
Abstract
Background
The objective of this review is to describe the diagnosis of neck vein obstruction and the possible role of chronic persistent Chlamydophila pneumoniae infection in producing the syndrome of chronic cerebrospinal venous obstruction.
Method
The normal patterns of flow in the neck veins are described and guidelines for interpretation of the quantitative duplex ultrasound examination of the extracranial neck veins are developed.
Result
An infective cause of neck vein obstruction is proposed and from a literature search of the role of the obligate intracellular bacterium Chlamydophila pneumoniae in vascular and chronic diseases, a diagnostic protocol for confirming chronic persistent Chlamydophila pneumoniae infection, which includes the quantitative duplex ultrasound examination and specific blood tests are suggested.
Conclusion
Further research to validate this diagnostic protocol is required.
Neck vein obstruction – Chronic cerebrospinal venous obstruction
Overview
CCSVI is a syndrome originally postulated by Zamboni 1 where abnormal flow of blood (reflux) in veins draining the brain and spinal cord is associated with multiple sclerosis (MS). In contrast, chronic cerebrospinal venous obstruction (CCSVO) is rarely associated with reflux flow and refers to cerebrospinal venous blood flow disturbances with venous obstructions in the major extracranial veins of the head and neck. Although reflux can occasionally be observed, the predominant pathology is chronic and constant obstruction of the major veins of the neck with resultant development of collateral flow and new pathways. The veins involved include the internal jugular veins (IJVs) and vertebral veins (VVs). While CCSVI was postulated to be associated with MS, CCSVO may be associated with a wide range of chronic vascular diseases, generally with manifestations in the head, neck, and chest. 2
