription factor Id2 and the common-γ chain (γc) cytokine receptor. In response to IL-25 and IL-33, ILC2s proliferate and produce high amounts of anti-inflammatory Th2 cytokines IL-5 and IL-13, thus, counterbalancing or even overriding the Th1/IFN-γ pro-inflammatory response. 41 In this respect, IL-33 exerts its effects predominantly, if not exclusively, on ILC2s, whereas macrophages, eosinophils or other cellular populations are not affected. The addition of exogenous IL-33 to WAT tissue samples results in the increased production of eosinophils and alternatively activated macrophages by ILC2s. Paradoxically, as it may appear, eosinophil numbers are decreased in the spleen and in the bone marrow, evidence of their partial redistribution which occurs in parallel to their increase in WAT. Conventional B or T lymphocytes do not participate in these IL-33-mediated activities in WAT. 42
However, in subjects who are inflicted with really monstrous forms of obesity without as much as trying to get it under control, IL-33, either via direct impact on respective genes or indirectly by way of innate helper type 2 cells, is unable to afford any relief exclusively by itself without the co-operation of such an individual. Large quantities of fat is stored predominantly in endothelial cells in these persons. Adipose tissue microcirculation advances with the engrossing adipose tissue itself, disseminating with each step, however tiny it may be for one course, the low-grade sterile inflammation. If left unaided or, more precisely, if they refuse any appropriate help, such people usually die young. 43
Summed up, IL-33 performs the role of a metabolic regulator in WAT. A Th1/Th2 shift can be inferred from the above findings, backed up by the increase of Treg cells. The latter present another important facet of IL-33’s beneficial activities in atherosclerosis, as will be discussed later.
Last, but not least, the addition of exogenous IL-33 to just-differentiating murine SVF cells (stromal vascular fraction cells, a part of WAT which is made up by interstitial and vascular cells with sparsely scattered adipocytes) significantly inhibits lipid accumulation in the above-described tissue sample. The mechanisms of decreased lipid content in a primarily non-lipid tissue are attributable to down-regulation of mRNA for C/EBPα (CAAT enhancer-binding protein-α), SREBP-1c (sterol regulatory element-binding protein 1c) and liver X receptors (LXR)-α, LXR-β and PPAR-γ, all of them presenting transcription factors needed for proper (and healthy!) adipogenesis to take place.
IL-33 in myocardial infarction and/or solid organ transplantation: the V-day for ischemia-reperfusion injury
Apart from IL-33’s beneficial activities in atherogenesis and in adipose tissue remodeling, this cytokine also seems to exert protective effects in anoxia-reoxygenation or ischemia-reperfusion injury. If confirmed by human studies, exogenous IL-33 may expand its therapeutic impact on myocardial infarction. In reality, just the same as in atherogenesis, the favorable activities of endogenous IL-33 may be incessantly at work without our knowing it. If such is the case, the outcome of IL-33’s effects in ischemia-reperfusion would depend on the addition of the individual’s conditions on the local (organ) level as well as of his/her conditions on the systemic (whole body) level, not to dismiss, among other variables, the quality of the admitting hospital.
Reperfusion of an ischemic or even anoxic tissue/organ brings about death by the apoptotic route to many cells that have as yet survived despite the previous inaccessibility of oxygen. In the heart, vascular endothelial cells and cardiac muscle cells are more susceptible to this mode of death than fibrocytes making up the interstitial tissue. Apoptotic cellular death differs from necrotic death, among other features, by the persistence of the integrity of the outer cellular membrane in the former case. Thus, intracellular components, including nuclear IL-33, are not liberated out of the dying cell. Instead, its total volume shrinks and the shrunken dead cell is removed from its original site within the tissue by the immune system without eliciting, in contrast to the necrotic cell, an inflammatory reaction. As it is, an apoptotic cell is eliminated by an “immunologically silent” mode, although the whole process is far more complicated than that, but its details cannot be dwelt upon in this article.
Morphological signs of apoptotic death are defined primordially by internucleosomal DNA fragmentation. Under quiescent conditions, the DNAse responsible for DNA cleavage dwells within the cytoplasm in an inactive form. After the pro-apoptotic stimuli have set in, this apoptosis-specific enzyme is activated by way of proteolytic caspases. Caspase 3 is probably the most important enzyme isoform which is activated during reperfusion. Another pro-apoptotic enzyme is c-Jun N-terminal kinase (JNK), a member of the mitogen-activated protein kinase (MAPK) family. The whole of the MAPK enzyme family is readily activated by reactive oxygen species which are generated in excess during reperfusion. Third, the protein kinase C (PKC) constitutes a serine/threonine PKC family that consists of at least 11 isoforms. From among these, the conventional (c-PKC) isoform PKCβ is preferentially activated by ischemia/reperfusion, itself generating a trigger for the activation of the MAPK kinase JNK. Genetic ablation of PKCβ obliges its oxygen-deficient tissue with heightened resistance to reperfusion injury. Recent findings might open a new vista in the treatment or an amelioration of results in human myocardial infarction by diminishing PKCβ activities via the introduction of exogenous IL-33. IL-33, itself, is richly expressed in the human heart. The mechanism by which IL-33 safeguards the heart from ischemia-reperfusion injury resides in the down-regulation of apoptosis-eliciting enzymes mechanisms. 44 Experimentally, the total exclusion of IL-33 from investigated animals results in larger areas of infarcted/necrotic myocardial tissue, notwithstanding whatever preventive measures have been taken. The devastating effects of IL-33 deficiency are extraordinarily manifest in mice with type 2 diabetes mellitus. 45 As can be expected, an IL-33-treated heart exhibits less myocardial necrosis, improved cardiac contractile performance and a better overall survival rate than hearts of untreated animals under the same ischemia-reperfusion conditions. 46 Of note, the presence of the membrane receptor ST2L is indispensable for these favorable effects to occur. We will encounter this simple sentence again and again since, in ST2L−/− mice, no salutary effects of IL-33 are apparent during reperfusion despite immeasurable, apparently never-ending quantities of IL-33 being available. The soluble receptor isoform, sST2, when administered in excess to neutralize IL-33’s biological potential, ablates the beneficial activities of IL-33 down to the level of untreated animals. This is another quasi-eternal truth in myocardial infarction acknowledged by Seki et al., 47 otherwise confirmed by multiple authors in different situations in which IL-33 and sST2 were confronted. 48
