
Abstract
Background:
The subcutaneous route is widely used in both palliative care and geriatrics. Numerous compounds are administered by this route, including paracetamol. However, there is no recommendation on which to base this latter practice and, in the absence of published evidence, nothing is known regarding its local tolerability in palliative care patients.
Aim:
The main objective of this study was to assess the local tolerability of paracetamol when administered subcutaneously for analgesic or antipyretic purposes in patients hospitalized in the palliative care unit. The secondary objective was to identify the factors favoring the occurrence of local adverse events.
Design:
This is a prospective multicenter observational study (NCT02884609).
Participants:
Study conducted in 160 patients hospitalized in the palliative care units of three hospitals in metropolitan France from 2014 to 2017.
Results:
Of the 160 patients, 44 (28%) presented at least one non-serious local adverse event (edema in 29, erythema in 5, pain in 15, hematoma in 2, pruritus in 1, and local heat in 2). No serious adverse events were observed. Factors associated with the occurrence of local adverse events were younger age, administration in the arm and thorax, and a high number of daily administrations.
Conclusion:
This first ever study carried out on this subject reveals that subcutaneous administration of paracetamol in palliative care patients was well tolerated locally.
Keywords
The subcutaneous route is widely used in palliative care and geriatrics.
Paracetamol is currently not recommended for subcutaneous use in the absence of validated references but is nonetheless classified in the category of drugs “used by some teams.”
No serious local adverse events were observed after subcutaneous administration of paracetamol.
Of the 160 patients, 44 (28%) had at least one non-serious local adverse event during the follow-up.
Edema represented the main non-serious adverse event followed by pain erythema.
Factors associated with the occurrence of local adverse events were younger age, administration in the arm and thorax, and a high number of daily administrations.
Subcutaneous administration of paracetamol seems well tolerated in palliative care patients.
However, further studies are needed to show that this practice, while well tolerated, is also effective.
Introduction
The subcutaneous route is widely used in both palliative care and geriatrics.1,2 This mode of administration offers many advantages when other administration routes (oral, sublingual, rectal, or venous) are unreliable, unavailable, or unacceptable for the patient.1,3,4
Several studies have recently reviewed the drugs that can be used subcutaneously.1,3
Paracetamol is not mentioned 1 or is referred to as “non-evaluated.” 3 The subcutaneous administration of paracetamol is nonetheless practiced since it is classified in the category of products that “some teams use.” 1 Moreover, a survey regarding the subcutaneous use of paracetamol in palliative care units (PCUs) in France revealed that it is used by 40% of the physicians responsible for these entities. 5
Paracetamol has indeed appealing analgesic and antipyretic properties for the management of frail patients.6,7 It allows limiting the use of aspirin, nonsteroidal anti-inflammatory drugs, or morphine, which displays significant side effects.8,9
Given the existence of subcutaneous administration of paracetamol, and that such practice is not documented in the literature, we conducted a study the objectives of which were (1) to evaluate the local tolerability of subcutaneous-administered paracetamol in patients hospitalized in PCUs and (2) to identify the factors favoring the occurrence of local adverse events.
Materials and methods
Study design and population
This prospective multicenter observational study was conducted between 2014 and 2017 in patients hospitalized in the PCUs of three hospitals located in France.
According to French legislation applicable during the implementation of the study and consistent with a European directive, 10 this observational study was approved by the French Advisory Committee on Information Processing in Health Research (CCTIRS, No. 16-083), authorized by the French National Data Protection Commission (CNIL, DR-2016-302), and registered at ClinicalTrials.gov under registration number NCT02884609.
Each patient was informed of the course of the study and gave verbal consent for inclusion and use of the data. In instances where the patient was unable to communicate, his or her support person (personne de confiance according to French legislation) or relatives were informed and gave verbal consent.
To participate in the study, the PCUs were required to already routinely use paracetamol by subcutaneous administration.
All adult patients hospitalized in the PCUs and requiring subcutaneous administration of paracetamol for analgesic or antipyretic purposes were included. Patients were not included in instances of (1) opposition to the use of the data collected for clinical research, refusal expressed either by the patient or by his or her relatives when the patient could not give his consent and (2) subcutaneous administration of paracetamol in the week prior to arrival in the PCUs.
Assessment criteria
The primary assessment criteria were the occurrence of local adverse events (pain, pruritus, edema, erythema, warm skin, hematomas, and vesicles) or serious local adverse events (skin ulcers, abscesses, and skin necrosis).
