
Abstract
Regulators of G protein signaling are proteins that accelerate the termination of effector stimulation after G protein-coupled receptor activation. Many regulators of G protein signaling proteins are highly expressed in the brain and therefore considered potential drug discovery targets for central nervous system pathologies; for example, here we show that RGS12 is highly expressed in microdissected mouse ventral striatum. Given a role for the ventral striatum in psychostimulant-induced locomotor activity, we tested whether Rgs12 genetic ablation affected behavioral responses to amphetamine and cocaine. RGS12 loss significantly decreased hyperlocomotion to lower doses of both amphetamine and cocaine; however, other outcomes of administration (sensitization and conditioned place preference) were unaffected, suggesting that RGS12 does not function in support of the rewarding properties of these psychostimulants. To test whether observed response changes upon RGS12 loss were caused by changes to dopamine transporter expression and/or function, we prepared crude membranes from the brains of wild-type and RGS12-null mice and measured dopamine transporter-selective [3H]WIN 35428 binding, revealing an increase in dopamine transporter levels in the ventral–but not dorsal–striatum of RGS12-null mice. To address dopamine transporter function, we prepared striatal synaptosomes and measured [3H]dopamine uptake. Consistent with increased [3H]WIN 35428 binding, dopamine transporter-specific [3H]dopamine uptake in RGS12-null ventral striatal synaptosomes was found to be increased. Decreased amphetamine-induced locomotor activity and increased [3H]WIN 35428 binding were recapitulated with an independent RGS12-null mouse strain. Thus, we propose that RGS12 regulates dopamine transporter expression and function in the ventral striatum, affecting amphetamine- and cocaine-induced increases in dopamine levels that specifically elicit acute hyperlocomotor responses.
Introduction
Psychostimulant drugs of abuse such as (+)-amphetamine (AMPH) and cocaine are among the most widely abused substances in the USA (Center for Behavioral Health Statistics and Quality, 2015). These psychostimulants impose severe liabilities of high abuse potential, capacity to induce psychoses, considerable neurotoxicity, and mortality (Ciccarone, 2011; Heal et al., 2013). The locomotor-activating and rewarding properties of these psychostimulants are dependent on the mesolimbic dopamine (DA) system of the brain (Adinoff, 2004; Kauer and Malenka, 2007; Nutt et al., 2015; Thomas et al., 2008), defined as the projections from DAergic cell bodies in the ventral tegmental area (VTA) to presynaptic terminals in the nucleus accumbens (NAc) of the ventral striatum (vSTR). Dopaminergic signaling within the vSTR is central to psychostimulant-induced locomotor activity (Ikemoto, 2002): lesioning of DAergic terminals within the NAc–but not in the dorsal striatum (dSTR)–by local injection of 6-hydroxydopamine markedly reduces the hyperlocomotor responses to the dopaminergic indirect agonists AMPH and cocaine (Kelly and Iversen, 1976). Similarly, microinjections of AMPH or cocaine into the NAc–but not dSTR–increase locomotion and rearing behavior in rodents (Carr and White, 1987; Delfs et al., 1990; Kelley et al., 1989; Staton and Solomon, 1984).