IL-33 associating with its ST2L membrane receptor eventually steers the activities of the transcription factor NF-κB. Canonically, NF-κB takes place in many pathologic processes damaging the heart muscle. Most often, nuclear NF-κB is activated by multiple stimuli which converge on the cellular IL-1R/TLR receptor complex, which is used by the IL-33/ST2L axis as well. Intracellular transmission of the signal occurs by way of multiple check-points, the most important and best known being MyD88, IRAK, TNF receptor–associated factor–6 (TRAF6), TAK1 and/or MAPK. 49 This is by no means an exhausting enumeration. Dependent on the actual route taken, NF-κB (more often) behaves according to expectations in a pro-inflammatory manner. This behavior also includes the prevention of loss of viable cardiomyocytes or it arrives at an alternative program, partially contrary to the former, but, most importantly, in its final result, producing either anti-apoptotic proteins or acting against angiotensin II and/or ROS. It even seems that this endogenous program at least (and probably for a limited time) equals the activities of statins. Be that as it may, these effects of activated NF-κB are of vital importance in myocardial infarction. By virtually the same way, IL-33 confers protection to a re-oxygenated liver during hepatic transplantation. 50
IL-33’s anti-apoptotic activity relies on up-regulation of the following genes: XIAP, cIAP1, cIAP2, survivin, Bcl-2 and Bcl-xL, all of them being NF-κB-regulated proteins which promote cell survival instead of death. IL-33 was shown to increase the expression of cIAP1, XIAP, survivin, Bcl-2 and Bcl-xL in the infarct area, while sST2 excluded this effect.
IL-33 and two cardiac cells: the cardiomyocyte and the myofibroblast
Besides the ischemia followed by reperfusion when IL-33 abrogates superfluous apoptotic death of potentially viable cells, there is another setting in which IL-33 impacts NF-κB to counteract its well-tried activities. This setting is the failing heart, either acutely or chronically, owing to an excessive loss of functional cardiomyocyte fibres, which are either being or have been replaced by dysfunctional fibrocyte fibres. Not so easy to write down, even less to read out, and far from easy to live with. The contractile performance of the heart inevitably declines, so that, in the end, the heart is made up predominantly of fibrocytes aided by rapidly accumulating fibroblasts, with the functional cardiomyocytes having vanished, either by apoptotic or necrotic death. The heart chambers lose their original, functionally advantageous elliptical shape which is, instead, replaced by a pathologic spherical shape which, by no means, can generate an expulsive force to propel the blood into the body comparable with the initial, healthy chamber. This is a relatively frequent condition in elderly people after a myocardial infarction. Clinically, it is known as myocardial remodeling. 51
Underlying causes may be manifold and, frankly, in this progressive stage their origin, is of no practical relevance to IL-33’s action. It is the result that counts. IL-33, if permitted to intervene in time and with needed intensity, might down-regulate or even extinct, the latter only rarely, worse luck, due to fragile personalities of thus afflicted patients, the literally diabolic effects of angiotensin II. 52 The heart is now delivered to the mercy of it. Ang II acts on the IL-33/ST2L myocardial cell membrane with a closely related intracellular signal transduction. Some negligible deviations on its intracellular road (compared AngII and IL-33) do not represent any grave obstacle(s) to reach the transcription factor NF-kB which, upon AngII’s order, will set off proteosynthesis which may include accumulating fibroblasts and a rich mixture of predominantly malevolent cytokines. Thus, a vicious circle is set up, ever more deteriorating the broken heart’s function. By contrast, IL-33 behaves like a virtual endogenous angiotensin II-converting enzyme inhibitor (individual pharmacologic compounds are collectively referred to as ACE-Is) and an angiotensin II receptor antagonist (individual pharmacologic compounds collectively called as sartans). Endogenous catecholamines are also increased in cardiac remodeling and join Ang II in its would-be praiseworthy effects. However, if the damage inflicted to the heart is serious, irrespective of its nature, the salutary effects of IL-33, modest already from the beginning, give up their trials to reverse the situation and the heart ultimately fails. 53 Accumulation of the IL-33 cytokine without proper access to its membrane receptor, which is occupied with ever less IL-33 and ever more angiotensin II, would present an accumulation of biologically active mediator which lacks its corresponding partner.54-58 Although caspases would shorten its molecule and, thus, diminish its overall activities, the number of “orphaned” IL-33 molecules would present a problem all the same. Luckily, in such a case, there is the decoy receptor sST2 which, in its function, is fully comparable to, or even identical with, the IL-1Ra (interleukin-1 receptor antagonist). Mutual binding of IL-33 + with sST2 gives rise to functionally inactive heterodimers whose value is seemingly close to null. However, here we encounter another both interesting and important phenomenon: what do the heightened levels of sST2 (the IL-33 part escapes measurment and, frankly, knowing its volume precisely is of no practical use) do?
