
Abstract
Recent studies have reported that the ischemia/reperfusion (I/R) myocardium may act as an immune system where an exaggerated inflammatory reaction initiates. With activation of the immune system, damage-associated molecular patterns migrate and adhere into the I/R region and, consequently, induce myocardial injury. Emerging data have indicated that the adaptor proteins Crk are thought to play essential roles in signaling during apoptosis and cell adhesion and migration. Accumulated data highlight that Crk proteins are potential immunotherapeutic targets in immune diseases. However, very few studies have determined the roles of Crk on myocardial I/R injury. This mini review will focus on the emerging roles of Crk adaptors during myocardial I/R injury.
Introduction
Myocardial ischemia/reperfusion (I/R) injury is still thought to be an unsolved puzzle with a great diversity of investigational approaches. Injury occurs when the onset of acute myocardial ischemia is followed by the restoration of blood flow to the ischemic tissue.1,2 Ischemic myocardial injury is due to deprivation of oxygen and nutrients supply.1,3 It is reasonable to consider that rapid and early restoration of blood flow to the ischemic area prevents further damage. Yet, numerous studies have reported myocardial injury is further aggravated after the blood flow to the heart is restored.1,3,4Activation of the innate immunity is an essential component of the acute inflammatory response in the setting of ischemia/reperfusion (I/R).5,6 Myocardial I/R injury occurs when “danger” signals are released from the activation of the immune response.5–7
The inflammatory response in myocardial ischemia/reperfusion injury
It is widely recognized that neutrophils, monocytes, macrophages and dendritic cells are involved in innate immunity. However, recent experimental evidence has suggested that ischemia/reperfusion cardiomyocytes are involved in an excessive innate immune and inflammatory response.8,9 A moderate innate immune response may facilitate tissue repair and limit the extent of cardiac injury, whereas an excessive response is likely to cause cardiac dysfunction. 10
Role of endoplasmic reticulum (ER) and mitochondrial dysfunction in myocardial I/R injury
Recently, studies have reported that mitochondria play a critical role in the initiation and progression of myocardial I/R injury. A close association between ER and mitochondria was describe in I/R injury11,12 They are early responders to hypoxia and then re-oxygenation, initiating responses that lead to changed metabolic and bioenergetic status, autophagy, inflammation and the induction of cell death pathways.13,14 Besides, mitochondria are both the sources and target of reactive oxygen species (ROS) production. 15 Cardiomyocytes are the main producers of ROS and are later accompanied by activated leucocytes, that is, oxidative burst related to inflammation during I/R injury. 16 They participate in the initiation and progression of myocardial I/R injury, linking oxidative stress, inflammation and cell death. ROS, through interactions with small metabolites as well as proteins, lipids and nucleic acids, might irreversibly destroy or alter the function of these target molecules and associated organelles and cells. 17 ROS can also serve as homeostatic signaling molecules, which primarily depends on the magnitude and duration of the provoking stimuli for ROS production. 18 The administration of antioxidants has been used as a way to prevent and treat I/R injury for a long time. 19
Role of lymphocytes in myocardial I/R injury
Previous studies have supported evidence that CD4+ T-cells contribute to myocardial injury, most likely by regulating a local innate immune activation, especially the infiltration of proinflammatory monocytes after I/R20,21 Using lymphocyte-deficient RAG1 KO mice models, researchers examined myocardial infarct size after transient ischemia. CD4+ T-cell-depleted mice had significantly smaller infarcts compared to controls, but CD8+ T-cell-depleted mice had no differences. On the contrary, RAG1 KO mice transfection with CD4+ T-cells reversed the protection seen in RAG1 mice in the I/R model. 22 These experiments strongly indicate that CD4+ T-cells contribute to myocardial I/R injury. However, there are few data explaining how CD4+ T-cells are activated.23–25 Some studies consider that CD4+ T-cells are activated via non-classical T-cell activation pathways, for example, by pattern recognition receptors, such as toll-like receptors or mediated alarmin recognition.26,27 Toll-like receptor 2 is thought to be activated by high-mobility group box-1, which is released by ischemic tissues.28,29 The RAGE receptor resembles another kind of pattern recognition receptor; it not only regulates high-mobility group box-1, but also leads to inflammasome activation. 30 Inflammasome activation after tissue injury induces strong IL-1β expression that significantly amplifies the inflammatory response by recruiting inflammatory cells and directly effects leukocytes, as in the stimulation of cytokine expression and matrix-metalloproteinase activity. 30 Thus, pattern recognition receptor activation on T-cells might be a relevant mechanism in the rapid proinflammatory lymphocyte activation seen in myocardial ischemia/reperfusion injury.