AMPH and cocaine exert their effects by increasing extracellular DA levels in the vSTR via their action at the dopamine transporter (DAT; refs. (Federici et al., 2014; Giros et al., 1996)), which acts to translocate released DA back into the presynaptic DA terminal, thus terminating neurotransmission at post-synaptic DA receptors. However, the mechanisms by which AMPH and cocaine modulate DAT are mechanistically distinct. AMPH acts as an indirect dopaminergic agonist in two ways: first, the drug acts as a substrate for (and hence, a competitive inhibitor of) DAT (Sitte and Freissmuth, 2010), whose role is to re-uptake dopamine after its release (Gainetdinov, 2008). Upon entry into dopaminergic terminals, AMPH also acts as a substrate for the vesicular monoamine transporter, whose role is to transport synaptoplasmic dopamine into vesicles for release (Sulzer, 2011). Once inside dopaminergic vesicles, AMPH causes release, through reverse transport of the vesicular monoamine transporter, of free dopamine into the synaptoplasm (Sitte and Freissmuth, 2010). In addition, AMPH causes release of that free DA through DAT, thereby increasing synaptic levels of dopamine (Sitte and Freissmuth, 2010). Cocaine acts more simply: it is not a DAT substrate, rather, it is a competitive inhibitor of the protein (Rothman, 1990). Cocaine blocks the ability of DAT to re-uptake released dopamine, thereby increasing levels of dopamine in the synapse (Nestler, 2005). Thus, each drug (AMPH and cocaine) ultimately yields increased levels of synaptic dopamine in dopaminergic (DAT-expressing) synapses. These increased levels of dopamine in response to AMPH or cocaine act on post-synaptic, dopamine-responsive, G protein-coupled receptors (GPCRs) to elicit responses post-synaptically (Schultz, 2002).
DAT expression, subcellular trafficking, and DA uptake function are all tightly regulated by a host of protein interactants (Torres, 2006), such as receptors, kinases, and scaffolds. In particular, several groups have identified presynaptic GPCRs and scaffolding proteins that directly and/or functionally interact with DAT (Torres, 2006), suggesting that GPCR-dependent, multiprotein networks are critical to DAT function. RGS12, a member of the ‘regulators of G protein signaling’ (RGS) protein superfamily (Snow et al., 1997, 1998), has not previously been associated with DAT regulation. RGS proteins are known to accelerate the inactivation of GPCR-activated Gα subunits (i.e. guanine nucleotide triphosphate-bound (GTP-bound) Gα subunits) by stimulating their intrinsic GTPase activity (Berman et al., 1996). This inactivation is followed by the reassociation of GDP-bound (i.e. inactive) Gα subunits with Gβγ subunits, thereby terminating signaling originally evoked by free GTP-bound Gα subunits (e.g. inhibition of adenylyl cyclase; (Clark and Traynor, 2004)) and/or free Gβγ subunits (e.g. stimulation of G protein-coupled inwardly rectifying potassium (GIRK) channels; (Zhou et al., 2012)).
As many RGS proteins are expressed in the central nervous system (CNS; e.g. ref. Gold et al., 1997), they are suspected to play key roles in neuronal function and behavior (Neubig and Siderovski, 2002). For example, RGS14, the closest related paralog to RGS12, is integral to hippocampal synaptic plasticity underlying learning and memory (Lee et al., 2010). Several RGS proteins, including RGS12 and RGS14, are endowed with functional domains other than the RGS domain (Kimple et al., 2011). In addition to being a Gα-directed GTPase-accelerating protein (GAP) (Snow et al., 1998), RGS12 contains four additional functional motifs: a post-synaptic density (PSD95)/drosophila disc large tumor spressor/zona occludens-1 binding protein (PDZ) binding domain (common among scaffolding proteins), a phosphotyrosine binding domain, tandem Ras binding domains, and a GoLoco motif (which sequesters GDP-bound Gα subunits) (Kimple et al., 2002; Snow et al., 1998; Willard et al., 2007). Thus, the function of RGS12 in vivo need not be restricted to Gα-directed GAP activity in opposition to GPCR activation; for example, we have shown that RGS12 acts as a scaffold for Ras, Raf, and mitogen-activated protein kinase kinase (MEK) in prolonging nerve growth factor (NGF)-stimulated extracellular signaling related kinase (ERK) signaling and stimulating axonogenesis by dorsal root ganglion neurons, as detailed elsewhere (Willard et al., 2007). To explore more deeply the potential roles for RGS12 function in the CNS, we created both conventional and conditional mouse knockout strains and conducted subsequent behavioral (locomotion, sensitization, conditioned place preference) and biochemical (tissue radioligand binding, synaptosome uptake and release) studies. Based on our findings, we propose that RGS12 modulates the expression and function of DAT in the vSTR, thus explaining decreased hyperlocomotor responses in RGS12-null mice to AMPH or cocaine compared with wild-type littermates.