Since myocardial fibrosis and the physical strain inflicted on the heart by the remodeling process enhance irrevocable heart failure, meticulous studies in human patients have shown that circulating sST2 in the setting of heart failure is heralding a messenger of a poor prognosis. Not only so, previously there had been many biomarkers advocating such and such prognosis. But, levels of sST2 proved to be slightly superior to those of brain natriuretic proteins, the indisputable royal family golden standard predictors of heart failure prognosis forecasters. Continuing concomitant measurements in parallel with the two biomarkers substantially augments their overall prognostic value, unequivocally surpassing that conferred by the most meticulous echocardiographic measurement, which offers a precise picture of the failing heart as a whole or as a cellular mosaic made up of different cardiomyocytes, some of them still relatively competent others already worn out. Echocardiography is (not yet?) able to measure physical forces which must withstand a single cardiac cell or the heart as a whole. 59
IL-33: a road back to the endothelial cells
Thus far, it might appear that IL-33 is the sought-after panacea for cardiovascular diseases. However, its scarcely foreseeable co-operation with microenvironmental cellular populations has already been described, although much remains to be added, at least what is foretellable at this point of knowledge. 60 It can be said already that skewing the Th1/Th2 immune response does not unequivocally guarantee the down-regulation of Th1-associated cytokines. The particular scenario may dress up unforecastable appearances which, into the bargain, may vary in line with the plasticity and the versatility of the action itself, including the players representing both the attack and the defense. One of the cornerstones of this novel comprehension is the knowledge that the IL-33/ST2 axis by itself can produce substantial amounts of IFN-γ, the master regulator cytokine in the pathogenesis of atherosclerosis. By contrast, the manifold IL-33 interactions can result, often irrevocably, in the down-regulation of the production of IL-10, the most important anti-inflammatory cytokine known so far. This last fact or effect is the more surprising in that the IL-33/ST2-mediated Th1/Th2 switch underlies enhanced production of other anti-inflammatory cytokines, IL-4, IL-5 and IL-13. 61 Furthermore, IL-33’s impact on the nuclear transcription factor NF-κB results in the preferential production of anti-apoptotic proteins which ultimately rescue apoptotic death-bound cells. Whenever IL-33’s intracellular signal is transmitted by way of little different cytoplasmic check-points, it eventually activates NF-κB in a “classical” pro-inflammatory fashion, in just the same way as do IL-33’s co-fellows from the IL-1 family. IL-33-borne information may conceivably target transcription factors other than NF-κB, usually with an indisputable pro-inflammatory effect. 62
On an individual cell level, this enhanced pro-inflammatory activation has been manifested when excess IL-33 was added to in vitro-cultivated endothelial cells. In cultured human endothelial cells, however, IL-33 was shown to elicit inflammatory activation which was evidenced by increased vascular permeability, increased production of pro-inflammatory cytokines, increased expression of adhesion molecules ICAM-1, VCAM-1, E-selectin and the chemokine MCP-1 (CCL2) and increased adhesion of leukocytes to the endothelium under both static and flow conditions.
Quite recently, it has been shown that IL-33 drives an inflammatory response in cultured human umbilical vein endothelial cells (HUVECs). 63 This response was highly similar to that elicited by IL-1β. However, there was one prominent difference: while IL-1β activated both quiescent and non-quiescent endothelial cells, IL-33 activated selectively and uniquely the latter, i.e., the non-quiescent cells. The respective response went hand in hand with decreased levels of the IL-33 receptor ST2L and, hence, decreased activation of the downstream mediators NF-κB, p38 and JNK in quiescent cells. The different molecular responses translated into a phenotypical divergence in quiescent and non-quiescent endothelial cells, which was evident in the amounts of target proteins expression. The indisputable result claims that quiescent endothelial cells are resistant to the activation by extracellular IL-33, being maintained in this respective state by cytokines and/or transcription factors which cannot be overcome by IL-33 itself. The authors concluded that, in endothelial cells, IL-33 elicited intracellular signal transmission involving kinase phosphorylation such as had been reported to occur in response to IL-33 in polarized Th2 cells and mast cells. On the whole, the authors showed that endothelial cell activation, manifest as phosphorylation of NF-κB, JNK and p38 mitogen-associated protein kinases, was higher in non-quiescent, i.e., proliferating cells, as compared with quiescent cells that were stimulated with IL-33 instead of IL-1β, whose effects were far less pronounced in this particular cellular population. Effectively, the action of IL-33 requires previous endothelial cell pre-activation by another pro-inflammatory mediator. Otherwise the endothelial cells remain unresponsive to IL-33 which, on the other hand, might bring about protection from bystander damage during inflammatory responses and, conversely, cells which have already entered the inflammatory reaction can contribute to. 64 Moreover, IL-33, thus, contributes to wound healing, including neovessel formation.
Non-responding cells, i.e., quiescent endothelial cells, keep high amounts of IL-33 in their nuclei, which is in line with prior observations. This nuclear IL-33 reserve, by way of dampening NF-κB-elicited pro-inflammatory signaling, might represent a “molecular brake” to prevent undue activation of endothelial cells in inflammation-exposed vascular segments. 65 The functional dichotomy of IL-33, thus, goes on.
IL-33 and T helper cells: a well-regulated mix-up
And yet, despite all these warnings, the IL-33/ST2 axis has maintained its originally surmised and later questioned endothelial- and athero-protective activity. It should be all the same, conceded that the Th1/Th2 switch in the overall immune responses probably plays a minor role in this process. According to up-to-date knowledge, IL-33’s athero-protective effects are carried out by its co-operation, i.e., induction and maintenance, with another CD4+ T cell lineage, namely the regulatory CD4+ T cells or Tregs.
