
Abstract
Erythropoietin (EPO) promotes neurogenesis and neuroprotection. We here compared the protection induced by two EPO formulations in a rodent model of Alzheimer’s disease (AD): rHu-EPO and a low sialic form, Neuro-EPO. We used the intracerebroventricular administration of aggregated Aβ25-35 peptide, a non-transgenic AD model. rHu-EPO was tested at 125–500 µg/kg intraperitoneally and Neuro-EPO at 62–250 µg/kg intranasally (IN). Behavioural procedures included spontaneous alternation, passive avoidance, water-maze and object recognition, to address spatial and non-spatial, short- and long-term memories. Biochemical markers of Aβ25-35 toxicity in the mouse hippocampus were examined and cell loss in the CA1 layer was determined. rHu-EPO and Neuro-EPO led to a significant prevention of Aβ25-35-induced learning deficits. Both EPO formulations prevented the induction of lipid peroxidation in the hippocampus, showing an antioxidant activity. rHu-EPO (250 µg/kg) or Neuro-EPO (125 µg/kg) prevented the Aβ25-35-induced increase in Bax level, TNFα and IL-1β production and decrease in Akt activation. A significant prevention of the Aβ25-35-induced cell loss in CA1 was also observed. EPO is neuroprotective in the Aβ25-35 AD model, confirming its potential as an endogenous neuroprotection system that could be boosted for therapeutic efficacy. We here identified a new IN formulation of EPO showing high neuroprotective activity. Considering its efficacy, ease and safety, IN Neuro-EPO is a new promising therapeutic agent in AD.
Introduction
Alzheimer’s disease (AD), the most common dementia in the elderly, is characterized by a progressive cognitive deterioration, resulting from synapse and nerve cell destruction in the brain. AD symptoms include memory loss, failure to communicate or perform routine tasks, alteration of the personality and, finally, vegetative state. The histopathological characteristics found in AD patient brains are the presence of extracellular senile plaques, intracellular neurofibrillary tangles, reduction of synapses, neuronal degeneration and reduction in brain volume. Senile plaques are composed of insoluble aggregates of amyloid-β (Aβ) proteins, which are generated by enzymatic cleavages of the amyloid precursor protein (APP), while neurofibrillary tangles are the result of hyper- and abnormal phosphorylation of the microtubule-stabilizing protein Tau (Mattson, 2004; Selkoe, 2001). Progressive accumulation of Aβ species, under the form of soluble monomers and oligomers, fibrillar deposits and mature plaques, contribute to AD physiopathology (Walsh and Selkoe, 2007). Althougḥ A
Here, we took advantage of the Aβ25-35 mouse model, in which most of these neuropathological hallmarks are observed. Indeed, the toxicity induced after Aβ25-35 injection in rodents was repeatedly shown to result in neuroinflammation and reactive gliosis, activation of pro-apoptotic caspases, oxidative stress, neuronal loss in hippocampal pyramidal cell layers, loss of cholinergic neurons, and memory deficits (Chavant et al., 2010; Delobette et al., 1997; Klementiev et al., 2007; Maurice et al., 1996; Meunier et al., 2006; Stepanichev et al., 2003, 2004, 2006; Villard et al., 2009, 2011; Zussy et al., 2011).
Several pharmacological strategies are currently being investigated to enable significant neuroprotection against Aβ toxicity and AD neurodegeneration. A new alternative strategy could be to boost endogenous neuroprotection systems, and notably haematopoietic cytokines, as they exert therapeutic potentials on neurological disorders such as AD. Cytokine treatments are able to prevent and restore cognitive deficits, increase microglial cells number and decrease both brain Aβ deposition and soluble Aβ accumulation by enhancing Aβ phagocytosis by microglia (Majumdar et al., 2007; Mitrasinovic and Murphy, 2003; Mitrasinovic et al., 2003). Among haematopoietic cytokines, erythropoietin (EPO) is receiving increasing attention since, besides its erythropoietic function, it has been shown to promote cell protection and regeneration in a number of indications (for reviews, see Bartesaghi et al., 2005; Rabie and Marti, 2008). EPO is a 165 amino-acids (30 kDa) glycoprotein belonging to the cytokine type I superfamily. It regulates erythropoiesis by inhibiting programmed cell death in erythroid cells, thus allowing the maturation of erythrocytes (Rabie and Marti, 2008). EPO and its receptor are expressed in the liver, and also in tissues not involved in erythropoiesis, such as reproductive tract, lung, spleen, heart, and brain, in neurons and astrocytes (Nadam et al. 2007; Sanchez et al. 2009). The cytoprotective effects of EPO have been established in several organs and particularly the central nervous system (for review, see Maiese et al., 2008). EPO and its receptor are upregulated upon neuronal injury and neurodegeneration, such as in the temporal cortex and hippocampus of patients with mild cognitive impairment or AD (Assaraf et al., 2007). In addition, experimental injection of EPO can improve neurological function and reduce brain damage following cerebral ischaemia (Bernaudin et al., 1999; Sakanaka et al., 1998;), intracerebral haemorrhage (Hoynck van Papendrecht et al., 1992; Lee et al., 2006), traumatic brain injury (Brines et al., 2000), spinal cord injury (Celik et al., 2002), experimental autoimmune encephalitis (Li et al., 2004), status epilepticus (Nadam et al., 2007), neonatal hypoxia-ischaemia (Kumral et al., 2004), and amyotrophic lateral sclerosis (Grunfeld et al., 2007). In AD models, both in vitro and in vivo evidence for a neuroprotective effect of EPO has been obtained. Using in vitro cell culture models, recombinant human EPO (rHu-EPO) protected neurons from neurodegeneration induced by Aβ peptides (Chong et al., 2005) and particularly Aβ25-35 oligomers (Li et al., 2008; Ma et al., 2009). In aged Tg2576 mice, EPO improved contextual memory and enhanced, particularly, endothelial proliferation, synaptophysin expression and capillary density in the brain (Lee et al., 2012).
A new EPO formulation containing low sialic acid has been designed. This formulation, named Neuro-EPO, is devoid of haematopoietic effects and able to rapidly reach the brain after intranasal (IN) delivery (Garcia-Rodríguez and Rodríguez-Cruz, 2012; Garcia-Rodríguez and Sosa-Testé, 2009). The formulation has the required composition to remain for an appropriate time in the nasal cavity, avoiding natural elimination or cleaning by the mucal cilia. pH, volume and concentration were optimized to allow Neuro-EPO release through the nasal passage into the cerebrospinal fluid and the brain. Previous data with Neuro-EPO confirmed its protective efficacy in gerbils submitted to focal ischaemia (Genc et al., 2011; Rodríguez Cruz et al., 2010; Sosa Testé et al., 2006).
In the present study, we used the rapid, pathomimetic AD model induced in mice by intracerebroventricular (ICV) injection of Aβ25-35 oligomers (Maurice et al., 1996) to confirm and compare the protective activity of rHu-EPO, administerable intraperitoneally (IP) and Neuro-EPO administered IN. rHu-EPO and Neuro-EPO were repeatedly administered between day 1 and day 4 after injection of Aβ25-35. Memory deficits were analysed between day 7 and 12, using the spontaneous alternation, passive avoidance, place learning in the water-maze, and novel object recognition procedures. In hippocampus extracts prepared at day 7, the level of peroxidation of membrane lipids was analysed as a marker of oxidative stress. Bax expression and the pAkt/Akt ratio were analysed as markers of apoptosis and endogenous protection pathways, respectively. A histochemical approach was used to address brain morphological damage, and inflammation was evaluated by measuring cytokine release.
Material and methods
Animals
Male Swiss mice, 6 weeks old and weighing 31 ± 2 g, were purchased from Depré (Saint Doulchard, France). Mouse housing and experiments took place within the animal facility of the University of Montpellier 2. Animals were housed in groups with access to food and water ad libitum. They were kept in a temperature and humidity controlled facility on a 12 h/12 h light/dark cycle (lights on at 7:00 h). Behavioural experiments were carried out between 9:00 h and 17:00 h, in a sound-attenuated and air-regulated experimental room, to which mice were habituated for 30 min. All animal procedures were conducted in strict adherence to the European Union Directive of September 22, 2010 (2010/63/UE).
Drugs and injections
Male Swiss mice were treated intracerebroventricularly (ICV) with either Aβ25-35 amyloid peptide (9 nmol/mouse), or scrambled Aβ25-35 amyloid peptide (Sc.Aβ, 9 nmol/mouse), according to the previously described method (Maurice et al., 1996; Meunier et al., 2006; Villard et al., 2009, 2011). Both peptides were from Genepep (Saint Jean-de-Védas, France). Homogeneous oligomeric preparation of the Aβ25-35 peptide was performed by incubating the peptide solution for 4 days at 37°C. The peptides were administered ICV in a final volume of 3 µL/mouse (final concentration 3 mM). Recombinant human erythropoietin (rHu-EPO) and EPO modified to display low sialic acid content (Neuro-EPO, patents PCT/cu2006/000001 and 20050138 to CIDEM, Havana, Cuba) were supplied by the Center of Molecular Immunology (Havana, Cuba) through CIMAB (Havana, Cuba) and diluted in phosphate buffer saline (pH 7.0) at 0.15 mM. rHu-EPO was injected intraperitoneally (IP). Neuro-EPO was applied IN, as described previously (Sosa Testé et al., 2006).
Experimental series
rHu-EPO was tested at 125, 250 and 500 µg/kg IP, which corresponded to 1250, 2500 and 5000 U/kg respectively. Peptide injection was performed at day 0. rHu-EPO was injected IP once a day on days 1–4 after peptide injection. Animals were tested at day 7 and later. Neuro-EPO was tested at 62, 125 and 250 µg/kg IN, which corresponded to 62, 125, 250 IU/kg, respectively. Neuro-EPO was applied IN three times a day (at 08:00 h, 12:00 h and 16:00 h) on days 1–4 after peptide injection. Animals were tested at day 7 and later. Control groups included vehicle solution-treated animals (IP or IN) and Sc.Aβ-treated animals.