Methods
Animals
The generation of two independent strains of RGS12-null mice (constitutive Rgs12Δ5-8/Δ5-8 and Cre-dependent Rgs12Δ5-6/Δ5-6) is described in Figure 1 and Supplementary Material, Figure S1. Both mouse strains are in the C57BL/6J background. Mice were housed under standard temperature, humidity, and lighting conditions (12 h light/12 h dark). All behavioral experiments performed during the light cycle using mice between 8–10 weeks of age. All mice were provided food and water ad libitum. All experiments were conducted in accordance with West Virginia University Animal Care and Use Committee and followed the National Institute of Health guidelines for use of animals in research. The procedures of the behavioral assays are described below and were conducted according to previously published methods (as cited); mice were used for only one behavioral assay involving either acute or chronic drug dosing (i.e. all drug-based studies were performed with drug-naïve mice; there is no sequence of multiple, drug-based experimental trials to define for individual mice used in this study).
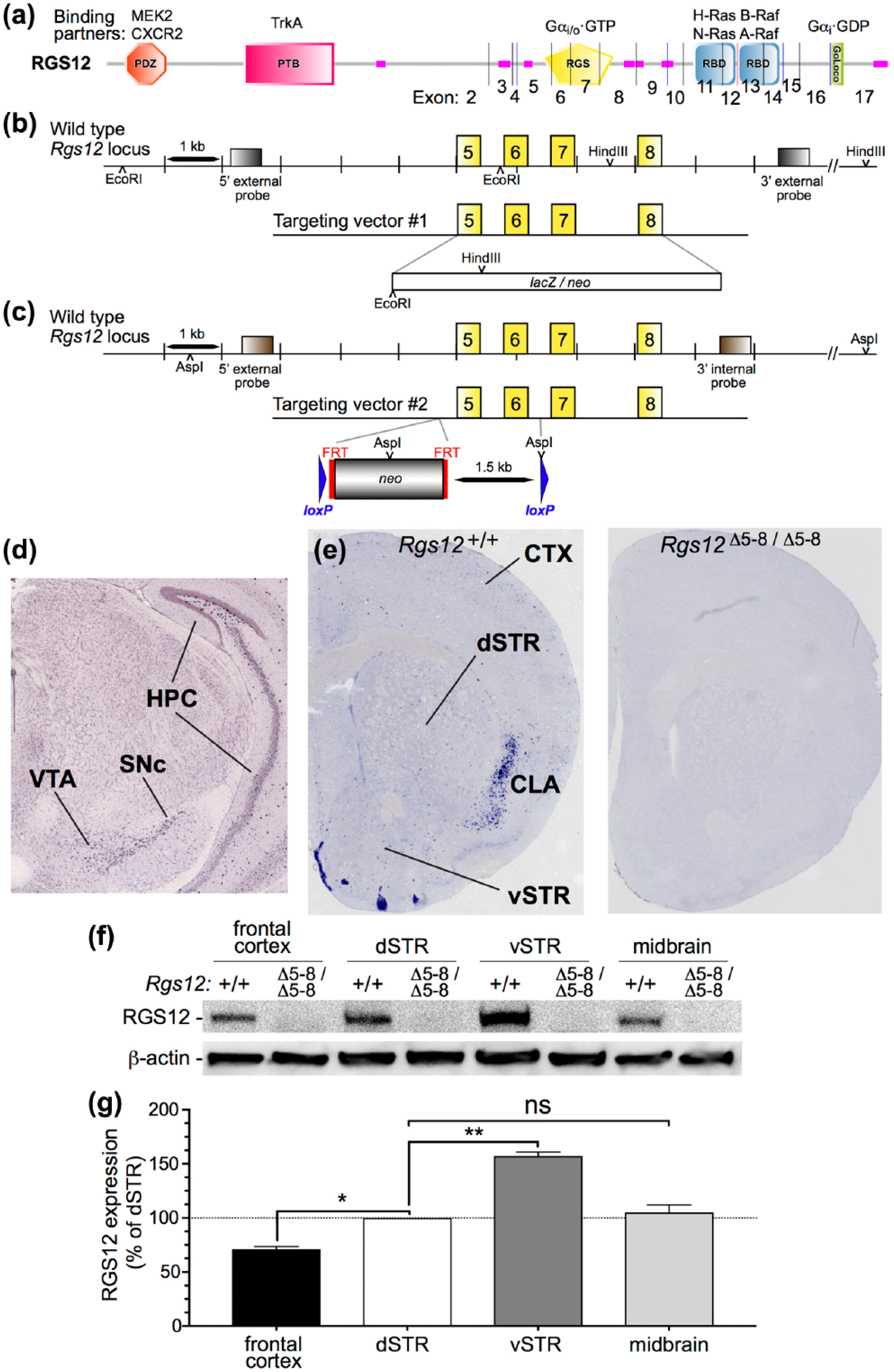
Independent targeting of two separate ablations to the mouse Rgs12 locus. (a) Multiple, protein-protein interaction domain architecture of the encoded regulators of G protein signaling (RGS)12 protein, including a PSD-95/Dlg/ZO-1 (PDZ; orange) domain capable of binding the C-termini of the mitogen-activated protein kinase (MAPK) middle-tier kinase MAPK-kinase (MEK2) and the interleukin-8 receptor CXCR2 (Snow et al., 1998; Willard et al., 2007), a phosphotyrosine-binding (PTB; pink) domain thought to bind the NGF receptor TrkA (Willard et al., 2007), an RGS domain (yellow) which binds and inactivates heterotrimeric G protein Ggdα subunits of the Gi/o subfamily (Snow et al., 1998), a tandem repeat of Ras-binding domains (RBDs; cyan) that bind indicated members of the Ras guanine nucleotide triphosphate (GTP) ase and MAPK first-tier (Raf) kinases (ref. Willard et al., 2007), a GoLoco