Local adverse events were assessed prior to the first injection as well as 30 min, 2 and 4 h after the first injection, and on a daily basis until the day after the discontinuation of use of the first subcutaneous administration site of paracetamol, regardless of the reasons for discontinuation of use of this first administration site and whether or not paracetamol subsequently continued to be administered on a different administration site.
Local adverse events were assessed as follows:
Pain was assessed using the numeric rating scale (NRS) for communicating patients and the Algoplus scale for non-communicating patients. 11
Pruritus was self-assessed by communicating patients, and scratching lesions were assessed by nurses for non-communicating patients.
Other local adverse events were investigated by the nurses.
Statistical analysis
The number of patients to be included was contingent on the need to be able to demonstrate rare adverse events. According to the literature,12–16 an adverse event after subcutaneous drug administration was considered rare when observed in 2% of cases. Hence, a minimum of 149 patients were deemed necessary to obtain at least a 95% probability of observing at least one adverse event with an expected frequency of 2%. This sample yielded an accuracy of ±8% on the outcome measures.
Several factors potentially influencing the occurrence of local adverse events were gathered: age, gender, presence of other products at the administration site, mean number of other products, duration of use of the administration site, total number of injections of paracetamol, mean number of injections of paracetamol per day, injection site, cancerous or non-cancerous nature of the disease, paracetamol manufacturer, and inclusion center.
All compounds used at the same injection site as paracetamol were recorded and classified into three groups: (1) products not presenting an identified risk of local adverse events, (2) products with identified risk of local adverse events, and (3) products for which the risk of local adverse events was unknown.