The venous obstructions reduce the flow in the neck veins and can result in complete occlusion of these veins, most commonly affecting the vertebral veins that pass down through the spinal vertebrae of the neck. Thibault 3 has suggested that these venous obstructions are due to a chronic persistent venulitis caused by the obligate, intracellular parasite, Chlamydophila pneumoniae (Cpn). This parasitic bacterium has also been associated with other vascular diseases including coronary artery disease, cerebrovascular disease, and aortic aneurysms. 4
This article describes the ultrasound methods to diagnose CCSVO, and the diagnostic markers for chronic persistent Cpn vasculitis.
The normal patterns of flow in the neck veins
A unique feature of cerebral venous drainage is its dependence on posture. While in supine position the IJVs are the main drainage pathways, in upright position the IJVs generally collapse with cerebrospinal venous system 5 consisting of the VVs, intraspinal veins, and paravertebral veins compensating to a large extent. 6 However, the IJVs are not always the main drainage veins in the supine position. In a minority (6%) of healthy subjects, the extra-jugular drainage pathways are at least similarly important for cerebral venous drainage. 7 In addition, duplex ultrasound studies have shown that the drainage of the cerebral blood is asymmetric with a preferential outflow via the right IJV and VV.8,9
Objective and quantitative duplex ultrasound assessment of the neck veins