A first series of animals was tested in the spontaneous alternation test at day 7 after peptide injection and in the passive avoidance test at days 8–9. A second series of animals was tested for place learning in the water-maze at days 7–12 after peptide injection and then in the novel object recognition test at days 13–15. A third series of animals was sacrificed at day 7 after peptide injection for hippocampus dissection, lipid peroxidation measurement, and western blot analyses. A fourth series of animals was sacrificed at day 7 after peptide injection by transcardiac perfusion with paraformaldehyde solution for histology and immunohistochemistry.
Spontaneous alternation in the Y-maze
Animals were tested for spontaneous alternation performance in the Y-maze, an index of spatial working memory. The Y-maze is made of grey PVC. Each arm is 40 cm long, 13 cm high, 3 cm wide at the bottom, 10 cm wide at the top, and converged at an equal angle. Each mouse was placed at the end of one arm and allowed to move freely through the maze during an 8 min session. The series of arm entries, including possible returns into the same arm, were checked visually. An alternation was defined as entries into all three arms on consecutive occasions. The number of maximum alternations was therefore the total number of arm entries minus two and the percentage of alternation was calculated as (actual alternations / maximum alternations) × 100. Parameters included the percentage of alternation (memory index) and total number of arm entries (exploration index).
Step-through passive avoidance
The apparatus for testing step-through passive avoidance consisted of an illuminated compartment with white polyvinylchloride walls (15 × 20 × 15 cm high), a darkened compartment with black polyvinylchloride walls (15 × 20 × 15 cm high) and a grid floor. A guillotine door separated each compartment. A 60 W lamp positioned 40 cm above the apparatus lit the white compartment during the experimental period. Scrambled foot shocks (0.3 mA for 3 s) were delivered to the grid floor using a shock generator scrambler (Lafayette Instruments, Lafayette, MA, USA). The guillotine door was initially closed during the training session. Each mouse was placed into the white compartment. After 5 s, the door was raised. When the mouse entered the darkened compartment and placed all its paws on the grid floor, the door was gently closed and the scrambled foot shock was delivered for 3 s. The step-through latency, i.e. the latency spent to enter the dark compartment, and the level of sensitivity to the shock were recorded. The latter was evaluated as: 0 = no sign; 1 = flinching reactions; 2 = flinching and vocalization reactions. None of the treatments used in the present study affected the step-through latency or shock sensitivity during training sessions (data not shown). The retention test was carried out 24 h after training. Each mouse was placed again into the white compartment. After 5 s, the door was raised. The step-through latency was recorded up to 300 s. Results were expressed as median and interquartile (25–75%) range.
Place learning in the water-maze
The water-maze was a circular pool (diameter 140 cm, height 40 cm). The water temperature, 23 ± 1°C, light intensity, external cues in the room, and water opacity were rigorously reproduced. A transparent Plexiglas non-slippery platform (diameter 10 cm) was immersed under the water surface during acquisition. Swimming could be recorded using Videotrack® software (Viewpoint, Champagne-au-Mont-d’Or, France), with trajectories being analysed as latencies and distances. The software divides the pool into four quadrants.
Acquisition
Between days 7 to 11 after the peptide injection, training consisted of three swims per day for 5 days, with 20 min intertrial time interval. Start positions, set at each limit between quadrants, were randomly selected and each animal was allowed a 90 s swim to find the platform. Swimming latency was measured using a stopwatch. Animals were left on the platform for 20 s. Animals that did not find the platform after 90 s had elapsed were placed on it manually and left for 20 s. The median latency was calculated for each training day and expressed for the experimental group as mean ± S.E.M.
Retention
A probe test was performed 24 h after the last acquisition session, on day 12. The platform was removed and each animal was allowed a free 60 s swim. The start position for each mouse was corresponding to one of two positions remote from the platform location in counterbalanced order. The swimming was videotracked and the time spent in the training (T) quadrant was analysed.
Novel object recognition memory
The apparatus consisted of four squared open-fields (50 cm × 50 cm × 50 cm high) made from white plexiglas and placed on a floor equipped with infrared (IR) light emitting diodes. The locomotor activity of the animal and position of their nose was captured through an IR-sensitive camera and analysed using the Videotrack® and Nosetrack® software (Viewpoint). On day 13 after peptide and drug injections (session 1), animals were allowed to acclimate for 10 min to the open-field. On day 14 (session 2), two identical objects (50 mL plastic vials with caps) were placed at defined positions, at one-quarter and three-quarters of one diagonal of the arena. Each mouse was placed in the open-field and the exploratory activity and nose position was recorded for 10 min. The activity was analysed in terms of number of contacts with the objects and duration of contacts. On day 15 (session 3), the object in position 2 was replaced by a novel one (a soft plastic chair-foot protector) differing in colour, shape and texture from the familiar object. Each mouse was placed again in the open-field and the exploratory activity recorded for 10 min. The activity was analysed as described earlier. The preferential exploration index was calculated as the ratio of the number (or duration) of contacts with the object in position 2 over the total number (or duration) of contacts with the two objects. As the analyses in terms of number of contacts or duration of contacts led to very similar results, only the number of contacts is presented. Animals showing less than 10 contacts with objects during the sessions 2 or 3 are usually discarded from the calculations. No animal was discarded accordingly in the present study.
Western blot analyses
For determination of Bax, phospho-Akt (pAkt), Akt or EPO receptor expression, mice were decapitated 7 days after Aβ25-35 peptide injection. The hippocampus was removed onto an ice-cold glass plate and stored at -80°C. The hippocampus tissues were homogenized in ice-cold extraction buffer containing 2% sodium dodecylsulfate (SDS) and protease inhibitors (Roche Diagnostics, Mannheim, Germany). Homogenates were heated at 70°C for 10 min and centrifuged at 14,000 g for 30 min at 4°C. Protein concentration was determined using the Pierce BCA assay (Pierce Biotechnology, Rockford, IL, USA) according to the manufacturer’s instructions. Equal amounts of protein, 40 µg, were resolved in a 12% SDS–polyacrylamide gel electrophoresis, then transferred electrophoretically onto a nitrocellulose blot membrane (0.45 mm, Schleicher Schuell). The membranes were then blocked for 30 min at room temperature with 5% skim milk in Tris-buffered saline 20 mM, pH 7.6, containing 0.1% Tween 20 (TBS-T). The membranes were incubated at 4°C overnight with a rabbit monoclonal anti-Bax antibody (2772S, dilution 1:2000, Cell Signaling, USA), mouse anti-pAkt (4060P, 1:2000, Cell Signaling), or mouse anti-Akt antibody (9272S, 1:2000, Cell Signaling), rinsed for 30 min in TBS-T and then incubated for 2 h with a goat anti-mouse or anti-rabbit secondary antibody (1:2000 each, Sigma-Aldrich). Peroxidase activity was revealed by using enhanced chemiluminescence reagent (Millipore). The intensity of peroxidase activity was then quantified using a Li-Cor Odyssey Fc imaging system (Li-Cor, Lincoln, NE, USA). Results were normalized to control values (anti β-tubulin; 1:5000 Sigma-Aldrich).
Lipid peroxidation measures
Mice were sacrificed by decapitation 7 days after Aβ25-35 injection and brains were rapidly removed, the hippocampus dissected out, weighed, frozen in liquid nitrogen and stored at −80°C until assayed. After thawing, the hippocampus was homogenized in cold methanol (1/10 w/v), centrifuged at 1000 g for 5 min and the supernatant collected. Homogenate was added to a solution containing FeSO4 1 mM, H2SO4 0.25 M, xylenol orange 1 mM and incubated for 30 min at room temperature. Absorbance was measured at 580 nm (A5801), and 10 µL of cumene hydroperoxide (CHP) 1 mM was added to the sample and incubated for 30 min at room temperature, to determine the maximal oxidation level. Absorbance was measured at 580 nm (A5802). The level of lipid peroxidation was determined as CHP equivalents according to: CHP eq. = A5801/A5802 × [CHP (nmol)] × dilution, calculated as CHP eq. per wet tissue weight, and presented as percentage of control group value.
TNFα and IL-1β ELISA assays
Animals were sacrificed 7 days after peptide injection, their hippocampus dissected out on ice, weighed, frozen in liquid nitrogen and stored at -80°C until analyses. After thawing, the hippocampus was homogenized in 50 mM Tris-150 mM NaCl buffer, pH 7.5, and sonicated for 20 s. After centrifugation at 16,100 g for 15 min at 4°C, supernatants were used for TNFα and IL-1β ELISA assays according to the manufacturer’s instructions (TNFα: ref. EMTNFA, ThermoScientific, Courtaboeuf, France; IL-1β: ref. E90563Mu, USCN, Wuhan, P.R. China). For each assay, absorbance was read at 450 nm and sample concentration was calculated using the standard curve. All samples were assayed in duplicate. Results are expressed as pg of cytokine per mg of wet tissue.
Histology
Each mouse was anaesthetized by intramuscular injection of ketamine, 80 mg/kg, and xylazine, 10 mg/kg, and quickly transcardially perfused with 50 mL of saline solution followed by 50 mL of paraformaldehyde 4%. Brains were removed and kept overnight in the fixative solution. They were cut in coronal sections (30 µm thickness) using a vibratome (VT1000S, Leica, Germany). Serial sections were selected to include the hippocampus formation and placed in gelatin-coated glass strips. Sections were stained with 0.2% cresyl violet reagent (Sigma-Aldrich), then dehydrated with graded ethanol, treated with toluene and mounted with DePeX medium (BDH Laboratories, Poole, UK). Examination of the CA1 area was performed on fields located between antero-posterior coordinates −1.90 and −2.30 from Bregma and lateral coordinates ±1.50 to ±1.75 (Paxinos and Franklin, 2004), using a light microscope (Dialux 22, Leitz). Slices were digitalized through a CCD camera (Sony XC-77CE) with the NIH ImageJ® software (NIH, Bethesda, MD, USA), to easily process CA1 measurement and pyramidal cells counts. Data were calculated as average of 6–11 slices per animal and 6–8 animals per group, and expressed as number of viable CA1 pyramidal cells per millimetre for each group.