motif which binds inactive Gα subunits of the Gi subfamily (ref. Kimple et al., 2002), and regions of predicted low complexity (rose). Vertical lines indicate exon-exon junctions within the open-reading frame encoded by the Rgs12 mRNA. (b) and (c) Mice lacking functional RGS12 (Rgs12Δ5-8/Δ5-8) were obtained from the Texas A and M Institute of Genomic Medicine (TIGM) in a 129/Sv×C57BL/6 mixed background. Because congenic C57BL/6 mice are standard for neuropsychopharmacological behavioral assays, we back-crossed the TIGM Rgs12Δ5-8/Δ5-8 mice with C57BL/6J mice for more than ten generations. Henceforth, Rgs12Δ5-8/Δ5-8 mice will refer to congenic Rgs12Δ5-8/Δ5-8 mice on the C57BL/6J background. Exons 5–8, encoding the entire polypeptide sequence of the RGS domain of RGS12 (yellow; panel (a)), were targeted for replacement with a neomycin resistance, drug-selectable marker. (d) Coronal in situ hybridization (Allen Brain Atlas) reveals marked Rgs12 mRNA labeling in the ventral tegmental area (VTA) and substantia nigra pars compacta (SNc), beds of the mesolimbic and nigrostriatal dopaminergic soma. Analyses also reveal hippocampus (HPC) Rgs12 mRNA, particularly in the dentate gyrus. (e) Coronal in situ hybridization from our group shows sparse Rgs12 mRNA in dorsal striatum (dSTR) and ventral striatum (vSTR) and cortex (CTX). Marked labeling was observed in the claustrum (CLA). (f) Immunoblotting for RGS12 protein levels in microdissected frontal cortex, dSTR, vSTR, and midbrain in wild-type (WT) and RGS12-null mice. â-Actin immunoreactivity served as a loading control. (g) Densitometric analysis demonstrates that RGS12 in vSTR is higher than frontal CTX, dSTR, and midbrain and, specifically is 157% more abundant than in dSTR. Data are means±standard error of the mean (SEM) (n=6 mice per group) relative to dSTR set to100%. One-way analysis of variance (ANOVA) with Tukey’s multiple comparisons: dSTR vs frontal CTX p=0.018 (*p<0.05), dSTR vs vSTR p=0.0014 (**p<0.01), dSTR vs midbrain p=0.724 (not significant (ns), p>0.05).