Thibault and Lewis 9 have developed a quantitative duplex ultrasound assessment (QDUA) of the neck veins. The method of the QDUA has been described in detail. 9 The determination as to whether a vein is obstructed depends primarily on comparison of the venous blood volume flow (VBVF) measurements for the different segments of vein examined (Figure 1) in the supine and the sitting position. Using venography as the gold standard, the sensitivity and specificity of the QDUA examination for identification of stenoses in the IJVs was calculated as 85% and 100%, respectively. 9
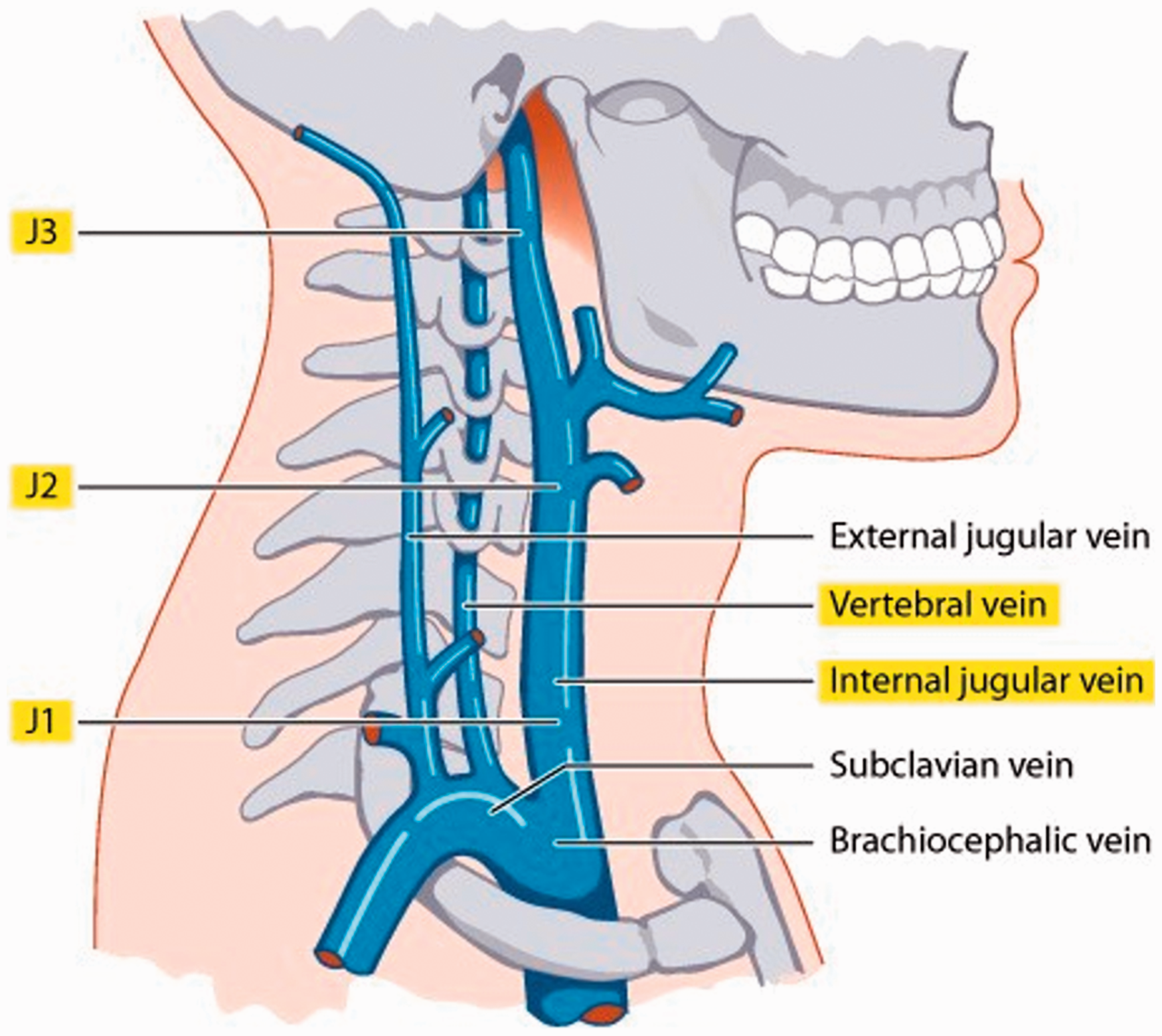
Schematic diagram demonstrating venous blood volume flow (VBVF) measurement sites.
Chambers et al. 8 evaluated early onset MS patients and “normal” controls using this QDUA and have published the results (Table 1).
Median (and interquartile range) volume flow values (mL/s) in early MS patients and controls. 8
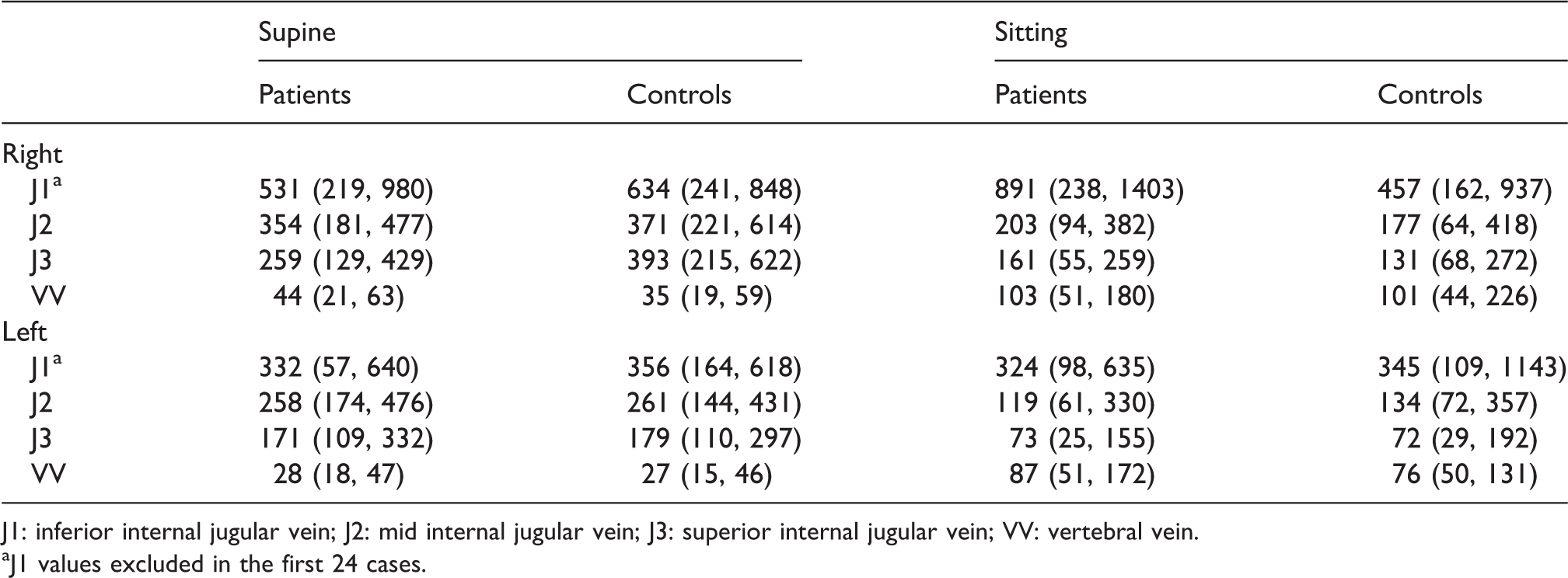
J1: inferior internal jugular vein; J2: mid internal jugular vein; J3: superior internal jugular vein; VV: vertebral vein.
aJ1 values excluded in the first 24 cases.
Note that the right side VBVFs is consistently higher than the comparative segment on the left side, and there is a progressive reduction in VBVFs going from proximal to distal in the IJVs and that the VBVFs in the VVs are higher in the sitting position, whereas the VBVFs in the IJVs are higher in the supine position. The VBVF reading in the J1 segment in the IJVs is generally discounted in the assessment due to excessive turbulence near the valve in this segment. 8
The loss of normal postural change in the VBVF reading is suggestive of obstruction in one of the four major extracranial draining veins, i.e. IVF or VV. If a VV is obstructed there will usually be increased collateral flow through the IJV on the ipsilateral side and occasionally through the VV of the opposite side. Similarly, if there is unilateral obstruction of an IJV, there will be increased VBVF through the IJV of the