In order to establish this classification, existing literature data were used.1,2,12,17,18
Results
A total of 160 patients were included in this study, aged between 27 and 100 (median: 80) years. The main characteristics of the patients are summarized in Table 1.
Description of the study population (N = 160).
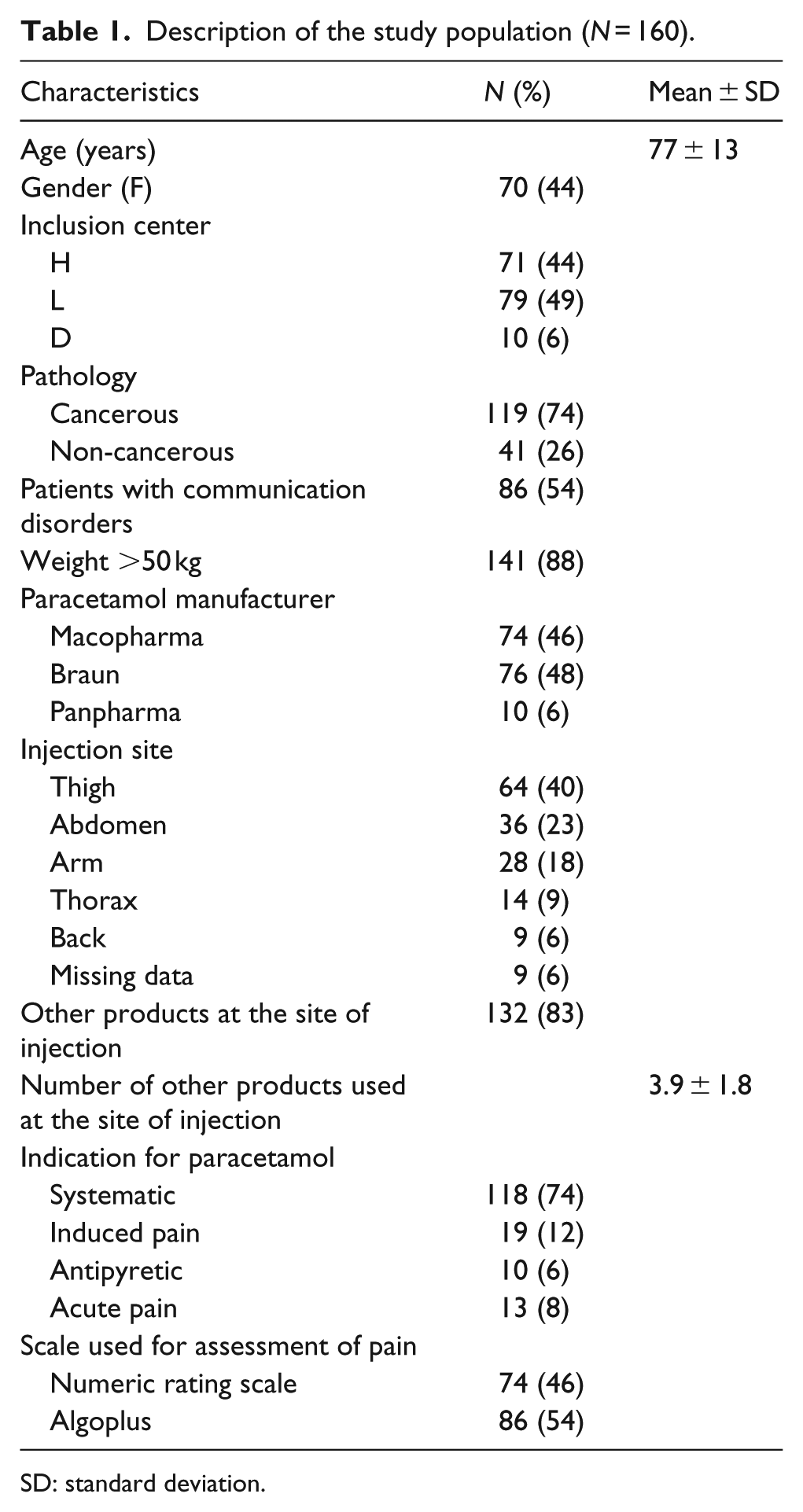
SD: standard deviation.
Paracetamol was administered at a dose of 1000 mg/injection if the patient’s weight was higher than 50 kg (141 patients); otherwise, the dosage was 500 mg/injection (19 patients).
Paracetamol was administered, either with other products on an existing subcutaneous access port (132 patients) or in isolation on a specific access port (28 patients).
The mean duration of use of the administration site for paracetamol was 2.3 ± 1.9 days and the mean assessment time for the occurrence of local adverse effects was 2.7 ± 2 days. The reasons for discontinuing the use of the first site of subcutaneous administration of paracetamol were as follows: change of injection site (60 patients, 38%), patient deceased (45 patients, 28%), appearance of a local adverse event (17 patients, 11%), change in the administration route (13 patients, 8%), accidental withdrawal of the catheter (8 patients, 5%), and return home (1 patient, 1%).
Over the course of the study, none of the patients presented serious local adverse events. Of the 160 patients, 44 (28%) had at least one non-serious local adverse event, either within the first 4 h following the first injection or during the monitoring of the administration site. Edema was observed in 29 patients, erythema in 5 patients, pain in 15 patients, hematoma in 2 patients, pruritus in 1 patient, and local heat in 2 patients. Table 2 shows the distribution of the local adverse events recorded during follow-up.
Local adverse events (at the injection site) during follow-up.
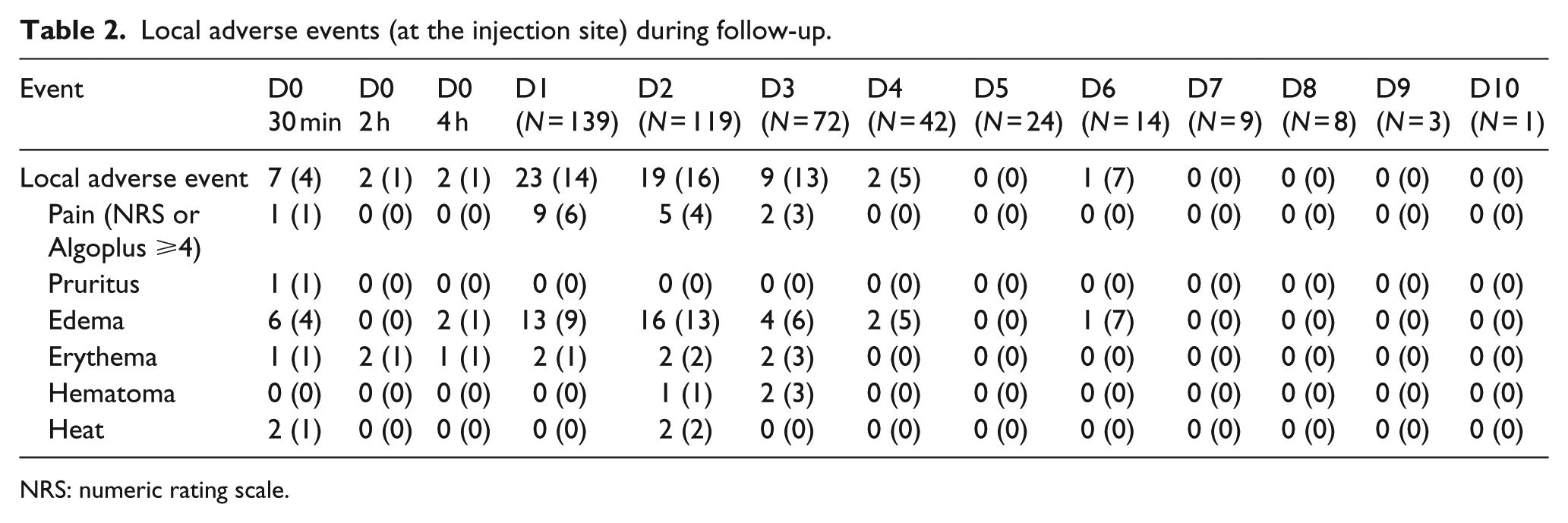
NRS: numeric rating scale.