After the thymic output, where they are relatively excluded from the environmental signals, CD4+ T cells undergo activation in the periphery when they encounter various antigens, most of them in the form of a digested peptide in association with the major histocompatibility complex (MHC) molecules. The original conception of the steps necessarily leading to a T-cell lineage commitment consisted of a single fate model pointing out the direction of T cells to several helper (Th), follicular helper (Tfh) or regulatory (Treg) phenotypes. 66 However, although a single lineage-committed T cell may best perform a single function, this conception of a single fate for T cells has been challenged. According to the new paradigm, the CD4+ T cells are much more versatile, with the capability to transit between seemingly steady phenotypes, which have been just described, such as helper phenotypes, helper and follicular helper, and helper and regulatory functions.
This last quality directly affects our theme, given the plethora of complex events impacting the differentiation of CD4+ T cells, evoked either by individual pathogens or a clusters of sterile noxae. 67 There are three signals, occurring either sequentially or concurrently, which activate transcription factors (TFs) and translocate them from the cytoplasm into the nucleus. Once in their destination, TFs bind to cis-regulatory elements, such as promoters, enhancers, insulators and silencers, within the gene promoter region so as to translate extracellular signals to downstream transcriptional programs. Target gene transcription and translation convert naive CD4+ T cells into mature cells with distinct features, such as the expression of specific surface receptors and adhesion molecules, cytokine/chemokine producing capacity and the activation of distinct metabolic pathways.
Differentiated or mature CD4+ T helper (Th) cells can be both defined functional individuals and discerned from one another by their prevailing cytokine-producing capacity, interferon-γ (IFN-γ)-producing Th1 cells, interleukin-4 (IL-4)-producing Th2 cells, interleukin-17A-producing Th17 cells and interleukin-9-secreting-producing Th9 cells. 68
Having attained the state of their respective maturation, Th cells can mobilize and activate antigenic-naive cells, re-enforce other CD4+ Th cell commitment and conduct local tissue immune responses, e.g., through lymphokine secretion. 69
In addition to a cell helper phenotype, naive CD4+T cells can also transform into follicular helper T cells (Tfh) which are specialized for B-cell help. On the other hand, naive CD4+T cells can acquire a regulatory (Treg) function which brings about important immunosuppressive capacities. Tregs are as versatile and heterogenous as are all the other T-helper cells, since both of these cellular groups’ origins are in highly comparable, if not identical, conditions. Step by step, several functionally distinct subgroups are constituted: Foxp3+ natural Treg (nTreg) which rise in the thymus where they are instigated by several self-antigens; inducible Foxp3+ (iTreg) cells, which rise in the extra-thymic milieu in response to exogenous antigens.
In order to complete this section, we will also mention non-Foxp3-expressing Treg cells, which include TGF-β-secreting or Th3 Treg, IL-10-secreting or Tr1 Treg and IL-35-secreting or Tr35 Treg.70,71
The transcriptional activities of CD4+T cells to ensure their maturation and differentiation into Th, Tfh and Treg cells, are composed of a suite of TFs and signal transducer and activator of transcription (STAT) molecules. The most illustrative examples of their requirements are listed as follows: the TF T-bet and STAT-1 and STAT-4 for Th1 differentiation, the TF GATA-3 and STST-5 for Th2 differentiation, the TF RORγt and STAT-3 for Th17 differentiation, the TF Foxp3 and STAT-5 for nTreg and iTreg, PU-1 for Th9 differentiation and Bcl6 for Tfh differentiation. PU-1 (Th9) and Bcl6 (Tfh) require an additional transcriptional regulator of an as yet unknown nature to coordinate full transcription.
Foxp3 appears to be restricted to Treg cells, hence, it is called “the master regulator” of Treg cell development and function. Notwithstanding, there are data which support the idea that Foxp3 by itself is not capable of maintaining the Treg phenotype in the ever-changing environment of inflammation. Therefore, a highly regulated network of transcriptional co-factors cooperating with Foxp3 is urgently needed to support Treg cell differentiation and activity. For a relatively long time, the paradigm that CD4+T cells are destined to a particular fate (Th1, Th2, Th9, Th17, Tfh, Treg) seemed fully justified. Recent studies, however, have cast doubt on this single-fate model. Step by step, a significant extent of plasticity between the respective T-cell destinies has come out. In our text, it will be conspicuous in Th1 versus Th2-biased patients afflicted by atherosclerosis.72,73 That is why we embodied this, maybe a little tedious, overture to the concluding parts which otherwise might be hardly understandable for some readers.
IL-33 and regulatory T cells: united we stand
It has been already evidenced that Th1 and Th2 cross-regulate one another. Th1-associated transcription factor T-bet inhibits Th2-related GATA-3 and vice versa. 74 It is of much interest that lymphocytic choriomeningitis virus (LCMV)-polarized Th1 or Th2 murine lymphocytes included comparable numbers of IFN-γ (Th1-specific) cells and that Th2-polarized cells included substantial amounts of both IFN-γ and IL-4 co-expressing cells. These are only the most outstanding examples of interactions among presumably unequivocally committed cells. It stands to reason that some substances from other origins modify these results in vivo. All the same, multiple TFs are co-expressed in Foxp3+ Treg cells whereby they are essential for the latter’s function. 75 Since these new findings are highly interesting, but they deviate too far from our basic theme, we must, with deep regret, leave them out.