In situ hybridization
In situ hybridization with an antisense riboprobe directed against Rgs12 was performed in order to assess the distribution of Rgs12 mRNA in brain. cDNA from the mouse Rgs12 open reading frame (full-length) was subcloned into a pBluescript II SK vector by restriction enzyme digestion. In vitro transcription with T7 RNA polymerase was conducted to produce a digoxigenin-labeled antisense riboprobe. In situ hybridization was then performed by the University of North Carolina Neuroscience Center In Situ Hybridization Core using standardized protocols (UNC-Chapel Hill).
Immunoblots
Immunoblot analyses of microdissected dopaminergic brain regions (frontal cortex, dorsal and ventral striatum, and midbrain) were performed in order to identify which regions contained RGS12 protein, to compare the relative abundances of RGS12 protein across selected regions, and to verify complete knockout of RGS12 protein in Rgs12Δ5-8/Δ5-8 mice. Mice were injected with a lethal dose of pentobarbital (200 mg/kg, intraperitoneal (ip)), decapitated, rapidly dissected, and frozen on dry ice. dSTR, vSTR, and midbrain were sonicated in 10 volumes of radioimmunoprecipiation assay buffer containing 25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS) (Pierce) supplemented with protease inhibitors (Halt protease inhibitor cocktail, Thermo Fisher Scientific, Waltham, Massachusetts, USA). Lysates were cleaned by centrifugation at 20,800×g for 20 min and pellets were discarded. Proteins were then separated by SDS-polyacrylamide gel electrophoresis on a gradient 4–15% gel (BioRad), electrotransferred to nitrocellulose membranes, and incubated overnight (4°C) with a monoclonal primary antibody directed against the N-terminus of RGS12 (generated in-house) or beta-actin (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Blots were then probed with horseradish peroxidase-conjugated goat anti-mouse secondary antibodies and visualized by chemiluminescence (Pierce). Data were derived from a sample size of n=6–9 per genotype per experiment.
Locomotor assays
AMPH- or cocaine-induced locomotor activity assessments were carried out in order to evaluate behavioral phenotypes in RGS12-null mice. Open field locomotor activity was evaluated in a 16×16 photobeam activity system (PAS) under standard environmental conditions (San Diego Instruments, San Diego, California, USA). Horizontal locomotor activity was measured as the sum of the total x and y coordinate beam breaks collected every five minutes. Acute psychostimulant-induced locomotor activity experiments were carried out over three days. On Day 1, mice were habituated to locomotor chambers over 150 minutes to reduce the influence of novelty. On Day 2, all mice we allowed to habituate to locomotor chambers for 30 min before ip administration of saline (0.9% NaCl) at a volume 10 mL/kg and locomotor activity was measured over 110 min.
On Day 3, mice were habituated to locomotor chambers for 30 min and then injected with d-AMPH (1, 3, or 5 mg/kg, ip) (Chen et al., 2007; Salahpour et al., 2008; ) or cocaine (10, 20, or 40 mg/kg, ip) (Jung et al., 2013; Ramsey et al., 2008) dissolved in saline (ip, at a final volume of 10 mL/kg). Psychostimulant-induced hyperlocomotion was then measured over 110 min. All drugs were obtained from Sigma-Aldrich (St Louis, Missouri, USA). Data were derived from a sample size of n=7–23 per genotype per experiment.