In nine patients (6%), adverse events occurred within 4 h after the first subcutaneous injection of paracetamol. Of the 139 patients followed at least 1 day after the first subcutaneous administration of paracetamol, 38 (27%) had at least one non-serious local adverse event.
The management of these non-serious local adverse events was always straightforward, with occasionally a change in administration site, but never by discontinuing the subcutaneous use of paracetamol.
Factors associated with the occurrence of a non-serious adverse event after subcutaneous injection of paracetamol were as follows: administration in the arm or thorax, higher number of daily injections, and younger age (Table 3).
Factors associated with the occurrence of a non-serious local adverse event (AE) during follow-up after subcutaneous injection of paracetamol.
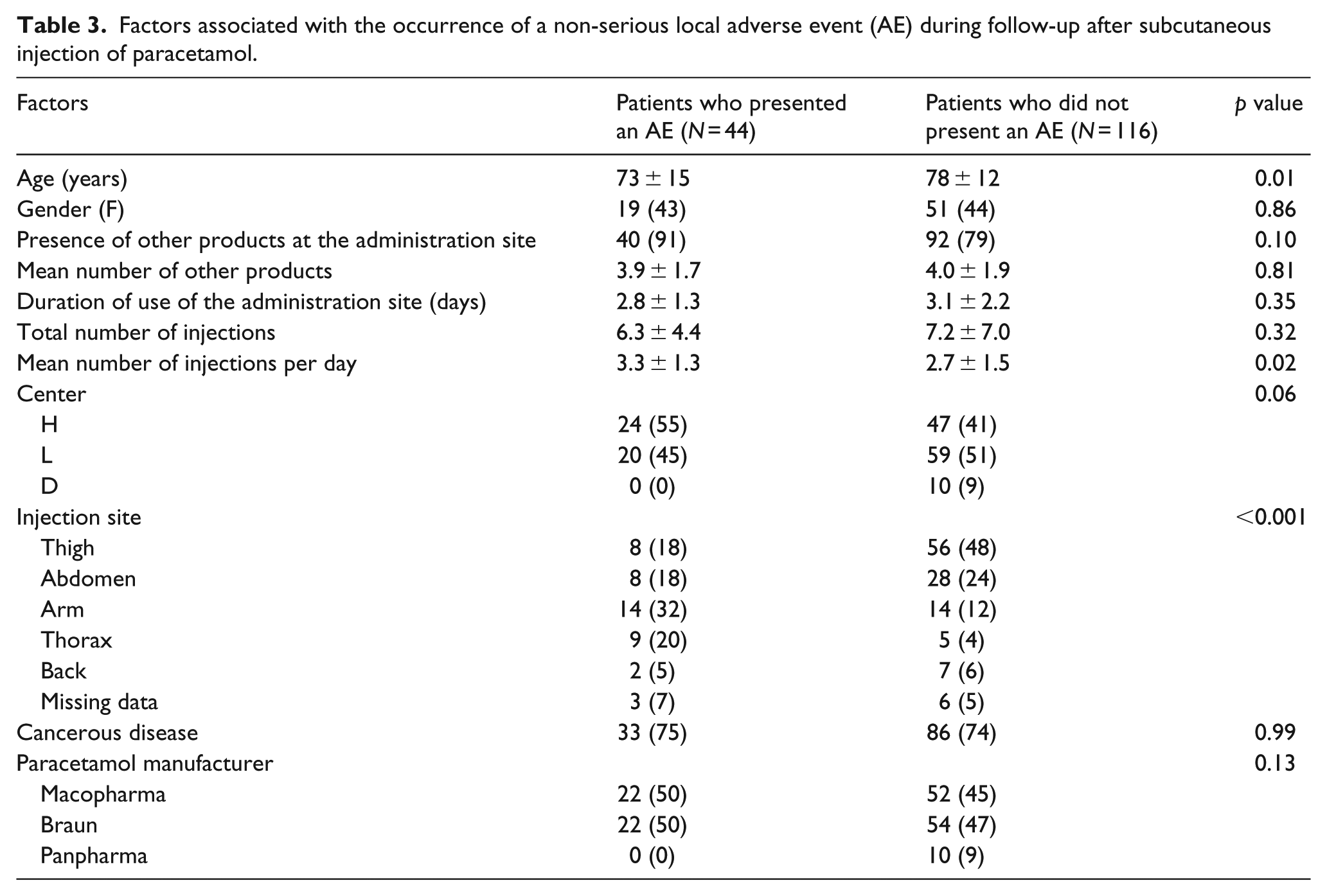
Presence of other products at the administration site was not associated with the occurrence of a non-serious adverse event. The products used at the same subcutaneous administration site as paracetamol are summarized in Table 4 and classified according to the level of risk of local adverse events.
Drugs used on the same subcutaneous injection site as paracetamol, classified according to the level of risk of local adverse event.

Discussion
One of the key findings of this study is that, over the entire follow-up period, no patient presented a serious adverse event, whether ulceration, necrosis, or abscess.
Edema represented the main non-serious adverse event followed by pain and erythema, which is consistent with prescribers’ perception of this practice. 5 Such side effects are also often highlighted when various compounds are administered subcutaneously,14,19,20 including during hydration. 12 Subcutaneous administration of antibiotics can also be responsible for local reactions such as skin necrosis.21–23 Such complications were not observed in this study.
The greater occurrence of adverse events when injections were made in the arm or thorax can be explained by a lower volume of diffusion at these sites than in the thigh or abdomen. The effect of the number of daily injections may be explained by an increased volume of injected paracetamol. 24
The higher incidence of adverse events in younger patients is more difficult to explain. The clinical relevance of this statistical difference may nonetheless be questioned, with the mean age increasing from 73 to 78 years.