However, we cannot skip the finding that Foxp3+- expressing Tregs (notably – but not exclusively – human CD4+CD127lowCD25+Foxp3+ T cells) are able to produce IFN-γ. This is, of course, in direct contradiction with the mission of Tregs which resides in counterbalancing, in the first place, the pro-inflammatory activities of the Th1 CD4+ T cell subset; the paramount mediator of the latter is IFN-γ. The ultimate result will be shaped by the ratio of the respective CD4+ T cell subsets, including the character of the substances (pro- or anti-inflammatory in their net addition) whose secretion by other cells prevails both in the microenvironment and on the level of the whole body. 76
IL-33 and Tregs in atherosclerosis
And now, what about the Treg cells in atherosclerosis? It has been found that, in patients, both in a compensated, quiescent state and more so in acute cardivascular and cerebrovascular complications, the overall number of Tregs is substantially diminished compared to healthy controls.
The beneficial impact of Tregs is, thus, beyond any doubt. 77 Experiments were carried out to heighten their numbers in menaced population, but, so far, they collided with varied obstacles. When administered exogenously from a donor, difficulties, arising from white blood cells transfusions or for any other reason, must be solved, taking into account that each malady requires specific access. Mucosal vaccination by way of oral or nasal administration seems to be a more promising approach, yet there are still numerous failures of technical rather than of conceptual origin to broaden this method into daily clinical practice. 78
IL-33 and Tregs in laboratory animals
And yet, there have been some early encouraging results obtained on laboratory rodents. 79 What is in line with our topic is that administering exogenous IL-33 led to an increase of protective Tregs, both in “classical” acute coronary syndromes 80 and in substantially better graft tolerance in animals after cardiac transplantation. 81 In the former of these last two studies, the authors confirmed their earlier observation that Tregs, which support the Th1 to Th2 transition with some relief to the diseased arterial wall due to the down-regulation of the Th1-originating pro-atherogenic cytokines IFN-γ, IL-12 and IL-18, are really effective in healthy, but not in atherosclerosis-prone ApoE−/− mice.82,83 The diseased animals – i.e., those with established atherosclerosis and diminished levels of Tregs – manifested increased levels of the IL-33 decoy receptor sST2. One logical explanation, according to the authors, could reside in some form of damage or dysfunction to the membrane receptor ST2L, which otherwise would welcome the increased levels of IL-33 to enter the afflicted cells so as to secure, therein, as much order as could be achieved. However, due to the inaccessibility of diseased vessel cells by way of the IL-33/ ST2L complex – endothelial cells on top of the CD4+ Th2 lymphocytes, the “orphaned” IL-33 is quickly seized and eliminated from any action by the decoy receptor sST2. In this situation, sST2 behaves just in the same way as does the IL-1 receptor antagonist in relation to IL-1β. As it is, ApoE−/− mice with established atherosclerosis exhibit diminished numbers of ST2L+ Th2 CD+ lymphocytes with virtually no ST2L+ Tregs in vivo. An ex vivo experiment documented that this is not a final, unmoveable situation. Luckily, splenic Th2 lymphocytes, enriched exogenously in IL-33, acquired the qualities of IL-33 fully responsive cells.
Atherosclerosis and the Th1/Th2 balance: do not break up the sacrosanct equilibrium
It is ruthlessly repeated here that atherosclerosis is an inflammatory setting in which Th1 cells promote the underlying pathologic process whereas Th2 cells act in the opposite way, aided, when doing so, by the IL-33/ST2 complex. Its beneficial effects reside in skewing the Th1/Th2 balance, thus, increasing the levels of anti-inflammatory mediators, among them interleukin-5, taking care of the production of anti-atherogenic antibodies while concomitantly decreasing the levels of pro-inflammatory mediators, the net result being the down-regulation of foamy cell formation and, in increasing the generation of anti-inflammatory regulatory T cells or Tregs, due to the presence of the ST2L receptor on their plasmatic membrane directed unreservedly in the anti-atherogenic fashion. 84 The idea that developing or not developing atherosclerosis in a human being is conditioned by an individual genetic program with external injurious noxae striking against a more or less rather resistant or more susceptible organism has been conceived for some time. There were even some partial results of different studies available, concluded on the basis of both antagonistic mediators and/or of leukocyte subsets. 85 This idea was experimentally reinforced in 2008 by Schulte et al. 85 on the basis of a prevailing inborn Th1 or Th2 controversy reactivity. In their experimental set-up, there were C57BL/6 versus BALB/c ApoE−/− mouse strains, displaying an inborn strictly opposite T-cell subset polarization. Whereas the former strain corresponded fully to the Th1 orientation, the latter recapitulated spontaneously the Th2 bearings. Both groups were fed high-cholesterol diet. While the Th1-slanted group developed significantly more atherosclerosis, with a rapid progression accompanied by higher levels of “classical” inflammatory biomarkers, the latter strain remained relatively spared, with a substantially delayed onset and significantly retarded development of the disease. Inflammatory/immunological parameters that were measured coincided precisely with those followed in other T cell plasticity researches. Despite the scientific plausibility of these studies, including those which are not quoted here for the lack of space, it was difficult to draw any finite conclusions from animal studies, let alone to extrapolate them into human pathology. Engelbertsen et al. published, in 2013, results of a study which encompassed 700 participants followed for at least 15 years and they conclude that the prevailing CD4+ Th2 lymphocyte phenotype confers protection from potentially lethal complications of ischemic vascular diseases, such as myocardial infarction and stroke. 86 Tracy et al., on the other hand, showed that individuals inherently biased towards a Th1 immune response are more susceptible to developing atherosclerosis. 87 Thus, least of all, the Th1/Th2 immune controversy and the IL-33/ST2 axis cannot be ignored in estimating the pathogenesis of atherosclerosis and heart failure.