Crk proteins
Crk proteins are key regulators of adhesion and migration in many cell types. The Crk−/− embryonic data show that Crk is involved in cardiac and craniofacial development and plays an essential role in embryonic development. 31 This family of ubiquitously expressed adaptors comprises the alternatively spliced CrkI and CrkII isoforms, as well as the paralog Crk-like (CrkL) protein, which is encoded by an independent gene. CrkII is located on chromosome 17p13.3, while CrkL is located on chromosome 22q11.21 in humans. 30 The CrkL has a molecular mass of 36 kDa. CrkI (28 kDa) contains only one N-terminal Src homology 2 (SH2) and one Src homology 3 (SH3) domain while CrkII (40 kDa) and CrkL proteins contain one SH2 and two SH3 domains, namely, SH3N (N-terminal SH3 domain) and SH3C (C-terminal SH3 domain), respectively.32,33 In addition, CRKII and CRKL have a regulatory tyrosine (Y221 in CRKII, Y207 in CRKL) that is phosphorylated by ABL family kinases 34 (as shown in Figure 1).
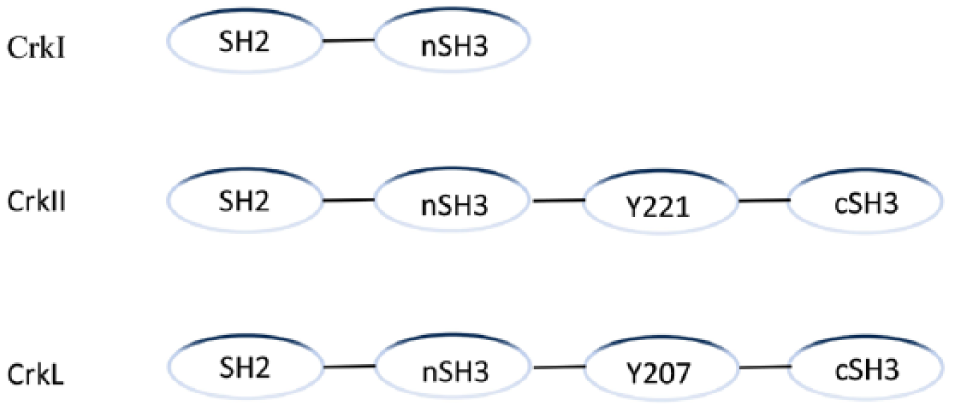
The structure of Crk adaptor proteins.
Generally, the intracellular signaling activated by Crk proteins is mediated by the interaction of the SH2 domain with phosphotyrosine residues (PRR), in conjunction with the interaction of the SH3 domain with PRRs as well. Widely expressed in various tissues, Crk proteins are important cell signaling adaptors without any enzyme activity. The regulation of Crk L and Crk II are mediated by phosphorylation at Tyrosine 207 (Tyr207) and Tyrosine 221 (Tyr221), respectively. 35 Phosphorylated Tyr221 binds to the SH2 domain; as a result, nSH3 is inhibited to integrate with other proteins or signal molecules. 36 The integration between pTyr207 and the SH2 domain, which is secondary to Crk L being phosphorylated by ABL kinase at Tyr207, will prevent the SH2 domain from interacting with other phosphorylated signaling molecules. 37 Phosphorylated Crk L may act as a negative regulator of the signaling pathway.