Our study shows that paracetamol was often administered in combination with strong opioids, although it may not provide added benefit above regular opioids25,26 mainly at high dose, 27 and although opioid-sparing effect of paracetamol in patients with cancer pain remains uncertain. 25 Paracetamol was also often used systematically, with the objective to alleviate pain due to cancer, although the role of paracetamol in the management of cancer pain still remains controversial. 28 It will be probably necessary to assess paracetamol indications in PCUs to avoid abusive generalization of use.
Our study has several limitations. First, some data were missing (injection site for 9 patients, reason for discontinuing the use of the first site for 12 patients, incomplete pain assessment during the first injection for 14 patients, lack of pain evaluation during daily monitoring for 14 patients, and lack of adverse event assessment the day after the discontinuation of first site use for 13 patients). As a result, the occurrence of local pain and delayed adverse events may have been underestimated.
Second, while no serious adverse events were observed in our study, it is possible that such events might occur after the subcutaneous injection of paracetamol without our study being able to highlight their occurrence if their frequency is below 2%, as this was the chosen frequency for defining a rare adverse event in this study.
Finally, the design of this study did not allow for the efficacy of subcutaneously administered paracetamol to be assessed because it was often administered in patients with little or no pain at the time of administration and with other analgesics. A study comparing the pharmacokinetics of paracetamol when administered subcutaneously versus intravenously in healthy volunteers would be of great interest.
Conclusion
Our study shows that the subcutaneous administration of paracetamol is well tolerated locally in palliative care patients hospitalized in PCUs. It would thus appear appropriate for teams already using this approach to continue this practice, while teams that do not could consider using paracetamol subcutaneously when no other route is available, reliable, or acceptable for the patient. However, further studies are needed to show that this practice, while well tolerated, is also effective.
Footnotes
Acknowledgements
The authors thank the following investigators who participated in this study: Raphaël Alluin, Anne-Cécile Bourjal, Stéphane Picard, Elise Piot, Claire Reneaux, and Jean-François Villard. They also thank Laura Saez for her feedback and revision of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.