The ST2 receptor: joins the scenario a close-up view
The ST2 receptor has been known since well before IL-33; even the former’s excessive occurrence in the bloodstream of heart failure patients, whether acute or chronic, was measured and evaluated as a valuable means to determine life prognosis. With the later advent of the ligand IL-33, these mutual relations were further refined so that today, in heart failure patients, circulating levels of sST2 affords as much and as exact prognostic information as do natriuretic peptides, i.e., B-type natriuretic peptide (BNP) and its amino-terminal cleavage fragment, N-terminal prohormone BMP (NT-proBNP). There is now a consensus that ST2 and BNP yield the most reliable prognostic information of all relevant biomarkers, with ST2, according to some authors, being even superior to BNPs in this respect. 88 BNPs are elevated in heart failure patients who are characterized by poor ventricular systolic and diastolic function, dilatation of the heart chambers, valvular incompetence inclusion in many cases due to the loss of essential heart chamber geometry and pulmonary hypertension of various degree due to the aforementioned affections.
Beyond the natriuretic peptides, growing interest has been attracted by biomarkers that are pathophysiologically implicated in the deleterious process subsequent to wall stretch, clinically known as ventricular cardiac remodeling. This process abolishes the original, ellipsoid shape of the heart chambers, predominantly of the left ventricle, and converts it to a pathological globular shape. The overall thickness of the remodeled heart chamber’s wall may be thickened, with a total inner dimension volume next to normal; or it may be get thinned, whereby, the afflicted chamber dilates. At any rate, the underlying substantial loss of functional cardiomyocytes is replaced by functionally valueless fibrous tissue. In the end, individual remaining cardiomyocytes are exposed to pathologically increased mechanical strain. Interestingly, if a tissue of fully competent cardiomyocytes is exposed to angiotensin II, a mighty pro-hypertrophic mediator, the latter evokes changes in the healthy myocardial tissue that are scarcely discernible from the remodeling process. The administration of IL-33 is able to rescue the myocardial phenotype to the healthy level, provided the cells are expressing the ST2L receptor in their membranes, which is the case in normal conditions.
IL-33: the good guy within the human heart
As a member of the IL-1 superfamily, ST2 was discovered when searching the screen of gene transcripts by mechanically stressed cardiomyocytes. 89 Under biomechanical stress, both forms of the IL-33 receptor, i.e., ST2L and sST2, have been found in cardiomyocytes and fibroblasts, respectively. In an in vivo model of pressure overload, endomyocardial biopsies from mice deficient in ST2 exhibited a more severe degree of cardiomyocyte hypertrophy and interstitial fibrosis along with a much worse fractional shortening, a sign of poor cardiomyocyte function, than in wild animals. 90 Sanada evoked much interest concerning the actual role of the IL-33/ST2 axis in the heart. 52 It could be concluded that IL-33/ST2 signaling may protect against adverse cardiac remodeling in vivo, a finding that was further corroborated by work of other authors. Furthermore, cardiomyocytes from ST2-deficient mice (ST2L−/− cardiomyocytes) displayed a higher expression of transcripts encoding for natriuretic peptides, which are, as yet, the most important endogenous protective mediators in hearts subjected to external pathologic forces and, subsequently, pathologic internal forces occurring within the heart wall. In this last case, the administration of exogenous IL-33 down-regulated expression of BNP transcripts, testifying of improved biophysical conditions implemented within the heart wall(s). As could be expected, IL-33 was helpless in ST2−/− cardiomyocytes. Current conception claims that intact ST2 signaling in the cardiomyocytes plays a deciding role in the ability of IL-33 to repair cardiac pressure overload states. And, just the same as was seen in atherosclerosis, an insufficient amount of IL-33, mainly relative to the available number of cellular ST2L receptors whose actual number in a critical moment exceeds that of IL-33 even if IL-33 is increased in absolute numbers, results in the delivery of “unemployed”, superfluous IL-33 into the custody of free sST2. With the utmost probability, it is this “IL-33-enriched” sST2 whose increases are measured and evaluated in heart failure patients.91,92
With this paradigm shift in our understanding of IL-33 from a mainly, if not exclusively, immunologic mediator to its increasing employment in primarily non-immunologic states, albeit the transition between these two conditions is always subtle in its nature, much new data on IL-33 have accumulated lately. As indicated in the headline of our paper, our attention will continue to focus on its latter position. Activated dendritic cells and macrophages are the only hematopoietic cells that exhibit some minor amounts of IL-33-specific mRNA. Endothelial cells, in contrast, seem to be the principal cellular population producing IL-33 in different human organs and tissues. Since the vascular tree is ubiquitous, so might be IL-33’s distribution, albeit with some preferences and some omissions. IL-33 is constitutively expressed in the nuclei of endothelial cells in both small and large blood vessels.93,94 In addition to endothelial cells, IL-33 is further expressed in epithelial cells, smooth muscle cells, fibroblasts and keratinocytes.
In human endothelial cells, IL-33 elicits inflammatory activation via the up-regulation of IL-6, IL-8 (CXCL8), monocyte chemoattractant protein-1 (MCP-1/CCL2), vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1) and endothelial selectin (E-selectin), increases vascular permeability and promotes angiogenesis.95,96
IL-33 within the human heart: even the good guy may evade obeisance hand in hand with pro-inflammatory cytokines
Expression and regulation of IL-33 in human adult cardiac myocytes and fibroblasts has been studied lately by Demyanets and colleagues. 88 They have furnished deeper knowledge concerning the occurrence of individual parts of the IL-33/ST2 complex in the cardiovascular system. We can disclose only their most important ones, according to our opinion.