Psychostimulant-induced locomotor sensitization
Sensitization experiments were performed in order to assess whether RGS12-null mice exhibited reduced responsiveness to repeated administration of AMPH or cocaine as was observed with acute hyperlocomotion induced by these psychostimulants. Experiments were performed as previously described (Steinkellner et al., 2014) with minor modifications. Briefly, mice were habituated to locomotor chambers on Days 1 and 2 as described in acute psychostimulant-induced locomotor activity experiments. Mice were then administered d-AMPH (2 mg/kg, ip) or cocaine (20 mg/kg, ip) (Ramsey et al., 2008) for six consecutive days (d1–d6) to induce sensitization under the conditions described for Day 3 of acute psychostimulant-induced locomotor activity experiments. After the sixth day of psychostimulant administration, the drug was withdrawn for 14 days. Following washout (d20), mice were challenged with d-AMPH (2 mg/kg, ip) or cocaine (20 mg/kg, ip) and horizontal locomotor activity was evaluated. Data were analyzed by two-way, repeated measures analysis of variance (ANOVA). Data were derived from a sample size of n=6–9 per genotype per experiment.
Conditioned place preference (CPP)
CPP was conducted with RGS12-null mice in order to evaluate whether RGS12 influences the rewarding properties of AMPH or cocaine. CPP studies were performed in a two-chambered apparatus (Med Associates Inc., ENV-3013-2, Latham, NY USA) with the paradigm described by Ramsey and colleagues (Ramsey et al., 2008) with minor modifications. The two chambers of the CPP apparatus contained distinguishable floor grating (grid floor vs rod floor) and colored walls (white vs black). Each experiment consisted of habituation, preconditioning, conditioning, and post-conditioning phases. Habituation (Day 1) was carried out to reduce the influence of apparatus novelty. Mice were allowed free access to either chamber for 30 min and measurements of time spent in each chamber was measured. For the preconditioning phase (Day 2), mice were again allowed free access to either chamber for 30 min and time spent in each chamber was measured to determine the least-preferred chamber of each mouse. Preference on Days 1 and 2 were comparable (data not shown). The next day (Day 3) began the conditioning phase. Mice were injected with either d-AMPH (2 mg/kg, ip) or cocaine (20 mg/kg, ip) and were restricted to the least preferred chamber for 30 min. On Day 4, mice were injected with saline (0.9% NaCl, ip) and restricted to the other chamber for 30 min. The remainder of the conditioning phase consisted of two more repetitions of alternating drug (Days 5 and 7) and saline (Days 6 and 8). For the postconditioning test phase, CPP was determined by allowing mice free access to either chamber and measuring the time spent for 30 min. No differences in chamber preference were identified between habituation (Day 1) and preconditioning day (Day 2). Preconditioning and postconditioning were calculated by subtracting the time spent (s) in the preferred chamber (as determined on Day 2) from the least preferred chamber (as determined on Day 2). Data were analyzed by two-way, repeated measures ANOVA. Data were derived from a sample size of n=7–8 per genotype per experiment.
Preparation of brain sections and immunohistochemistry for c-Fos
Psychostimulant-induced c-Fos immunohistochemistry was performed in order to determine whether RGS12-null mice showed a reduction in neural activity specifically within the NAc of the vSTR. Mice received ip injection of saline or d-AMPH (3 mg/kg). Following 90 min, mice were anesthetized with pentobarbital (200 mg/kg) and transcardially perfused with 4% paraformaldehyde. Perfused brains were post-fixed overnight and then placed in 30% sucrose solution until brains sank. Cryoprotected brains were then frozen overnight at −80°C and 40-μm free-floating sections were prepared. Sections were incubated with a polyclonal anti-c-Fos primary antibody (1:100; Santa Cruz, Dallas, Texas, USA) overnight at 4°C, followed by incubation of a flurophore-conjugated goat anti-rabbit secondary antibody (Thermo Fisher Scientific, Waltham, MA, USA) for two hours at RT. Psychostimulant-induced c-Fos expression was visualized on an Evos epifluorescent microscope and c-Fos-positive nuclei were counted in a 0.16-mm2 field of the dSTR and vSTR. Anatomical landmarks, such as the anterior commissure, corpus callosum, and septum, were utilized to demarcate the dSTR and vSTR. Three independent sections separated by 200 μm were counted per mouse, and presented data were determined by computing the average number of c-Fos-positive nuclei per field in 3–4 mice per genotype.