Statistical analyses
Data were analysed using a one-way analysis of variance (ANOVA, F value), followed by a Dunnett’s test. Passive avoidance or swimming latencies did not show a normal distribution, since a cut-off time was set. Passive avoidance data were expressed as median value and interquartile range and analysed using the Kruskal–Wallis non-parametric ANOVA (H values), group comparisons being made with Dunn’s non-parametric multiple comparisons test. Shock sensitivity was also analysed using non-parametric ANOVA. Acquisition profiles in the water-maze were analysed using the non-parametric repeated-measure Friedman’s ANOVA (Fr value), followed by a Dunn’s or Mann–Whitney’s test for post-hoc comparisons. Probe test data were presented as time spent in the quadrants. Presence in the T quadrant was analysed using a one-sample t-test vs. the chance level (15 s). The object preference, calculated from the number of contacts with the two objects, was analysed using a one-sample t-test vs. the chance level (50%). For these two last analyses, the ANOVA was however performed and is detailed in the figure legends. The level of statistical significance was p < 0.05. All statistical data are indicated in the figure legends.
Results
Protective effects of rHu-EPO and Neuro-EPO in Aβ25-35-treated mice – behavioural study
rHu-EPO was administered to Aβ25-35-treated mice in the 125–500 µg/kg IP dose range. Animals were then tested in a series of behavioural tests. First, animals were examined at day 7 after peptide injection for the spontaneous alternation performances, a spatial working memory test. Then, at days 8–9, they were tested in the passive avoidance procedure, a non-spatial long-term memory test. As shown in Figure 1, the Aβ25-35 treatment led to a significant decrease in alternation that was significantly blocked by rHu-EPO at the two highest doses tested (Figure 1(a)). No effect was noted in terms of total number of arm entries during the session, whatever the treatment (not shown). The Aβ25-35-treated group showed a highly significant decrease in step-through passive avoidance latency (Figure 1(b)). The rHu-EPO treatment highly significantly prevented the deficit at all doses tested (Figure 1(b)).
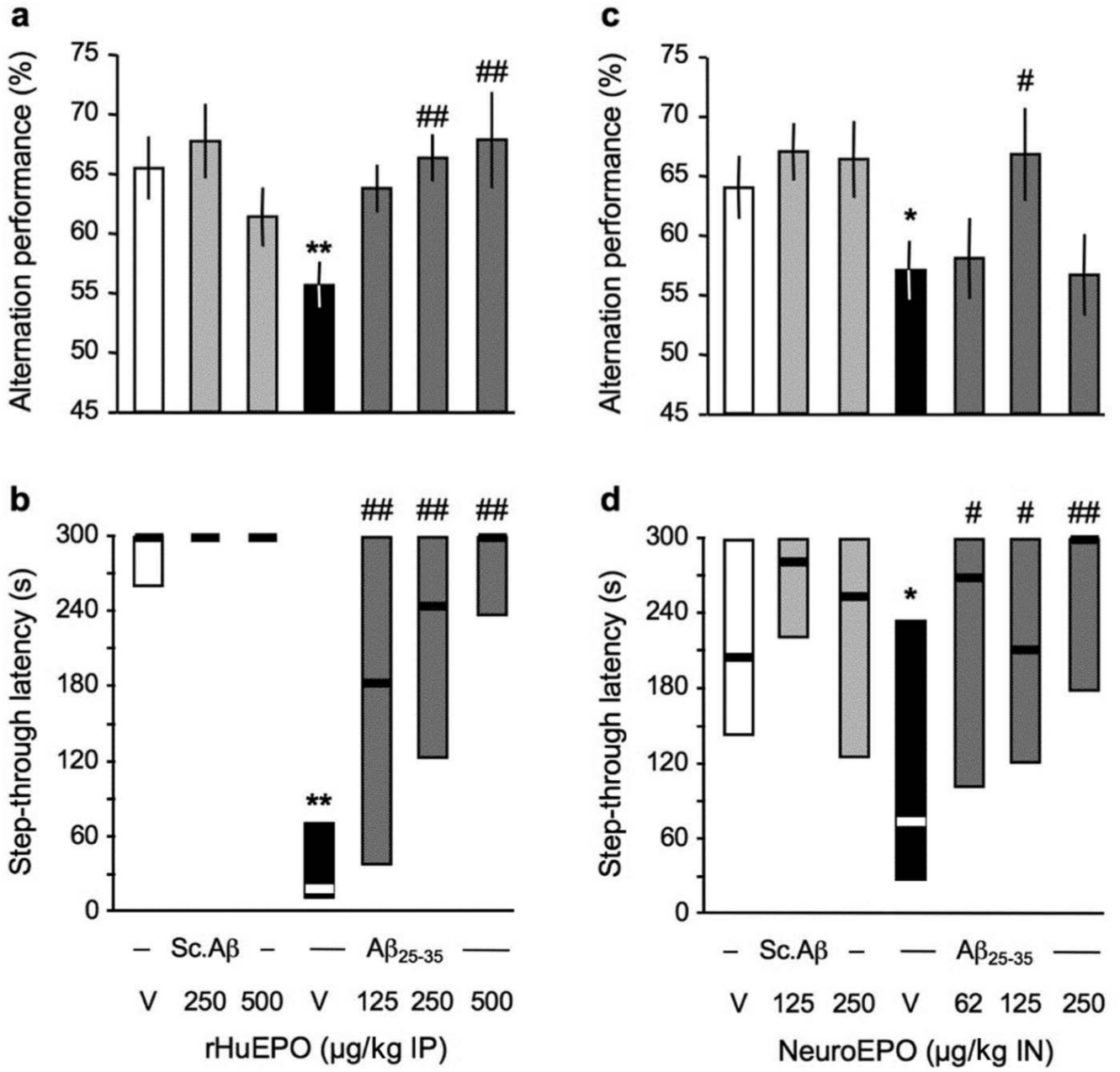
Protective effect of (a, b) rHu-EPO and (c, d) Neuro-EPO against the Aβ25-35-induced learning deficits in mice: (a, c) spontaneous alternation performances in the Y-maze and (b, d) step-through latency in the passive avoidance test. At day 0, mice were administered ICV Sc.Aβ or Aβ25-35 peptide (9 nmol). Between day 1 and day 4, they received vehicle solution (V) or rHu-EPO (125–500 µg/kg IP) once a day in (a, b) or vehicle solution (V) or Neuro-EPO (62–250 µg/kg IN) three times a day in (c, d). Mice were examined for spontaneous alternation at day 7, they were trained for passive avoidance at day 8 and retention was examined at day 9. One-way ANOVA: F(7,88) = 2.49, p < 0.05, n = 12–14 per group in (a), H = 35.8, p < 0.0001, n = 12–14 per group in (b), F(7,81) = 2.42, p < 0.05, n = 16–22 per group in (c), H = 31.3, p < 0.0001, n = 22–27 in (d). * p < 0.05, ** p < 0.01 vs. (Sc.Aβ+V)-treated group; # p < 0.05, ## p < 0.01 vs. (Aβ25-35+V)-treated group; Dunnett’s test in (a, c); Dunn’s test in (b, d).
Neuro-EPO was tested at the doses of 62, 125 and 250 µg/kg IN. As shown in Figure 1(c), the Aβ25-35 treatment led to a significant decrease in alternation that was significantly blocked by Neuro-EPO at the 125 µg/kg dose. The effect was bell shaped, and the 250 µg/kg dose appeared inactive. A marginal effect with the 63 µg/kg dose of Neuro-EPO was noted in terms of number of arms entered during the Y-maze session (not shown). The Aβ25-35-treated group showed a significant decrease in step-through passive avoidance latency (Figure 1(d)). The Neuro-EPO treatment significantly prevented the deficits at all doses tested (Figure 1(d)).
Hippocampus-dependent spatial memory was assessed in another batch of animals using place learning in the water-maze. The spatial reference memory of the animals was analysed through the learning of a fixed position of a hidden platform. rHu-EPO effect was examined at 250 and 500 µg/kg IP doses (Figure 2), and Neuro-EPO at 125 and 250 µg/kg IN doses (Figure 3). For rHu-EPO groups, the Friedman repeated-measure non-parametric ANOVA analyses of acquisition profiles showed that the latencies measured for the Sc.Aβ/V-treated group significantly decreased over training trials (Figure 2(a)). The Sc.Aβ/rHu-EPO 500-treated group also showed a significant learning, while the Aβ25-35/V-treated group failed to show a significant decrease in swimming duration (Figure 2(a)). In particular, swimming durations measured during trials 4 and 5 for Aβ25-35/V-treated animals were highly significantly higher than that measured for Sc.Aβ/V-treated mice. The rHu-EPO treatment, at 250 µg/kg, led to a decreasing profile in swimming durations, with a significant difference as compared with Aβ25-35/V-treated animals on day 5 (Figure 2(b)). The rHu-EPO treatment at 500 µg/kg also led to a significant decrease in swimming duration on day 5 as compared with the Sc.Aβ/V-treated group, although the overall acquisition profile was very similar to that observed for the Aβ25-35/V-treated group.