opposite side, increased flow through the VV of the ipsilateral side, or enlargement of other collaterals such as the ipsilateral external jugular vein. This explains why traumatic neck vein occlusion from cannulations will generally not result in any secondary neurological symptoms.
From the study of Chambers et al., 8 suggested normal results may be defined according to 10th and 90th percentiles (Table 2).
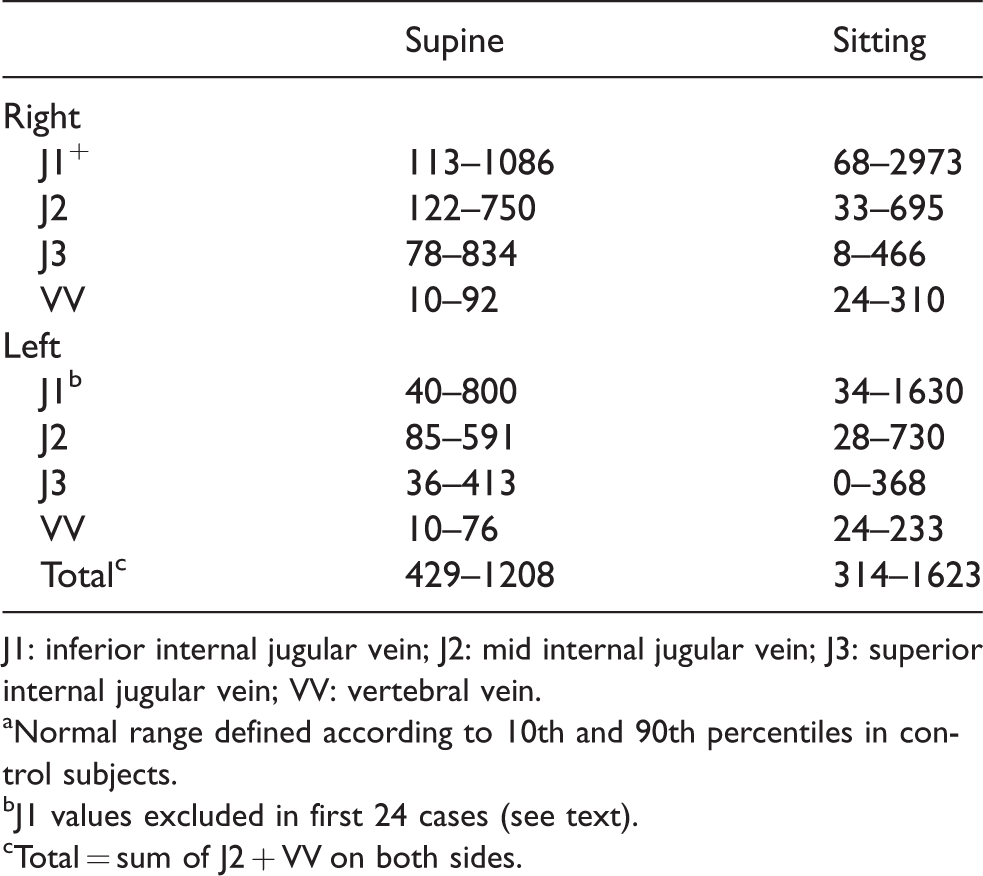
J1: inferior internal jugular vein; J2: mid internal jugular vein; J3: superior internal jugular vein; VV: vertebral vein.
Normal range defined according to 10th and 90th percentiles in control subjects.
J1 values excluded in first 24 cases (see text).
Total = sum of J2 + VV on both sides.
Guidelines to diagnosing abnormalities in neck vein blood flow.
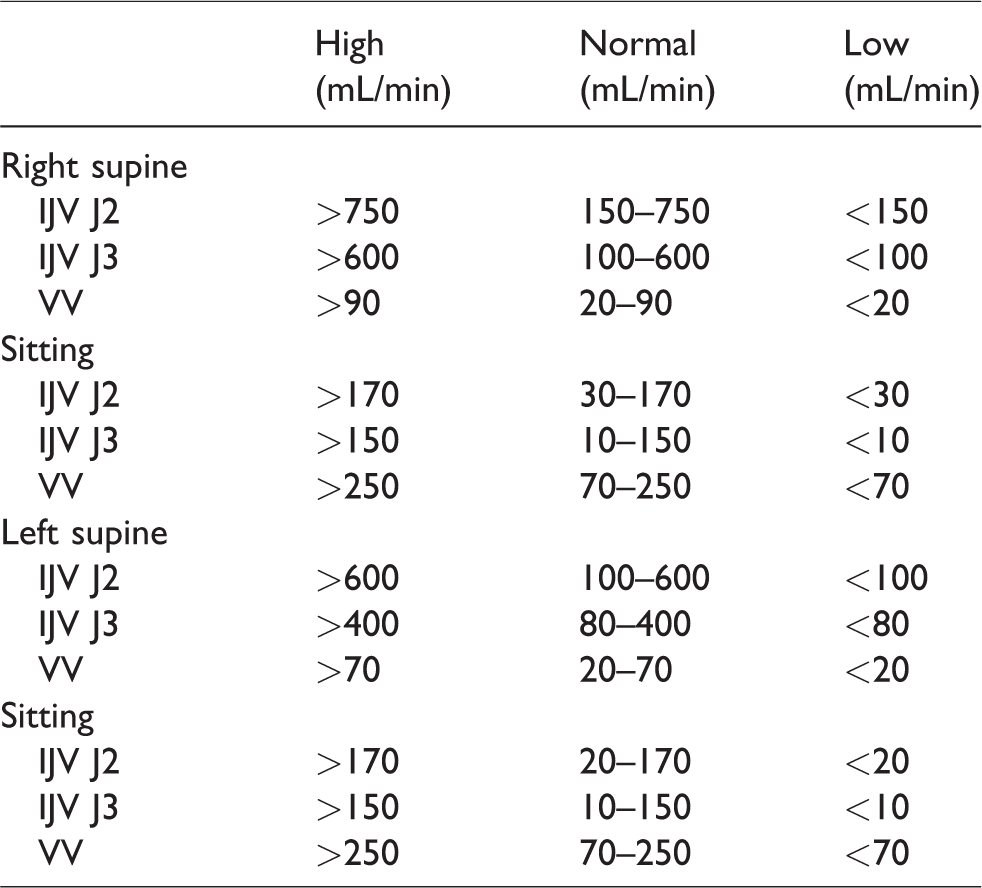
Note that according to Table 2, it is possible for a “normal” L IJV J3 segment to have no flow in the sitting position, whereas Zamboni 10 in his criteria for defining the presence of CCSVI stated that no flow in any segment in any position was abnormal. Therefore, the author recommends that in the situation of L IJV J3 segment showing no flow, the probability of abnormality should be based on the presence or absence of other evidence, in particular abnormal collateral flow. As variability in measurements is attributable to “within subject” factors (such as position, side, vein segment) as opposed to “between subjects” differences, 8 it is important to observe the patterns of varying VBVF measurements in the J2, J3, and VV segments in each patient to localize the probability of an obstruction in any part of the extracranial venous system. This pattern of flow (normal or abnormal) manifested by the VBVFs should be consistent over time in the same patient.