The authors showed that pro-inflammatory cytokines are known to initiate almost any inflammatory reaction and are also known as “proximal” cytokines, i.e., TNF, IL-1β and IFN-γ up-regulated IL-33, both mRNA and protein, in cardiac myocytes, fibroblasts and vascular smooth muscle cells. All three “proximal” cytokines were shown to induce the intracellular accumulation of IL-33, but not its extracellular secretion, from intestinal epithelial cells, synovial fibroblasts, stellate cells and adipocytes/pre-adipocytes. 97
These findings corresponded to those acquired earlier in human cardiac fibroblasts and myocytes. In order to identify cellular targets for IL-33 that was generated by cardiac myocytes and fibroblasts, the authors screened different cell types isolated from the human heart and its adjacent vascular beds for the presence of specific mRNA for ST2 isoforms and for the macrovascular endothelial cells isolated from the aorta and coronary arteries and for the microvascular endothelial cells isolated from the myocardial tissue. All these cells expressed total ST2, ST2L and sST2 mRNA and secreted sST2.
IL-33 within a single cell: save what can be saved; an example of a tight cage
On the other hand, cardiac myocytes, as well as cardiac fibroblasts and/or vascular smooth muscle cells from coronary arteries or the aorta, express only minor levels of total ST2, ST2L and sST2 mRNA and do not secrete detectable amount of sST2 antigen. Thus, the human heart falls in with the general concept of IL-33/ST2 axis function. Primarily, IL-33 in its fully active, i.e., full-length form, is expressed in nuclear chromatin where it represses gene transcription within its maternal cell. Under quiescent conditions IL-33 never leaves the cell of its production. Whenever its maternal cell dies by necrotic death, i.e., its outer membrane is disrupted to provide a gate for IL-33 out of the cell, this interleukin quits the cell of its production. Full-length IL-33, however, would be too dangerous for the adjacent cells. Therefore, its coiffure by caspases diminishes the size of its molecule, thereby, substantially down-regulating its inherent pro-inflammatory activities. Nevertheless, IL-33 is now active as an alarmin, alerting those cells of the Th2 immune system which are equipped with the ST2L receptor.
IL-33’s on the road: nuclear chromatin is not such a tight cage; it is an escapable dwelling
The possibility/impossibility of IL-33 to quit an undamaged cell with intact continuity of its outer membrane remained an enigma for a relatively long time, since there was no plausible explanation for the extracellular presence of IL-33 in a well-functioning organism. It is true that its nuclear localization and the lack of an export signal sequence maintained IL-33 within the cell, but its extracellular occurrence could not be denied all the same. Kakkar et al. were successful in showing that IL-33 localized simultaneously to nuclear euchromatin and to membrane-bound cytoplasmic vesicles. 91 Furthermore, the authors detected a dynamic nucleo-cytoplasmic flux of IL-33 within its parent cell (or within its cage, metaphorically spoken), which was dependent on nuclear pore complex function. Of note, in murine fibroblasts, both in vitro and in vivo, it was the mechanical strain which gave rise to the secretion of IL-33 in the absence of any cellular damage (necrosis). 98 This set of information fitly complements the adaptive/maladaptive program which is set in motion by heart failure. As such, these data confirm IL-33 dynamic inter-organelle trafficking as well as IL-33 release by way of an undamaged outer cellular membrane during mechanical overload. IL-33 release out of a living cell is now an occurrence beyond any doubt. The process is augmented in the setting of a non-fatal cellular stretch, particularly, in such mechano-sensitive cells such as the fibroblasts which, as we now know, will replace, to a varying degree, dead cardiomyocytes. It is hypothesized that IL-33 is trafficked for release from living cells via non-classical mechanisms upon sub-lethal biomechanical strain. In tracing IL-33’s intracellular fate, it has been found that IL-33, itself, is a multi-organelle protein that is released from mechanically stressed cells. Newly synthesized molecules are initially shuttled into the nucleus in order to associate with euchromatin. It is here that IL-33 makes use of the nuclear pore complex so as to transit into the cytoplasmic space and reside in membrane-bound vesicles. 