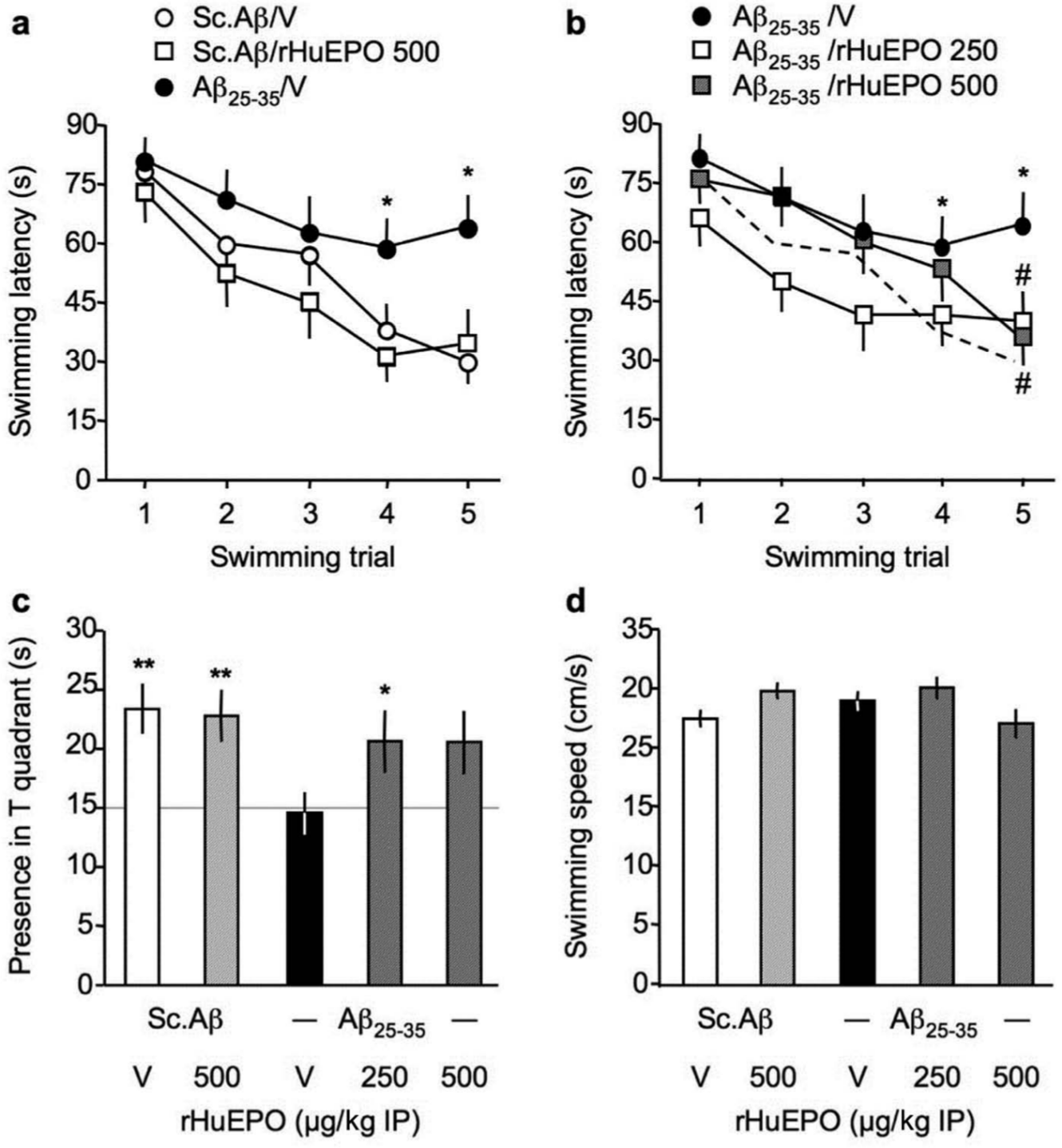
Protective effect of rHu-EPO against the Aβ25-35-induced place learning deficit in the water-maze in mice: (a, b) acquisition profiles, (c) probe test and (d) swimming speed. At day 0, mice were administered ICV Sc.Aβ or Aβ25-35 peptide (9 nmol). Between day 1 and day 4, they received vehicle solution (V) or rHu-EPO (250–500 µg/kg IP) once a day. Mice were trained between day 7 and 11 and the probe test was examined without platform on day 12. In (b), the profile of control animals (Sc.Aβ+V) is indicated by a dotted line. In (c), the chance level (15 s) is outlined. Repeated-measure non-parametric Friedman’s ANOVA: Fr = 29.1, p < 0.0001, n = 14 for (Sc.Aβ+V), Fr = 10.9, p < 0.05, n = 11 for (Sc.Aβ+rHu-EPO 500) in (a), Fr = 5.32, p > 0.05, n = 13 for (Aβ25-35+V), Fr = 12.1, p < 0.05, n = 14 for (Aβ25-35+rHu-EPO 250), Fr = 18.7, p < 0.0001, n = 13 for (Aβ25-35+rHu-EPO 500) in (b). * p < 0.05 vs. same trial (Sc.Aβ+V)-treated group; # p < 0.05 vs. same trial (Aβ25-35+V)-treated group; Dunn’s test in (a, b). F(4,64) = 2.79, p < 0.05; * p < 0.05, ** p < 0.01 vs. chance level (15 s), one-sample t-test in (c). F(4,64) = 2.39, p > 0.05 in (d).
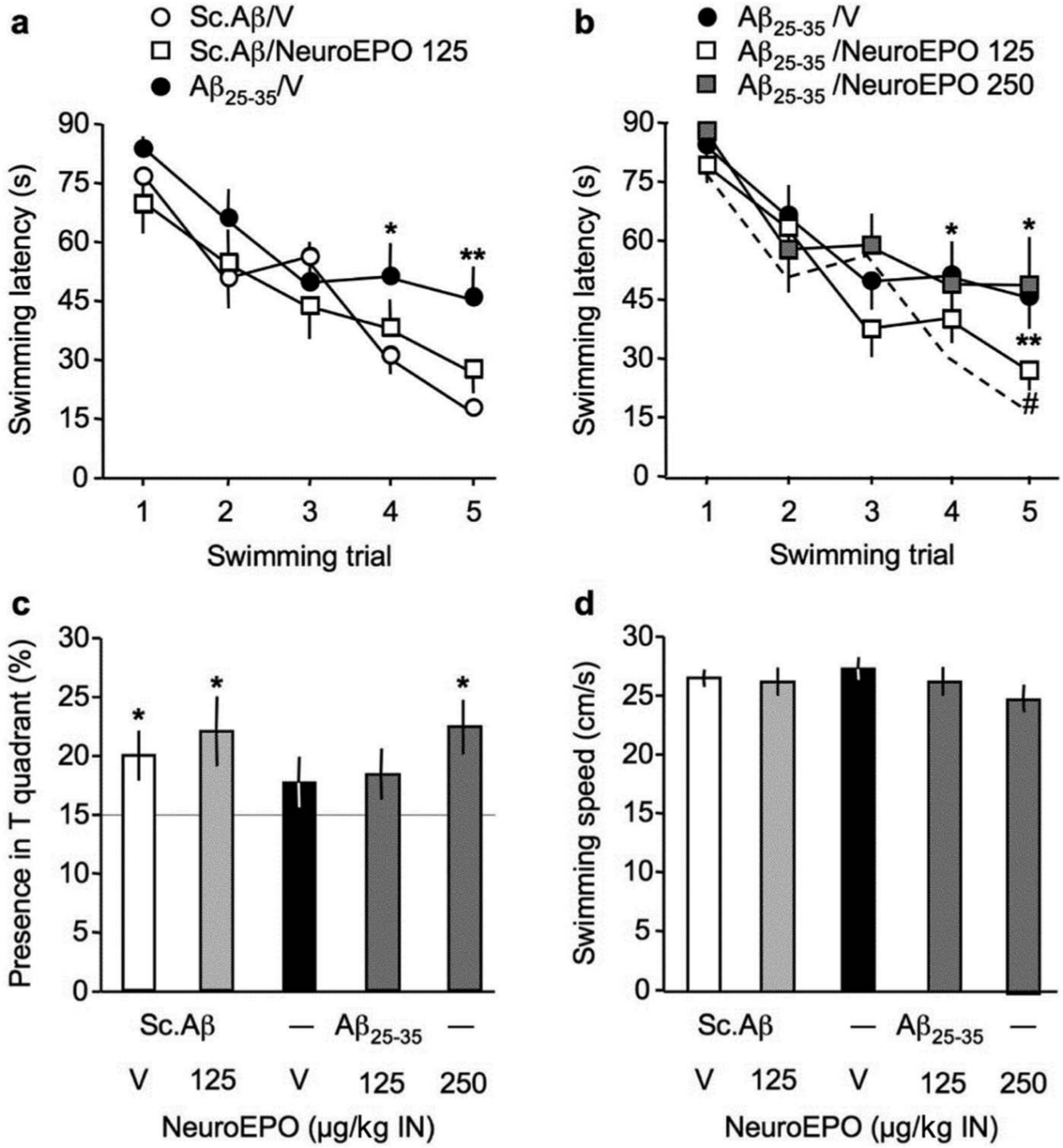
Protective effect of Neuro-EPO against the Aβ25-35-induced place learning deficit in the water-maze in mice: (a, b) acquisition profiles, (c) probe test and (d) swimming speed. At day 0, mice were administered ICV Sc.Aβ or Aβ25-35 peptide (9 nmol). Between day 1 and day 4, they received vehicle solution (V) or Neuro-EPO (125–250 µg/kg IN) three times a day. Mice were trained between day 7 and 11 and the probe test was examined without platform on day 12. In (b), the profile of control animals (Sc.Aβ+V) is indicated by a dotted line. In (c), the chance level (15 s) is outlined. Friedman’s repeated-measure non-parametric ANOVA: Fr = 33.4, p < 0.0001, n = 16 for Sc.Aβ+V, Fr = 13.9, p < 0.01, n = 16 for Sc.Aβ+Neuro-EPO 125 in (a), Fr = 20.8, p < 0.001, n = 16 for Aβ25-35+V, Fr = 33.4, p < 0.0001, n = 20 for Aβ25-35+Neuro-EPO 125, Fr = 13.0, p < 0.05, n = 10 for Aβ25-35+Neuro-EPO 250 in (b). * p < 0.05, ** p < 0.01 vs. same trial Sc.Aβ+V-treated group; # p < 0.05 vs. same trial Aβ25-35+V-treated group; Dunn’s test in (a, b). F(4,75) = 0.79, p > 0.05; * p < 0.05 vs. chance level (15 s), one-sample t-test in (c). F(4,75) = 0.52, p > 0.05 in (d).