In the clinical setting, the author has developed guidelines in interpreting the VBVFs in the QDUA of neck veins based on previously published data of “normal” subjects,6–8,11 and the author’s clinical experience in assessing abnormalities in neck vein venous flow using this examination (Table 3). It should be noted that if there is borderline flow in the VVs in the sitting position, the probability of abnormality is increased if there is associated loss of postural change (i.e. decrease) in the ipsilateral IJV when repositioning to sitting.
An infective cause of neck vein obstruction
An ascending infective venulitis theory involving chronic persistent infection with Cpn was first published in 2012. 3 The spread of Cpn from the lungs to the vasculature has been studied in New Zealand white rabbits by Geiffers et al. 12 Cpn infection of the lungs results in an interstitial and alveolar pneumonia with bronchiolitis that resolves spontaneously after 2–4 weeks. Histology reveals infiltrates of heterophilic granulocytes and mononuclear cells within the interstitium, alveolar space, and bronchiolar lumen. After three days, the granulocytic infiltrates are replaced by mononuclear cells. Sometimes, there is a mild vasculitis and perivasculitis within the first three days. Perivascular and peribronchiolar lymphatic hyperplasia is observed from day three until up to eight weeks from initial infection. Geiffers proposed that granulocytes act as a kind of Trojan horse for Cpn in the early stage of infection, granting access to the alveolar macrophages, which arrive later and can disseminate the pathogen through the lymphatic system. The ascending vasculitis theory postulates that infective Cpn organisms are then transmitted through peri-hilar lymph nodes within infected blood monocytes to the thoracic duct and right lymphatic duct. From these lymphatic conduits, the monocytes can transmit the Cpn elementary bodies (EBs) to the venous endothelium firstly through communications of the thoracic duct with the azygos vein in the chest, then finally at the respective confluences of the internal jugular, vertebral, and subclavian veins bilaterally (Figure 2(a) and (b)). Once blood borne, the Cpn can also “metastasize” to distant vascular sites whilst harboring within the infected blood monocytes. 13 Importantly, it has been demonstrated that Cpn-infected monocytes exhibit increased adhesion to vascular endothelial cells. 14 Furthermore, Cpn causes activation of chemokines in human endothelial cells and promotes the recruitment of leukocytes in vitro. 15
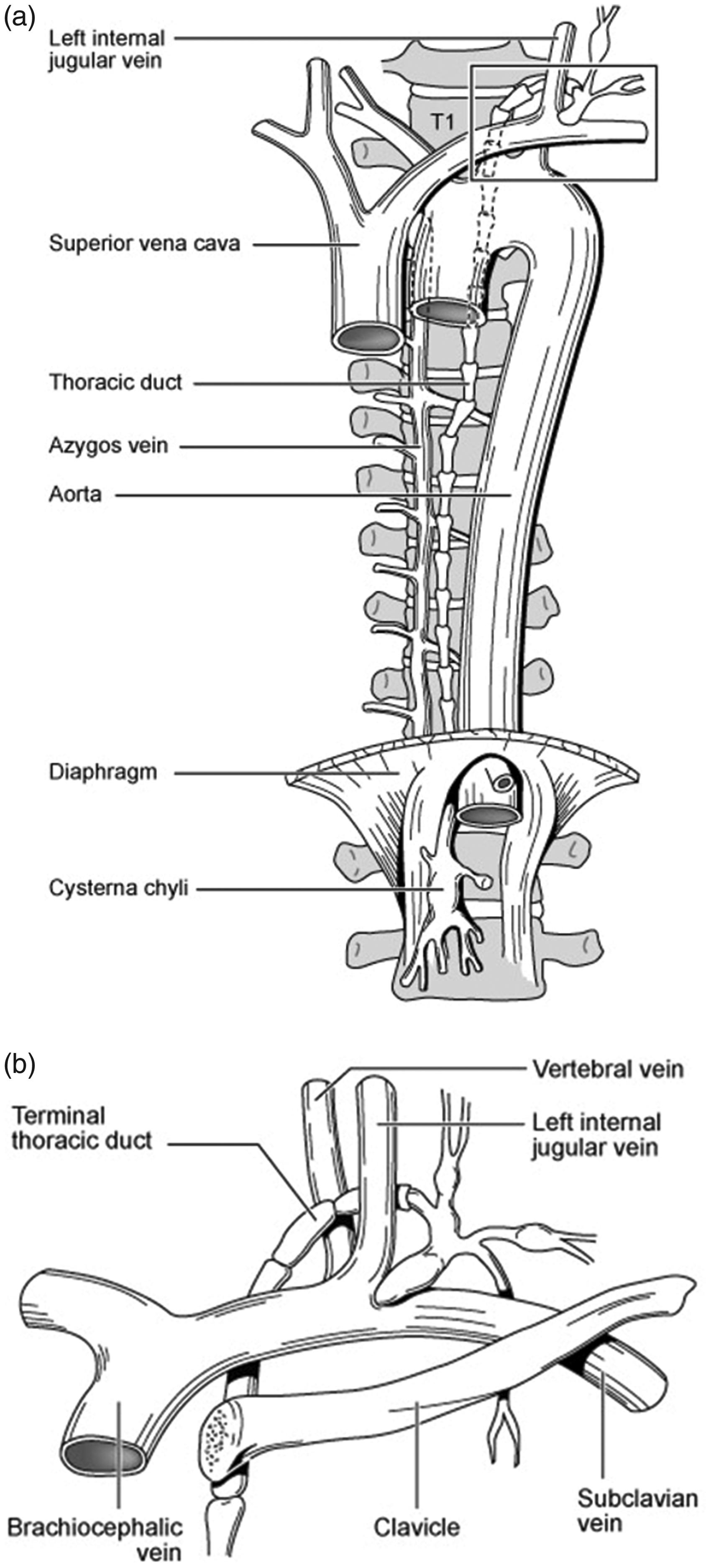
(a) Relative anatomy of the thoracic duct. Note the close association of the thoracic duct to the azygos vein on the thoracic spine. (b) The termination of the thoracic duct at the confluence of the subclavian vein, left internal jugular vein and left vertebral vein. Infected macrophages and lymphocytes with C. pneumoniae transmit the infection to the venous endothelium at this site, triggering a creeping venulitis to affect the cerebrospinal venous system (CSVS).