The role of Crk in regulating T-cell functions
Despite their importance in other cell types, the function of Crk proteins in T-cells is poorly understood.38,39 T-cell receptor (TCR) performs the role of antigen recognition and signal transduction in T-cell immune responses and plays an important role in viral infections, cancer, inflammation and autoimmunity. 40 Few studies have focused on the TCR signaling pathway. Using the small number of surviving Crkl–/– mice showed that thymocyte number was reduced, but T-cell differentiation and activation were intact. 41 In contrast, Nolz et al. used RNAi to suppress CrkL expression in Jurkat cells and ex vivo human T-cells and observed defects in integrin activation and cytokine production downstream of TCR engagement. 42 Crk proteins appear to mediate the initial steps of T-cells adhesion via its nSH3 domain binding to C3G, which is guanine-nucleotide exchange factors (GEFs) for the small GTPases RAP1. 43 RAP1 is a key molecule needed for adhesion to endothelial cells and subsequent diapedesis (as shown in Figure 2). Using conditional knockout mice, researchers found that, in the absence of adaptor proteins, Crk and CrkL exhibit reduced integrin-dependent T-cell adhesion, chemotaxis and diapedesis. In experimental myocardial I/R, T-cells lacking Crk proteins maintain effector function and the ability to home to lymphoid organs, but show selective defects in migration into I/R sizes. 44 Thus, we conclude that CrkI, CrkII and CrkL play partially overlapping roles in T-cell adhesion and migration. The function of Crk proteins in T-cell adhesion has been reported to form multiprotein complexes through T-cell receptor (TCR) stimulation, with a specific focus on the CD4+ T-cells line - the Jurkat cell line. Crk regulates TCR signaling via Cbl (an E3- ligase that catalyzes protein ubiquitination).45,46 When T-cell activation occurs, Crk binds to phosphorylated Cbl with the SH2 domain, 47 eventually interfering with its efficacy to interact with the other binding partner-C3G. C3G is a guanine exchange factor of Rap- 1 (Ras-related GTP-binding protein 1), 48 which indicates that Cbl functions as a negative regulator of CrkL-C3G-mediated Rap1 (a member of the Ras family of small GTPases) activation in TCR-stimulated T-cells. 49 In both inflammatory Jurkat and normal human peripheral blood-derived T-cells, TCR stimulation induces the association of Cbl with all the CrkI, CrkII and CrkL, most prominently with CrkL. 50 Therefore, the Crk family of proteins involve TCR signaling by forming multi-complexes with other signal molecules, such as C3G.
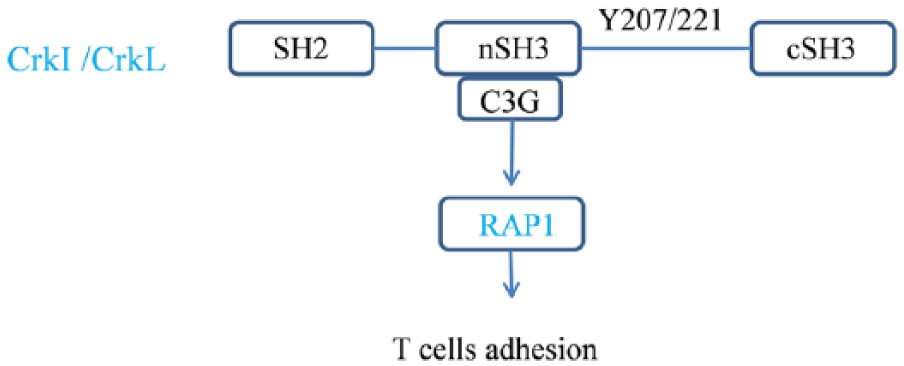
Crk proteins mediate T-cell adhesion mechanism.
T-cell migration into inflamed tissue exaggerates myocardial I/R injury. At the molecular level, Crk proteins function to promote chemokine-dependent integrin activation by RAP1, a key event needed for adhesion to endothelial cells and subsequent diapedesis.41,51 Crk proteins signal through the RAP1-GEF-C3G to regulate the RAP1 activation pathway, which is important for chemokine-induced T-cell adhesion and migration. 52 In many respects, migrating neurons lacking Crk fail to pass through cortical preplate cells and earlier-born neurons, resulting in disruption and apparent inversion of cortical layers. 53 As in T-cell migration, the Crk/CrkL-C3GRAP1 pathway is considered to play critical roles in Reelin-dependent neuronal migration. Therefore, it is possible that Crk and CrkL function to modulate the cell-cell interactions that allow migrating cells to pass through non-migrating cell boundaries. 54 Thus, Crk proteins act in regulating chemokine-dependent adhesion, migration and diapedesis of T-cells whereas those lacking Crk proteins show selective defects in migration into inflamed tissues.
The role of Crk in other immune cell functions
The role of Crk in other immune cells, such as B cells and NK cells, have been described previously in other reviews. 55
Concluding remarks
Crk proteins as highly promising therapeutic targets for disrupting deleterious T cell adhesion and migration to sites of inflammation in myocardial I/R injury. However many unanswered questions regarding their signaling pathways remain open to investigation, especially with regard to a detailed dissection of the molecular complexes that are formed. In addition, some of the pathways in which Crk adaptors have been implicated are still incomplete and disconnected. Further investigations are also required in order to determine the potential regulation of selected Crk proteins in T-cell adhesion and migration.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Nature Science Foundation of China (81270218, to Dr. Chen) and National Natural Science Foundation of China (81400190, to Dr. Wang).