Analysis of the animal presence in the training (T) quadrant of the pool during the probe test showed that Sc.Aβ/V- or Sc.Aβ/rHu-EPO 500-treated animals spent significantly more time in this quadrant, showing an effective learning of the platform location (Figure 2(c)). The Aβ25-35/V-treated group spent 15 s in the T quadrant, indicating a failure to acquire the platform location (Figure 2(c)). The rHu-EPO IP treatments allowed a reversion of this deficit, with the data measured for the Aβ25-35/rHu-EPO 250-treated group reaching significance (Figure 2(c)). In parallel, we checked that none of the treatment altered the swimming speed of the animals (Figure 2(d)).
For the Neuro-EPO groups, Friedman’s ANOVA analyses of acquisition profiles showed that the latencies measured for the Sc.Aβ/V-, Sc.Aβ/Neuro-EPO 125-, and Aβ25-35/V-treated groups significantly decreased over training trials (Figure 3(a)). However, Aβ25-35/V-treated animals appeared less efficient during the two last training trials, since the swimming duration appeared significantly higher as compared with Sc.Aβ/V-treated mice (Figure 3(a)). The Neuro-EPO treatment at 125 µg/kg allowed a significant improvement of the acquisition profile, as compared with Aβ25-35/V-treated mice, and statistical significance was reached for trial 5 (Figure 3(b)).
During the probe test, Sc.Aβ/V- and Sc.Aβ/Neuro-EPO 125-treated animals spent significantly more time in the T quadrant, showing an effective learning of the platform position (Figure 3(c)). The Aβ25-35/V-treated group failed to show a presence time significantly higher than the chance level (Figure 3(c)) and the Neuro-EPO treatment at 250 µg/kg, but not 125 µg/kg, attenuated the deficit (Figure 3(c)). In parallel, we checked that none of the treatments altered the swimming speed of the animals (Figure 3(d)).
The same batches of animals were used in the novel object recognition procedure (Figure 4). Analysis of object exploration showed no preference among the two similar objects in Aβ25-35-injected mice and after the rHu-EPO treatments during session 2 (Figure 4(a)). With an average of 188 ± 13 contacts during session 2, none of the treatments affected the exploratory activity of the animals (data not shown). However, Aβ25-35-treated mice failed to show a preferential exploration of the novel object, as compared with Sc.Aβ-treated mice, during session 3 (Figure 4(b)). The rHu-EPO treatment attenuated the recognition memory deficit, with a significant effect measured at 250 µg/kg (Figure 4(b)). With an average of 127 ± 10 contacts in this session, none of the treatments changed the exploratory activity of the animals in session 3 (data not shown).
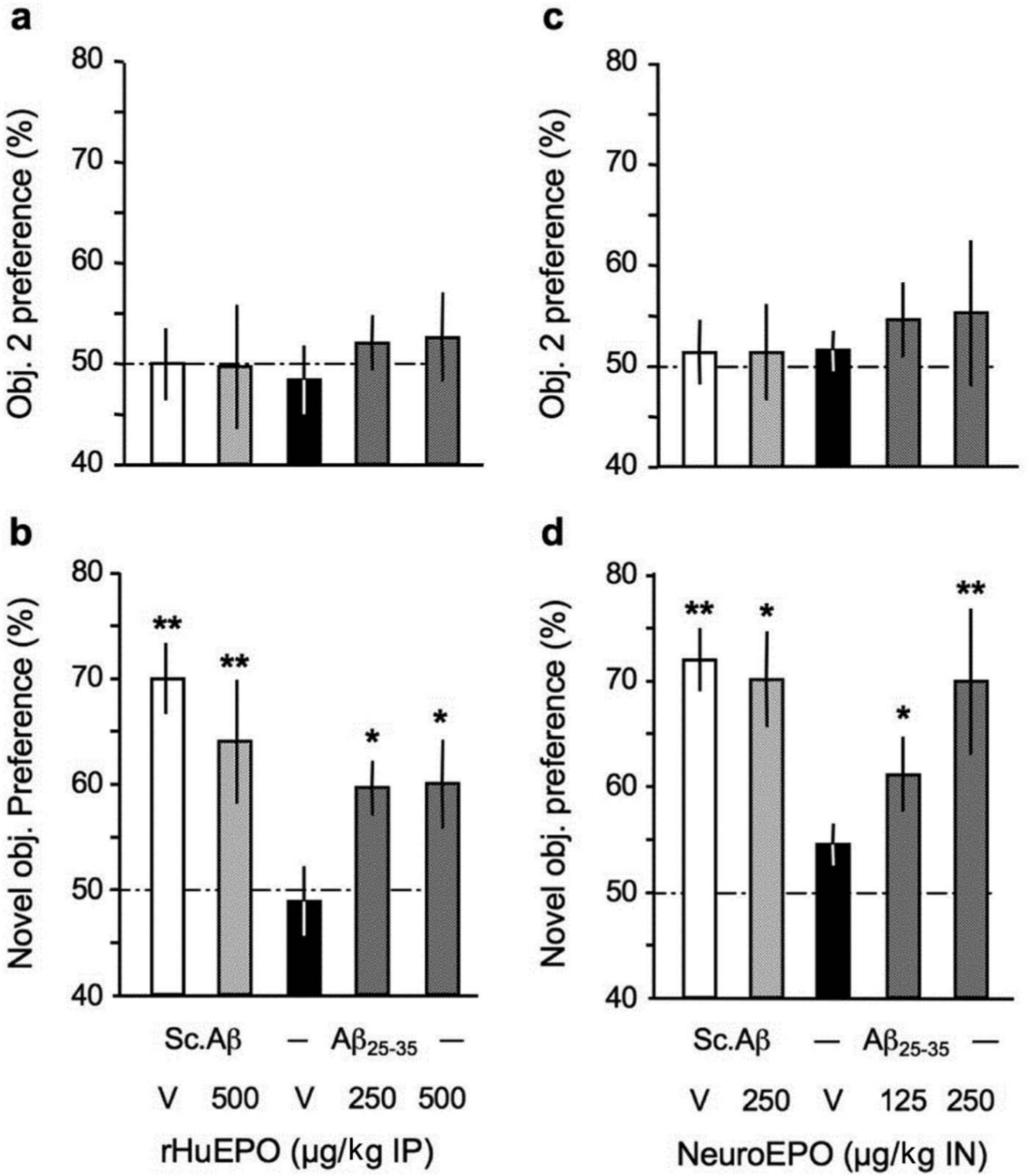
Protective effect of (a, b) rHu-EPO and (c, d) Neuro-EPO against the Aβ25-35-induced novel object recognition deficits in mice: (a, c) session 2 with two identical objects and (b, d) session 3 with a familiar and a novel object. At day 0, mice were administered ICV Sc.Aβ or Aβ25-35 peptide (9 nmol). Between day 1 and day 4, they received vehicle solution (V) or rHu-EPO (250, 500 µg/kg IP) once a day in (a, b) or vehicle solution (V) or Neuro-EPO (125, 250 µg/kg IN) three times a day in (c, d). Habituation was performed on day 6, session 2 on day 7 and session 3 on day 8. ANOVA: F(4,49) = 0.27, p > 0.05, n = 10 per group in (a), F(4,49) = 3.11, p < 0.05 in (b); F(4,50) = 0.19, p > 0.05, n = 9–11 per group in (c), F(4,50) = 2.80, p < 0.05 in (d). * p < 0.05, ** p < 0.01 vs. chance level (50%), one-sample t-test.
Animals treated with Neuro-EPO did not show difference in object exploration during session 2 (Figure 4(c)). With an average of 186 ± 12 contacts during session 2, the Neuro-EPO treatments did not affect the exploratory activity of the animals (data not shown). But the compound allowed a significant reversion of the Aβ25-35-induced recognition deficit during session 3, at both doses and with a dose-dependent effect (Figure 4(d)). The average number of contacts, 129 ± 8, was also similar among treatment groups in this session (data not shown).
Protective effects of rHu-EPO and Neuro-EPO in Aβ25-35-treated mice – biochemical measures
The impact of the EPO formulations on Aβ25-35-induced oxidative stress was analysed by measuring lipid peroxidation levels in hippocampus extracts. Aβ25-35 induced a highly significant 70–86% increase in lipid peroxidation (Figure 5(a), (b)). These increases were partially but significantly attenuated by rHu-EPO, at the 250 µg/kg dose (Figure 5(a)) and fully blocked by the Neuro-EPO treatment, at the two highest doses tested (Figure 5(b)).