99 The currently acknowledged paradigm says that endogenous IL-33 interacts with heterochromatin and mitotic chromosomes, but also in the nuclei of non-proliferating cells. Nuclear localization of ectopically expressed, full-length IL-33 critically depends on a homeodomain-like helix-turn-helix motif in its N-terminus, a region that also functions as a chromatin-binding motif (CBM). The CBM mediates binding of IL-33 to histone dimers H2A-H2B on the surface of nucleosomes and, hence, reorganizes chromatin architecture. As a result, IL-33 represses gene transcription. 100 Although gene silencing is usually associated with facultative or constitutive heterochromatin, it has been shown as well that the expression of genes within transcriptionally active euchromatin is usually modified by miscellaneous factors or protein-based transcriptional modifiers. 101
IL-33: a laudable poet for the heart
The evidence of nucleo-cytoplasmic traffic of IL-33 and the protein’s location in cytoplasmic vesicles set up an ideal route for IL-33 release in response to a biomechanical strain. It must be taken into account that the applied biomechanical stimulus stretching the target cell(s) may be biologically “pure” and exclusively physical in its nature. Nevertheless, if a whole heart is employed in the assay instead of cardiac tissue, the concurrent impact of manifold and anti-inflammatory mediators that are produced by the failing organ in a vain attempt to heighten contractile performance cannot be neglected. As it is, apart from the mechanical strain acting on the cardiac cells, there are numerous cytokine feedback loops at play involving, e.g., IL-18, IL-1.IL-6, TNF and TLR-4, to name only the most important ones. To what extent the release of IL-33 from living cardiomyocytes equals that of isolated cardiac tissue in vitro under increased mechanical stress remains to be established. (Figure 2)
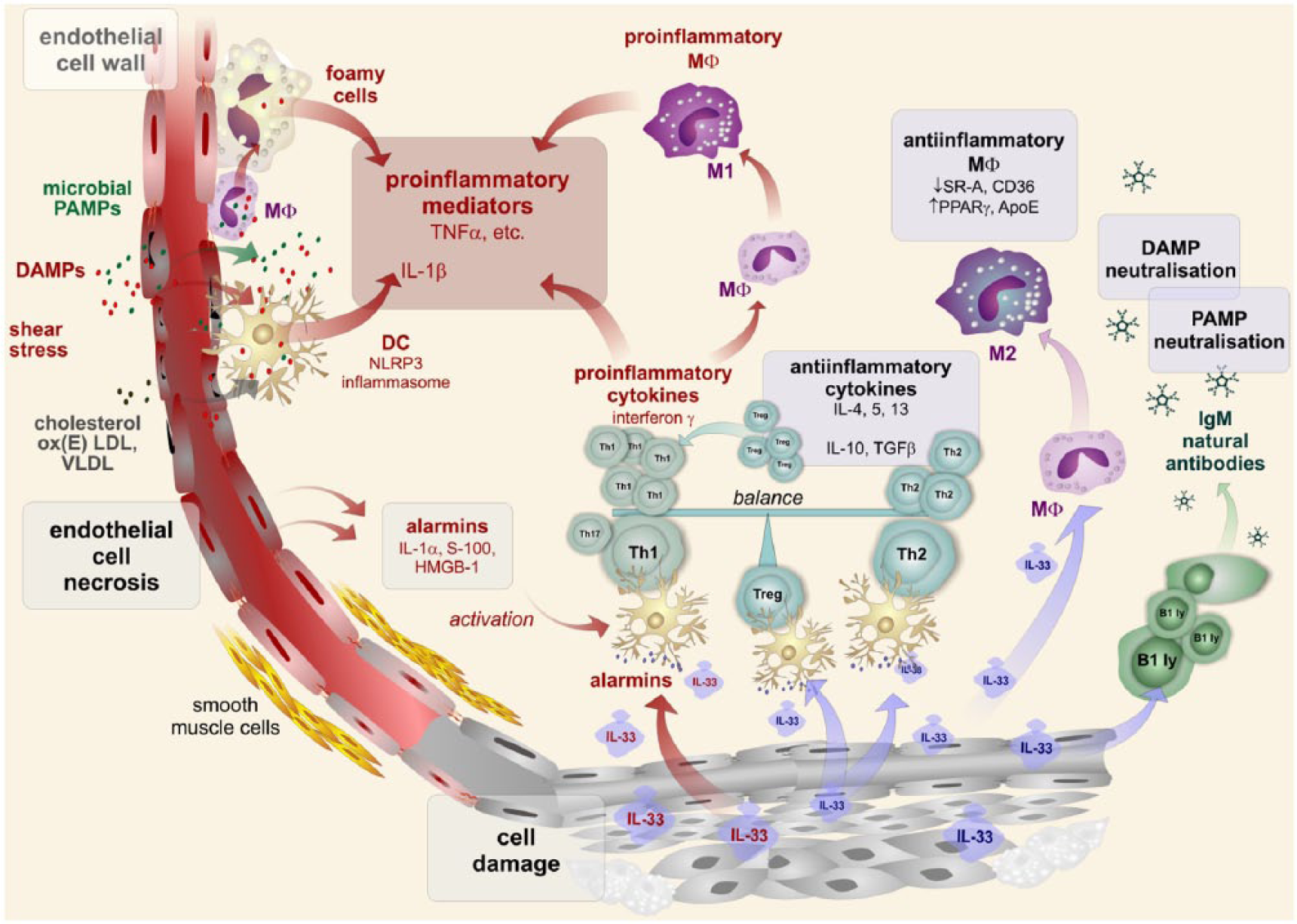
Arterial cell damage caused by either PAMPs or DAMPs is followed by the release of various pro-inflammatory mediators and the accumulation of inflammatory infiltrates, including foamy cell formation and smooth muscle proliferation. Dendritic cells are activated by alarmins released from injured cells. The Th1 subset of T cells is up-regulated and pro-inflammatory activities are further stimulated, especially by the interferon γ synthesis. This pro-inflammatory activity of dendritic cells is also induced by IL-33. Contrary to this, IL-33 is able to up-regulate the Th2 subset of T cells to down-regulate the Th1 subset. IL-33 can stimulated Treg subset activities to down-regulate the inflammatory response. IL-33 is inducing an anti-inflammatory subset of macrophages. In addition, B cells are stimulated by IL-33 to produce natural IgM antibodies, neutralizing both PAMP and DAMP and, thus, dampen their pro-inflammatory action.
Concluding remarks
On the basis of the results from the studies which have been reviewed here, it may be soundly confirmed that interleukin-33 is a “dual-acting” or “Janus-face” cytokine, originating from the IL-1 superfamily. Whether or not it exerts atheroprotective effects in humans and, more intriguingly, whether or not it might be used prophylactically or therapeutically are both “hot” questions, but about to be answered by further research. As in other segments of both basic and applied medicine, future surprise may be much nearer at hand than we anticipate. No matter in this moment how the IL-33 issue will turn out, we must be sure whether IL-33 either falls in with “Great Expectations” or, rather, we are “Waiting for Godot”.
Footnotes
Declaration of Conflicting Interest
The authors declare that there is no conflict of interest.
Funding
This work was supported by Charles University in Prague, Faculty of Medicine in Hradec Kralove, Czech Republic, project “PRVOUK“ P37/10.