The infective venulitis theory was originally developed to explain the neck vein abnormalities found in subjects with MS. 3 It has been demonstrated that Cpn rapidly binds to platelets causing platelet activation, aggregation, ATP secretion, and surface expression of P-selectin. 16 P-selectin mediates the recruitment and activation of leukocytes and thereby initiates an inflammatory response. 16 The ability of Cpn to activate platelets is concentration dependent so the maximum effect of Cpn–platelet–endothelial cell interaction would be expected to occur at the site of entry into the circulation, namely the termination of the thoracic and right lymphatic ducts. A creeping infective venulitis could then spread slowly and silently distally along the azygos vein in the chest, and internal jugular and vertebral veins in the neck. Conversely, the lymphatic ducts remain unaffected owing to the absence of platelets in lymph. Over time, the prothrombotic and inflammatory effects of the Cpn venulitis cause gradual obstruction of the VVs and due to their larger diameter the IJVs are less frequently obstructed. However, pathology studies of abnormal valves in IJVs in patients with MS has shown an absence of endothelial cells where a reticular and fibrotic lamina has replaced the endothelium suggesting a past, resolved inflammatory or thrombotic process that involved the wall of the IJV. 17
From the chest and neck, Cpn can also be transmitted to a wide range of other blood vessels throughout the body via infected monocytes to cause arterial and venous inflammation that could play a significant role in chronic vascular diseases. PCR testing of atheromatous vessels in the chest (aorta, coronary arteries, internal mammary arteries) and macroscopically abnormal great saphenous veins have been found to be positive for Cpn, whereas normal vessels in the same subjects have been negative indicating that Cpn has a role in both atheromatous changes in arteries and degenerative changes in veins.18,19 There is now a large number of studies that confirm the presence of Cpn in atheromatous coronary arteries and other major arteries. In addition, there are many serological studies confirming that the presence of Cpn antibodies in serum increases the risk of vascular disease. Moreover, the mechanisms by which Cpn can promote vascular diseases and stimulate immune and inflammatory responses are well understood, thereby establishing Cpn as a potentially modifiable risk factor in cardiovascular disease and other diseases characterized by chronic inflammation. 20
Diagnostic markers of chronic persistent Cpn infection
Serological aspects
Chlamydophila pneumoniae was first isolated by Grayston et al. 21 in 1965 but was not identified as a separate species of the genus Chlamydia until 1989. This primary respiratory obligate intracellular parasite exhibits characteristics that distinguish it from other chlamydiae with the capacity to infect and multiply within a wide range of secondary host cells including macrophages, lymphocytes, and vascular endothelial cells. 21 The primary infection with Cpn does not induce life-long immunity with most individuals having several infections during their lifetime. Subsequent re-infections with Cpn induce a greater IgG response than the initial infection. It is thought that small children do not frequently produce IgA antibodies as a response to primary upper respiratory tract infections with Cpn and that IgA responses are generally more common in re-infections, which are more common in adults.22,23 Anti-Cpn antibodies, unusual in children under 5 years, occur in up to 50% of individuals by age 20 years. The prevalence of antibodies continues to increase with age among adults, reaching a peak in seropositivity of 80% in men and 70% in women by age 65.24,25 Grayston 26 suggested that everyone is infected with Cpn.