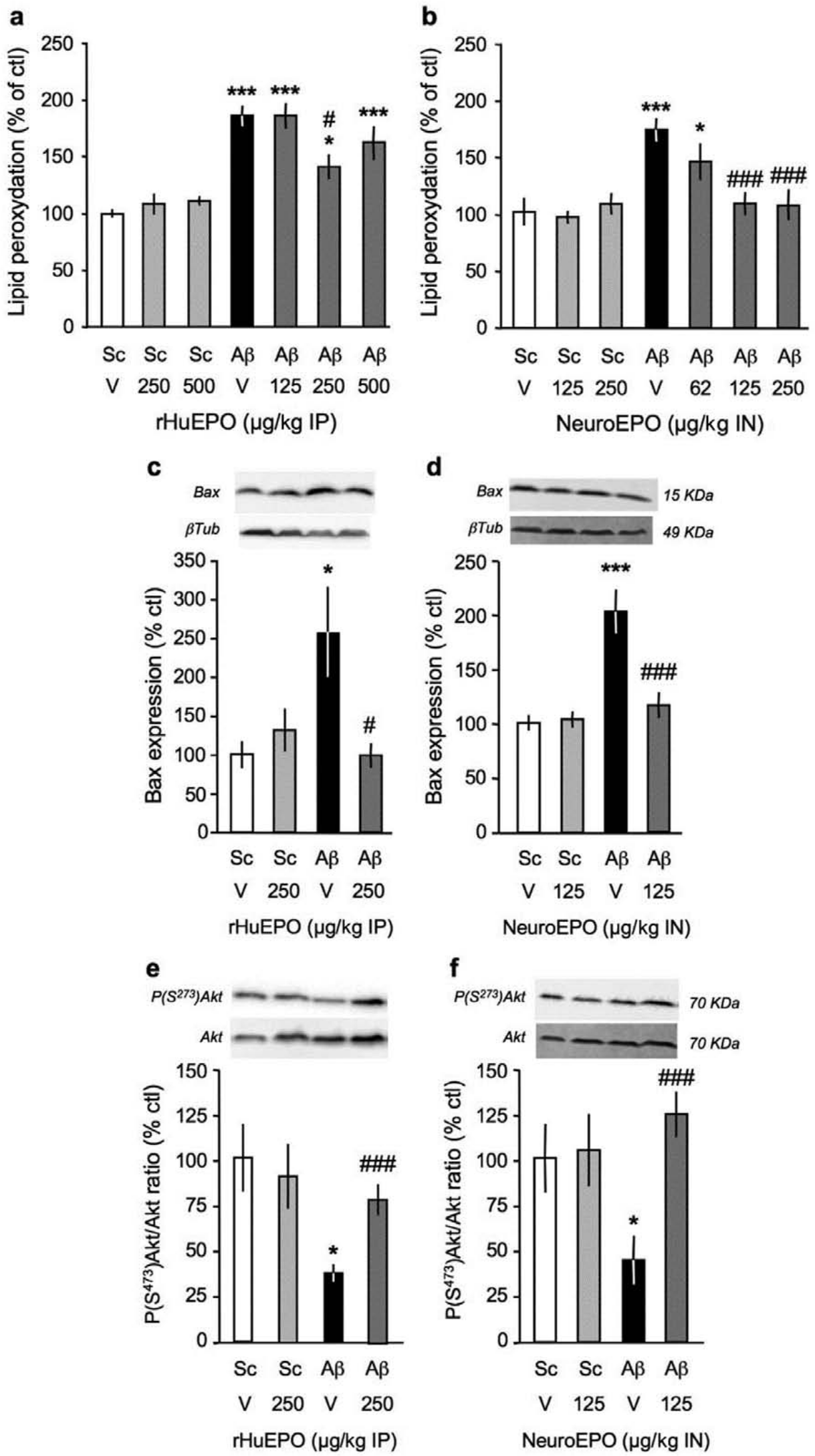
Neuroprotective effect of rHu-EPO (a, c, e) and Neuro-EPO (b, d, f) on: lipid peroxidation levels (a, b) Bax expression (c, d) and Akt activation (e, f) in the hippocampus, 7 days after Aβ25-35 injection in mice. At day 0, mice were administered ICV Sc.Aβ or Aβ25-35 peptide (9 nmol). Between day 1 and day 4, they received vehicle solution (V) or rHu-EPO (250 µg/kg IP) once a day in (a, c) or vehicle solution (V) or Neuro-EPO (125 µg/kg IN) three times a day in (b, d). Mice were sacrificed at day 9 for lipid peroxidation measures and at day 7 for western blot analyses. Typical blots are shown above the graphs in (c–f). Bax expression was calculated as Bax/β-tubulin ratio and Akt activation as phospho(S473)-Akt/total Akt ratio and all data expressed as percentage of control (Sc.Aβ+V) group data. One-way ANOVA: F(6,71) = 14.6, p < 0.0001, n = 8–12 per group in (a); F(6,76) = 5.47, p = 0.0001, n = 10–12 in (b); F(3,47) = 3.30, p < 0.05, n = 12 in (c); F(3,44) = 14.9, p < 0.0001, n = 11–12 in (d); F(3,47) = 3.49, p < 0.05, n = 12 in (e); F(3,45) = 4.39, p < 0.01, n = 11–12 in (f). * p < 0.05, *** p < 0.001 vs. the Sc.Aβ+V-treated group; # p < 0.05, ### p < 0.001 vs. the Aβ25-35+V-treated group; Dunnett’s test.
The induction of apoptosis in brain tissue was evaluated by a semi-quantitative method, the analysis of the pro-apoptotic Bax protein level by western blot (Figure 5(c), (d)). Aβ25-35 induced a highly significant 74–101% increase in Bax/β-tubulin ratio (Figure 5(c), (d)). rHu-EPO, tested at 250 µg/kg, or Neuro-EPO, at 125 µg/kg, blocked the Aβ25-35-induced increase in Bax level without any effect by themselves (Figure 5(c), (d), respectively).
The impact of amyloid toxicity on endogenous protection systems was examined on an effective signalling pathway, that involving the kinases PI3K/Akt and triggered by activation of several types of receptors, including receptors for trophic factor such as BDNF, neurotransmitters such as acetylcholine, or EPO. The activation of Akt was evaluated by quantifying the ratio between the active form phospho(Ser473)-Akt over total Akt (Figure 5(e), (f)). Aβ25-35 induced a significant -30% to -55% decrease in P(Ser473)-Akt/Akt ratio (Figure 5(e), (f)). rHu-EPO at 250 µg/kg, or Neuro-EPO at 125 µg/kg, fully prevented the Aβ25-35-induced decrease in P(Ser473)-Akt/Akt ratio without any effect by themselves (Figure 5(e), (f)).
The impact of the EPO formulations was also evaluated on Aβ25-35-induced neuroinflammation. The peptide has been reported to produce a strong astroglial and microglial reaction, with release of several cytokines (Chavant et al., 2010; Zussy et al., 2011). We analysed the TNFα and IL-1β contents in the hippocampus of Aβ25-35 injected mice (Figure 6). Aβ25-35 induced a highly significant 160–187% increase in TNFα levels (Figure 6(a),(c)). rHu-EPO 250 µg/kg, or Neuro-EPO 125 µg/kg, blocked the Aβ25-35-induced increase in TNFα levels without any effect by themselves (Figure 6(a), (c), respectively). Aβ25-35 also induced a highly significant 125–155% increase in IL-1β levels (Figure 6(b), (d)). rHu-EPO 250 µg/kg, or Neuro-EPO 125 µg/kg, blocked the Aβ25-35-induced increase in IL-1β levels without any effect by themselves (Figure 6(b), (d), respectively).
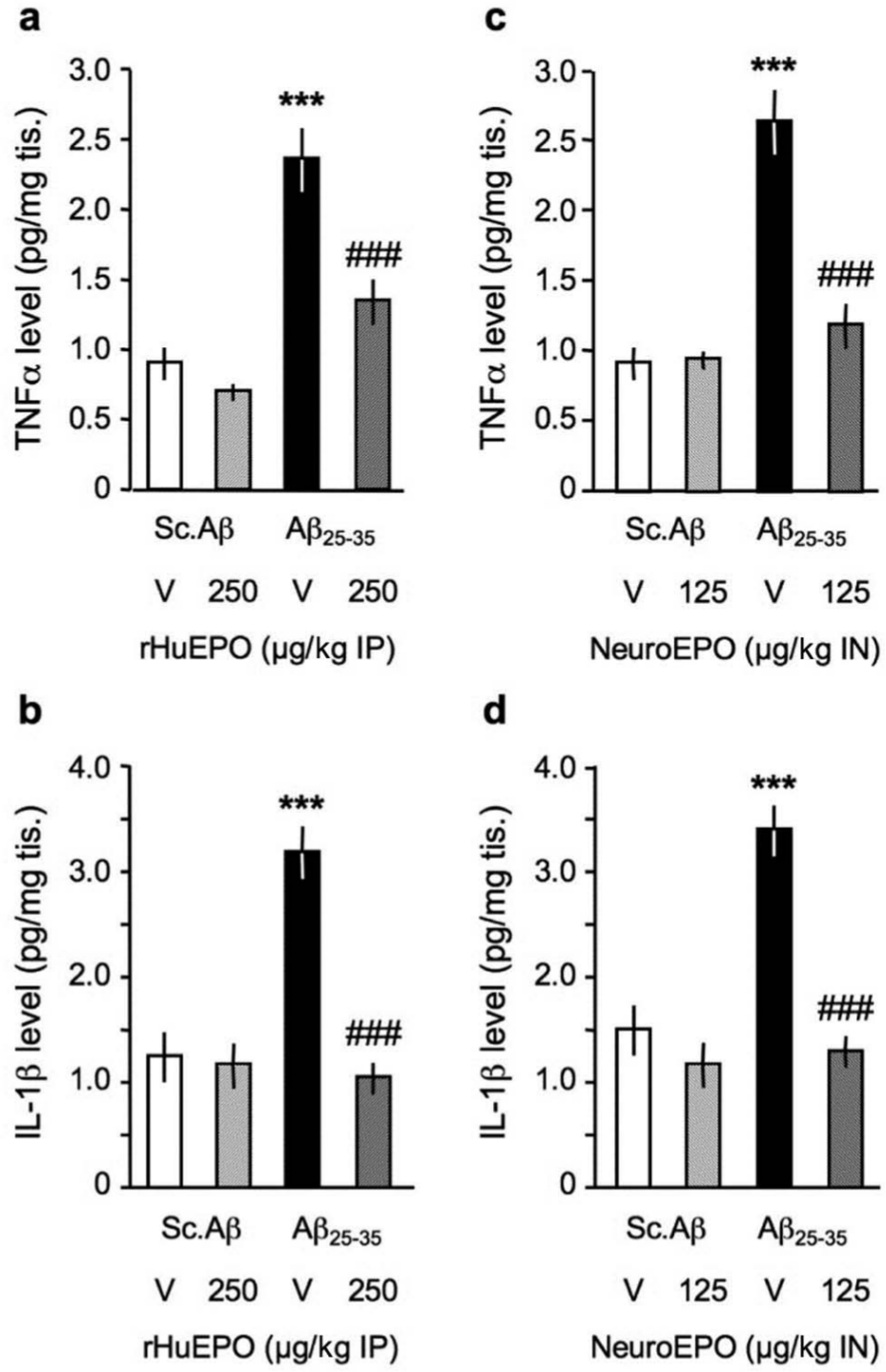
Protective effect of (a, b) rHu-EPO and (c, d) Neuro-EPO on (a, c) TNFα and (b, d) IL-1β levels in the hippocampus of Aβ25-35-injected mice. At day 0, mice were administered ICV Sc.Aβ or Aβ25-35 peptide (9 nmol). Between day 1 and day 4, they received vehicle solution (V) or rHu-EPO (250 µg/kg IP) once a day or vehicle solution (V) or Neuro-EPO (125 µg/kg IN) three times a day. Mice were sacrificed at day 7 for cytokine measures. n = 6 per group. One-way ANOVA: F(3,23) = 23.6, p < 0.0001 in (a); F(3,23) = 28.7, p < 0.0001 in (b); F(3,23) = 22.4, p < 0.0001 in (c); F(3,23) = 15.7, p < 0.0001 in (d). *** p < 0.001 vs. Sc.Aβ+V-treated group, ### p < 0.001 vs. Aβ25-35+V-treated group; Dunnett’s test.
Protective effects of rHu-EPO and Neuro-EPO in Aβ25-35-treated mice – histological measures
To assess the impact of the EPO treatments on brain cell integrity, we analysed the viable cells in the CA1 subfield of the pyramidal cell layer of the hippocampus, using cresyl violet staining. This area, which is highly sensitive to excitotoxicity, has been previously shown to be damaged by Aβ25-35 injection in the mouse (Villard et al., 2009) or rat (Stepanichev et al., 2004; Zussy et al., 2011). We observed a highly significant -22% to -24% decrease in the number of viable cells counted (Figure 7(b), (d)). The cell layer adopted a disorganized aspect with slightly swollen cells after Aβ25-35 injection (Figure 7(a), (c)). The rHu-EPO 250 µg/kg, or Neuro-EPO 125 µg/kg, treatments resulted in a highly significant blockade of cell loss (Figure 7(b), (d), respectively).
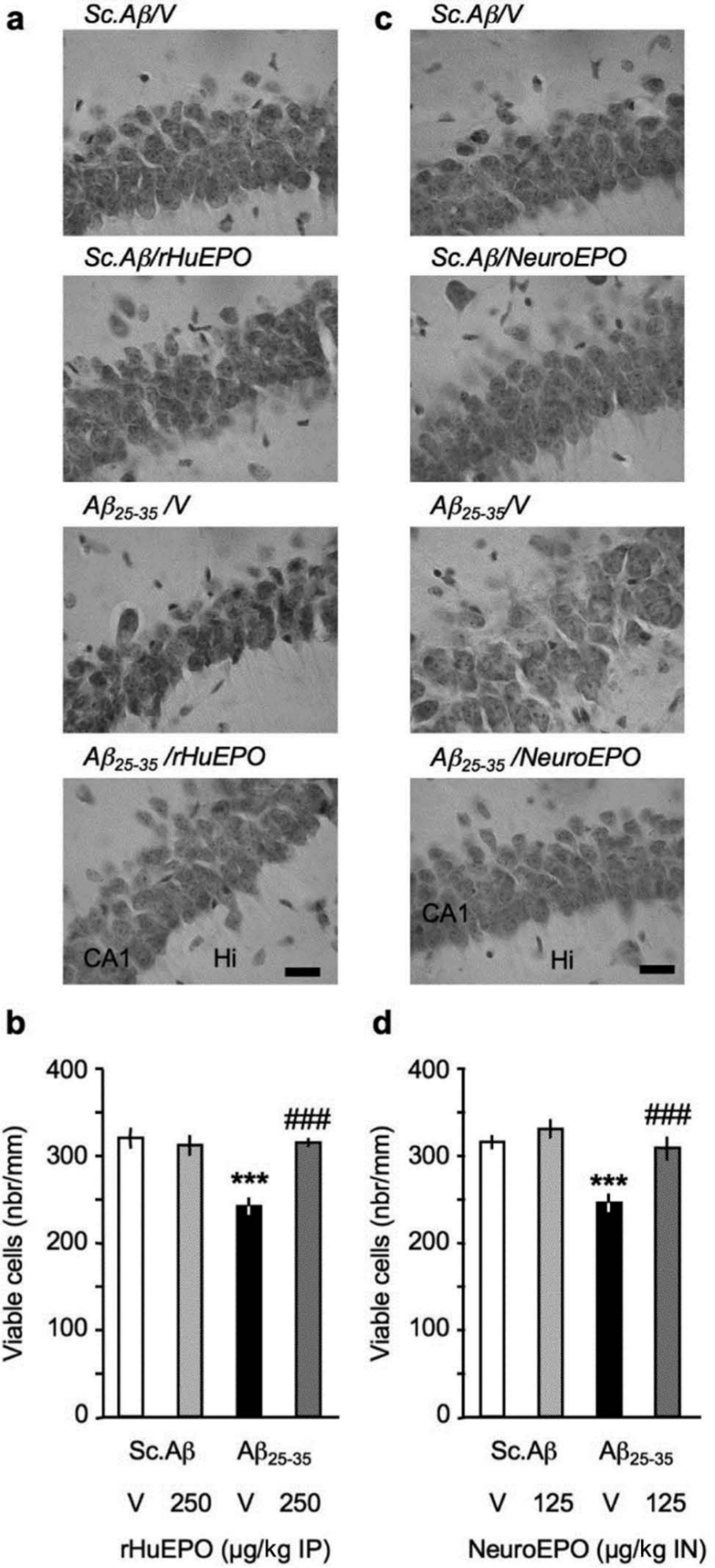
Neuroprotective effect of (a, b) rHu-EPO and (c, d) Neuro-EPO on Aβ25-35-induced toxicity in mice: pyramidal cell loss in the CA1 area of the hippocampal pyramidal cell layer, 7 days after Aβ25-35 injection. (a, c) Representative microphotographs of coronal sections of cresyl violet-stained hippocampal CA1 subfield. (b, d) Averaged levels of viable cells. At day 0, mice were administered ICV Sc.Aβ or Aβ25-35 peptide (9 nmol). Between day 1 and day 4, they received vehicle solution (V) or rHu-EPO (250 µg/kg IP) once a day in (a, c) or vehicle solution (V) or Neuro-EPO (125 µg/kg IN) three times a day in (b, d). Mice were sacrificed at day 7. In (a, b), scale bar = 50 µm; CA1, pyramidal cell layer; Hi, hilus. 6–11 slices were counted per mice and 6–8 mice per group. One-way ANOVA: F(3,28) = 12.8, p < 0.0001 in (b); F(3,26) = 12.0, p < 0.0001 in (d); *** p < 0.001 vs. ScAβ+V-treated group; ### p < 0.001 vs. Aβ25-35+V-treated group; Dunnett’s test.
Durability of the EPO protective effect
In a final experiment, we evaluated the duration of the Neuro-EPO effect in the passive avoidance test by performing a second retention session 1 week after the first retention session, i.e. 8 days after training, and a third retention session 3 weeks after the first retention session, i.e. 22 days after training (Figure 8). Sc.Aβ/V and Aβ25-35/V-treated groups (white and black columns, respectively) showed a time-dependent decrease in the step-through latency response during the second and third retention sessions, with a significant difference for the control group as compared with the first retention. Neuro-EPO-treated animals showed a particularly remarkable preservation of memory, with no alteration in the step-through latency during the second retention and a moderate, but significant, decrement during the third retention session (Figure 8). Three weeks after learning, the Neuro-EPO treatment, in Sc.Aβ as well as in Aβ25-35 animals, resulted in a clear cognitive-enhancing effect.
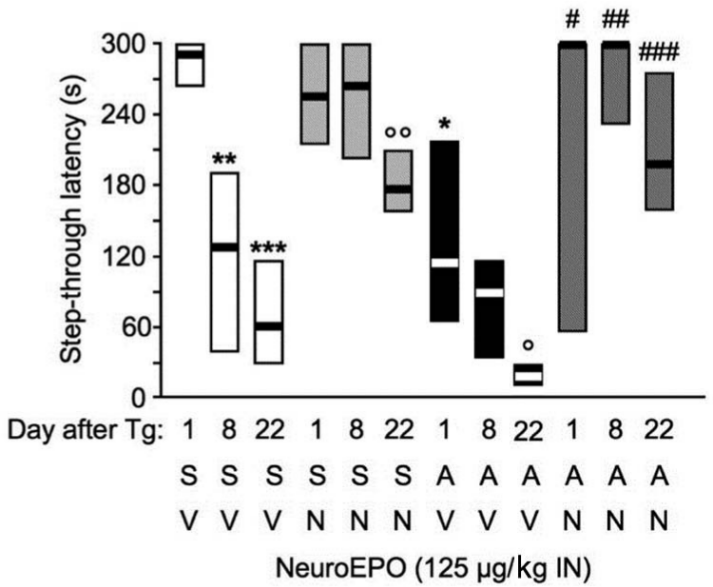
Sustained beneficial effect of Neuro-EPO against Aβ25-35-induced passive avoidance deficits in mice. At day 0, mice were administered ICV Sc.Aβ or Aβ25-35 peptide (9 nmol). Between day 1 and day 4, they received vehicle solution (V) or Neuro-EPO (125 µg/kg IN) three times a day. Mice were trained for passive avoidance at day 8 and retention examined at day 9 (1 day after training) and at day 16 (8 days after training). The number of animals per group was n = 10. H = 54.5, p < 0.0001; * p < 0.05, ** p < 0.01, *** p < 0.001 vs. Day 1 data for Sc.Aβ+V-treated group; ° p < 0.05, °° p < 0.01 vs. same retention day Sc.Aβ+V-treated group; # p < 0.05, ## p < 0.01, ### p < 0.001 vs. same retention day Aβ25-35+V-treated group; Dunn’s test.