Due to the high prevalence of antibodies present in the adult community, it will always be difficult to determine the relevance of persistent Cpn infection in any one clinical situation by serological testing alone. Persistently elevated IgG or the presence of IgA antibodies have been frequently used to identify persons with persistent or chronic infection. 27 It has been proposed that high IgA titers may be a better marker of chronic Cpn infection than are IgG titers because serum IgA has a half-life of 5–7 days, whereas IgG has a half-life of weeks to months. However, there is at present no validated serological marker of persistent or chronic infection, and the use of serological testing as a stand-alone test to define patients as “persistently infected” must await further validation. 28
Disturbed cholesterol metabolism and LDL
Cpn antibodies have been associated with an atherogenic lipid profile in men. 29 In particular, sero-positive subjects were found to have increased total cholesterol and decreased HDL cholesterol compared with sero-negative subjects after allowing for possible confounding factors. In another study of Finnish men who were nonsmokers, subjects positive for IgG had significantly higher triglyceride concentrations and lower HDL than sero-negative subjects. However, the presence of IgA antibodies had only a minor association with lipid concentrations. 30
Animal studies have indicated that Cpn mouse liver infection induces dyslipidemia effects with significant modifications of genes involved in lipid metabolism. 31 Cpn-infected mice showed significantly increased cholesterol and triglyceride levels compared with negative controls and C. trachomatis infected mice. In Cpn-infected livers, cholesterol 7α-hydroxylase and low-density lipoprotein receptor (LDLr) mRNA levels were reduced, while inducible degrader of the LDLr expression was increased.
Cpn-infected macrophages ingest excess LDL to become cholesterol-laden foam cells, the hallmark of early lesions in athero-sclerosis. 32 Moreover, Cpn has been shown to induce monocytes to oxidize lipoproteins, converting them to highly atherogenic forms. 33 In addition to causing increased platelet aggregation, Cpn interaction with platelets results in reactive oxygen species production and oxidative damage on LDL. 34 Cpn-induced foam cell formation is mediated chiefly by lipopolysaccharide, whereas lipoprotein oxidation occurs mainly by chlamydial heat shock protein 60 (cHSP60), an inflammatory protein abundantly expressed by persistent chlamydiae. 20 In addition, cHSP60 may contribute to atherogenesis by triggering antibody-mediated cytotoxicity through an immunological cross-reactivity to HSP60 produced by the infected endothelial cell. 35 This is a similar mechanism to that proposed to implicate the involvement of Cpn in the demyelination lesions found in MS by direct toxic effects of HSP60 and the activation of innate immunity.36–38
Inflammatory markers
C-reactive protein (CRP) is an acute-phase protein that serves as an early marker of inflammation or infection. The protein is synthesized in the liver and is normally found at concentrations of less than 10 mg/L in blood. During infectious or inflammatory disease states, CRP levels rise rapidly within the first 6 to 8 h and peak at levels of up to 350–400 mg/L after 48 h. CRP is an independent risk factor for cardiovascular disease. The risk of developing cardiovascular disease is quantified as follows:
39
low: CRP level under 1.0 mg/L average: between 1.0 and 3.0 mg/L high: above 3.0 mg/L
CRP binds to phosphocholine expressed on the surface of damaged cells, as well as to polysaccharides and peptosaccharides present on bacteria, parasites, and fungi. This binding activates the classical complement cascade of the immune system and modulates the activity of phagocytic cells, supporting the role of CRP in the opsonization (i.e. the process by which a pathogen is marked for ingestion and destruction by a phagocyte) of infectious agents and dead or dying cells. When the inflammation or tissue destruction is resolved, CRP levels fall, making it a useful marker for monitoring disease activity. 40