Discussion
In the present study, we provide clear evidence that the IP injection of rHu-EPO or the IN application of Neuro-EPO leads to a significant protection against the toxicity and memory deficits induced 1 week after the ICV injection of an aggregated preparation of Aβ25-35 peptide in mice. We first analysed the learning deficits induced by the peptide and affecting several forms of memory. We examined spontaneous alternation, a spatial working memory response; step-through passive avoidance response, a non-spatial long-term memory procedure; place learning in the water-maze, a spatial reference memory test; and novel object recognition, a long-term recognition memory test. rHu-EPO protected against Aβ25-35-induced impairments, at doses of 250 and 500 µg/kg IP that corresponded to 2500 and 5000 U/kg. This dose range was in agreement with expectations from the literature, particularly in rodent models of stroke (Kumral et al., 2004; Sosa Testé et al., 2006). Moreover, in mice overexpressing human APP with the double Swedish mutation (Tg2576 line), rHu-EPO, injected at 5000 U/kg EPO over 5 days, allowed 13-month-old animals to recover fear-conditioning learning, a neurobehavioural procedure close to the passive avoidance response (Lee et al., 2012). In the present study, we show that the IN-administerable form of EPO, Neuro-EPO, was also effective but in a lower dose range, since the doses of 125 and 250 µg/kg were effective in most of the tests used. These doses were previously reported to provide neuroprotection in ischaemic gerbils (Rodríguez Cruz et al., 2010). In addition, the dose of 125 µg/kg exhibited a long-lasting effect until 22 days after the treatment, as illustrated in the last experiment of this study. The advantage of Neuro-EPO is that it has no haematopoietic activity and that its peripheral administration may not have as many effects as observed for rHu-EPO. Moreover, the central effect of rHu-EPO would only be observed when high doses are applied or when the blood–brain barrier is deficient, since it is hardly crossed by rHu-EPO (Rodríguez Cruz et al., 2010).
EPO has been reported to directly impact synaptic plasticity and transmitter release, particularly in the hippocampus (Adamcio et al., 2008; Kamal et al., 2011). A 3-week treatment of young mice with EPO enhanced long-term potentiation and altered short-term synaptic plasticity and synaptic transmission, shifting the balance of excitatory and inhibitory activity (Adamcio et al., 2008). The authors reported that networks of EPO-treated primary hippocampal neurons developed lower overall spiking activity but enhanced bursting in discrete neuronal assemblies. These effects resulted in an improvement of hippocampus-dependent memory by modulating plasticity, synaptic connectivity and activity of memory-related neuronal networks. Moreover, EPO perfusion in mouse brain hippocampal slices depressed significantly the slope of the excitatory postsynaptic potentials (fEPSP) and increased paired-pulse facilitation ratio (Kamal et al., 2011). In EPO-perfused slices significant larger responses were obtained following intermediate and high-frequency stimulation when compared with the control slices, suggesting that EPO decreases the excitatory neurotransmitter release probability and may in this way protect the synapses from toxic levels of glutamate (Kamal et al., 2011).
These direct effects could be related to the improvement of learning abilities observed after rHu-EPO and Neuro-EPO treatments in our experiments. However, a protective mechanism is also involved, and different biochemical and histological parameters were used in the study to analyse the extent of the neuroprotective activity of the EPO formulations. Oxidative stress was evaluated by measuring the Aβ25-35-induced increase in peroxidation of membrane lipids in hippocampus extracts (Meunier et al., 2006). The expression of Bax, a protein known to promote Aβ-mediated cell death (Paradis et al., 1996), and Akt activation were examined. Akt is activated in response to Aβ25-35 in a PI3K-dependent manner (Martín et al., 2001). Neuroinflammation was addressed by directly measuring the increases in cytokines in hippocampal tissue, induced by Aβ25-35. Since Aβ25-35 has repeatedly been shown to moderately but significantly alter cell viability in the pyramidal cell layer of the hippocampus (Stepanichev et al., 2004, 2006; Villard et al., 2009; Zussy et al., 2011), we analysed the cell loss in CA1 using cresyl violet staining. The rHu-EPO and Neuro-EPO treatments restored all parameters, but although the blockades of Aβ25-35-induced Bax induction, hippocampal cell loss or decrease in Akt activity were total, the IN formulation appeared more effective in decreasing Aβ25-35-induced oxidative stress than the IP rHu-EP formulation. The anti-oxidative properties of EPO were previously reported to account at least in part for its cytoprotective effect in several models. Thus, in rat cultured hippocampal neurons submitted to hypoxia or glutamate exposure, administration of EPO resulted in a significant protection (Chong et al., 2003a; Morishita et al., 1997). The purported mechanism likely sustaining the protective effect of EPO against amyloid toxicity has been suggested to involve NF-κB p65 translocation into the cytoplasm, inhibition of pro-apoptotic proteins, suppression of TNFα-generated apoptosis, modulation of growth arrest and DNA damage protein 45 (Gadd45β), and direct activation of Bcl-xL (Chong et al., 2005). As a consequence, EPO attenuated reactive oxygen species production, and stabilized mitochondrial membrane potential, Bcl-2/Bax ratio and caspase-3 activation (Li et al., 2008).
The effect of EPO is, however, dependent on the activation of several kinases, including Janus kinase 2 (JAK2), a tyrosine kinase that associates with EPO receptor, and protein kinase B/Akt. EPO prevented apoptotic injury to neuronal cells through the induction of JAK2 autophosphorylation (Kawakami et al., 2001). Moreover, EPO was shown to significantly enhance Akt activity during oxidative stress thus preventing inflammatory activation of microglia (Chong et al., 2003a,b). Since prevention of Akt phosphorylation blocked the cellular protection induced by EPO, the Akt activity upregulation appeared to be a necessary step in the mechanism. Through the regulation of the PI3K/Akt signalling pathway, EPO is able to regulate cellular apoptosis following hypoxic/excitotoxic insults and oxidative stress (Chong et al., 2003a,b).
In the present study, we observed that both EPO formulations blocked induction of the pro-apoptotic Bax protein and the cell death in CA1 neuronal layer, a glutamatergic neuron-rich area highly sensitive to excitotoxicity. The mechanism of EPO anti-apoptotic action is likely to rely on inhibition of the activity of glycogen synthase kinase-3β (GSK-3β) or the release of forkhead transcription factor (FOXO3a), both downstream targets of PI3K/Akt. Activation of FOXO3a results in apoptotic cellular degeneration and disrupts mitochondrial membrane permeability (Brunet et al., 1999). GSK-3β has several roles in brain signalling and pathology. It is in particular one of the major kinases responsible for Tau hyperphosphorylation in AD (Kosik, 1992). However, it is also involved in Aβ-mediated toxicity, since its over-activation is associated with cognitive impairments, Aβ production, neuronal death and neuroinflammation (Bhat et al., 2000; Hooper et al., 2008). GSK-3β activity is suppressed by EPO, and this effect can be associated with conformational changes and decreased expression of Bax to prevent cell death (Somervaille et al., 2001). Moreover, the protective effect of EPO against Aβ25-35 toxicity was shown to directly involve PI3K activity and GSK-3β activation, since its protective effect on a cell culture model was blocked by selective PI3K or GSK-3β inhibitors (Ma et al., 2009).
EPO has also been reported to promote an effective protection against inflammatory pathologies. We observed a very potent blockade by both rHu-EPO and Neuro-EPO of the TNFα and IL-1β releases provoked by Aβ25-35 toxicity. The neuroprotective ability of EPO is believed to mainly involve extrinsic cell homeostasis through modulation of microglial activation and control of cytokine release (Maiese et al., 2004). Activated microglia release large amounts of these pro-inflammatory and neurotoxic cytokines, including TNFα and IL-1β, but also free radicals such as nitric oxide or superoxide ion, and fatty acid metabolites, which could directly contribute to and amplify apoptotic cell death (Naert and Rivest, 2011). EPO has been shown to exert an anti-inflammatory action through pathways that involve phosphatidylserine exposure, microglial activation, Akt activity, and regulation of caspases, or more directly by inhibiting several pro-inflammatory cytokines, such as IL-6, TNFα and MCP-1 (Chong et al., 2003a,b, 2004).
The biological effect of EPO involves its interaction with the EPO receptor. The different forms of EPO appear to show different affinities for the receptor, and the low sialic acid form in particular may need higher concentrations to activate EPO receptors. They are expressed in neurons, astrocytes, oligodendrocytes, microglial and endothelial cells (Maiese et al., 2004) and their signalling pathways involve JAK2/PI3K/Akt activation, as discussed previously (Chong et al., 2003a,b). Modulation of their expression is based on the transcriptional regulation of two hypoxia-inducible factors, HIF-1 and HIF-2 (Rabie and Marti, 2008; Wenger, 2000). In contrast to EPO, the expression of EPO receptor in the brain does not appear to be sensitive to hypoxia (Digicaylioglu et al., 1995; Juul et al., 1998); rather, it is regulated by pro-inflammatory cytokines (Nagai et al., 2001), EPO itself (Chin et al., 2000) and probably other factors that have not been identified yet. The mechanism has been analysed in hypoxia models, but it may also apply to neurodegenerative conditions.
Finally, this study outlined the potential of the IN form of EPO. Contrary to the other EPO variants, Neuro-EPO is not chemically modified, making it biologically similar to endogenous EPO, with the advantage of less adverse reactions when the molecule is applied chronically. This constitutes a potential benefit of Neuro-EPO over other variants of EPO for the chronic treatment of neurodegenerative illnesses. Indeed, EPO applied intranasally acts in the brain in a paracrine/autocrine fashion, activating multiple signalling mechanisms to inhibit apoptosis, to reduce inflammation and local oedema, to induce the EPO receptor expression, to increase BDNF levels and glutathione-S-transferase (GST) enzyme levels – the main defence against xenobiotics of exogenous or endogenous origin – in cortical and subcortical regions. IN EPO induces the synthesis of the Neuroglobin protein selectively in the damaged regions, and promotes angiogenesis and protection of the vascular endothelium. It also contributes in a direct way to neurogenesis and neuroplasticity, which guarantees the new homeostasis and rescue of damaged or lost functions following brain insult. An IN form of insulin is currently under clinical trial in AD (Craft et al., 2012). IN EPO formulation is at present tested in stroke (Fletcher et al., 2009; Rodríguez Cruz et al., 2010), but other neurodegenerative pathologies must be envisaged.
Footnotes
Acknowledgements
This work is a scientific collaboration between the University of Montpellier 2 and the University of Medical Sciences of La Havana, Institute of Basic and Preclinical Sciences “Victoria de Girón”. J.C.G.R. thanks a visiting professor fellowship from the Scientific Council of the University of Montpellier 2.
Conflict of interest
The authors declare no conflict of interest.
Funding
This research was funded by resources from Inserm and University of Montpellier 2, but received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.