A number of studies have shown a correlation with elevation of serum CRP and the presence of Cpn in carotid and coronary artery atheromatous plaques.41–44 Similarly, there is a strong correlation between serum Cpn IgA and serum CRP levels in subjects with known vascular disease.41,42 In addition, specific antibiotic treatment for chronic persistent Cpn infection in subjects with vascular disease has resulted in a significant reduction in CRP levels at 6-month follow-up. 45
Liver disorders and Cpn Infection
Cpn is known to infect the liver, generally in association with the presence of cardiovascular disease. 46 In addition, Cpn has been implicated in primary biliary cirrhosis 47 and granulomatous hepatitis. 48 Animal studies have demonstrated that Cpn acute liver infection affects cholesterol and triglyceride metabolism as described previously. 31 Moreover, Cpn has been demonstrated to survive and replicate in Kupffer cells of the liver thereby creating an inflammatory microenvironment within the liver. 49
Because of the ability for Cpn to infect the liver, liver function tests would be expected to be abnormal in some with chronic persistent Cpn. The liver function test marker, ALT has been associated with a greater probability of a positive serology result for Cpn, therefore may be a useful diagnostic marker for the disease. 50
Iron homeostasis and chronic persistent Cpn
Many bacteria, including Cpn are dependent on iron (Fe) for their growth. One of the first lines of defense against bacterial infection is the withholding of nutrients to prevent bacterial multiplication. The most significant nutrient involved in this defense is serum Fe. 51 Accordingly, Fe restriction in cell culture inhibits growth of Cpn. 52 The circulating peptide hormone hepcidin, produced by the liver acts as a regulator of body Fe homeostasis. During infection and inflammation hepcidin production is induced, driving a decrease in Fe concentration by inhibiting the absorption of Fe and promoting the sequestration of Fe in macrophages and the liver. 53
It has been demonstrated that liver hepcidin levels in mice increase during acute Cpn infection and that this induction is associated with altered Fe levels. 54 A recent study has confirmed that serum Fe levels decrease during the course of a Cpn infection in mice. 55 In a chronic persistent Cpn infection, we are therefore likely to frequently observe low to normal serum Fe levels associated with mild to moderately elevated serum ferritin levels. If found to be elevated initially, serum ferritin levels then become a useful parameter to measure response to treatment of chronic persistent Cpn infection.
Discussion
Enhanced clinical and pathological diagnosis of chronic persistent Cpn
A multitude of epidemiological, microbiological, serological, and histological studies have suggested that the intracellular bacteria, Cpn may play a role in the pathogenesis of many chronic vascular and inflammatory diseases. 56
However, available diagnostic methods to detect or monitor chronic persistent Cpn infection lack sufficient reliability and standardization.
To enhance the predictive capacity of the diagnostic procedure, the author suggests a combination of assessing the clinical presentation with the investigative tools of duplex ultrasound of the neck veins, Cpn serology from a known reliable laboratory, fasting serum lipids with particular emphasis on LDL levels, serum CRP as an inflammatory marker, serum ALT and serum Fe studies. In most cases, the predictive power of a positive diagnosis will be attained if the triad of neck vein obstruction, positive serology (particularly if Cpn IgA is present) and elevated fasting LDL (above 2.5 mmol/L) is present. Secondary supporting evidence consists of mild-to-moderate elevation of serum CRP, elevated ALT, and elevated serum ferritin in the presence of low to normal serum Fe.
Successful treatment of chronic persistent Cpn is recognized to be difficult and entails a multimodal therapy including a prolonged antibiotic protocol, usually for at least six months, dietary measures, various supplements, and possibly life-long use of a “statin” drug. Therefore, certainty of diagnosis is essential. Further research to validate this diagnostic protocol is required.
Footnotes
Acknowledgement
The author acknowledges the assistance of Warren Lewis, vascular sonographer from Vascular One Ultrasound, New South Wales, Australia who helped devise the QDUA.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not applicable.
Guarantor
Paul K Thibault
Contributorship
None.