Preparation of synaptosomes and measurement of [3H]dopamine uptake and release
These biochemical experiments were performed to assess whether the observed reductions in acute psychostimulant-induced hyperlocomotion were correlated with changes in DA uptake and/or AMPH-induced DA release. For [3H]dopamine uptake experiments, dorsal and ventral striatal synaptosomes were prepared by homogenizing brain tissue from 3–4 mice per genotype in 10 volumes ice-cold sucrose buffer (10% sucrose, 5 mM HEPES, pH 7.4) with a Potter-Elvehjem homogenizer (10 strokes). Homogenates were then centrifuged at 1000×g for 10 min and supernatants containing crude synaptosomes were maintained on ice. Synaptosomes were then added to tubes containing Krebs-Ringer uptake buffer (120 mM NaCl, 20 mM Tris-HCl, 5 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl, 10 mM glucose, 1 mM ascorbic acid, and 0.1 mM pargyline) plus [3H]dopamine (33.5 μCi, Perkin-Elmer, Bridgeville, Pennsylvania, USA) and incubated for 10 min at room temperature (RT) for uptake to occur. Non-specific [3H]dopamine uptake was determined in the presence of 10 μM cocaine and subtracted from total uptake. Uptake was terminated by rapid filtration (Brandel harvester) through 0.1% polyethelyneimine-soaked Whatman GF/B filters with ice-cold Krebs-Ringer buffer and washed three times before radioactivity measurement via liquid scintillation counting. Data were analyzed with nonlinear fitting software (GraphPad Prism 7) to determine Vmax and Km. AMPH-induced [3H]dopamine release assays were performed as previously described (Rothman et al., 2001) with minor modifications. Briefly, dorsal and ventral striatal synaptosomes were prepared as described above but with ice-cold sucrose buffer supplemented with 1 μM reserpine. Synaptosomes were then incubated to steady-state with 5 nM [3H]dopamine (30 min) in Krebs-Ringer buffer supplemented with 1 μM reserpine. Following incubation, 900 μL of preloaded synaptosomes were added to 12×75 mm polystyrene tubes containing d-AMPH (1×10–8.5–1×10–5 M) in Krebs-Ringer buffer. The release reaction proceeded for five minutes, then was terminated by rapid filtration over Whatman GF/B filters with ice-cold Krebs-Ringer buffer, washed three times, and radioactivity was measured via liquid scintillation counting. Data were analyzed with nonlinear fitting software (GraphPad Prism 7) to determine EC50. Data were derived from a sample size of n=6–12 per genotype per experiment.
Preparation of brain tissue and measurement of [3H]WIN 35428 binding
DAT saturation binding experiments were conducted in order to determine whether the increased DA uptake in RGS12-null mice was associated with elevated DAT protein levels. Dorsal and ventral striatal brain regions were rapidly dissected and frozen on dry ice. Tissue was then homogenized with a Polytron homogenizer with 10 volumes of transporter binding buffer (50 mM Tris-HCl, 120 mM NaCl, and 5 mM MgCl, pH 7.4). Homogenates were then centrifuged at 20,800×g for 20 min. The resulting pellet was then resuspended in transporter binding buffer and added to tubes containing [3H]WIN 35428 (1.75 to 52.5 nM) and incubated for one hour at RT. Nonspecific binding was determined in the presence of 10 GBR12909. Binding was terminated by rapid filtration over Whatman GF/B filters with ice-cold transporter binding buffer, washed three times, and radioactivity was measured via liquid scintillation counting. Data were analyzed with nonlinear fitting software (GraphPad Prism 7) to determine Bmax and Kd. Data were derived from a sample size of n=6–9 per genotype per experiment.
High-performance liquid chromatography (HPLC) with dual-cell electrochemical detection
HPLC on microdissected dopaminergic brain regions was carried out to assess whether tissular DA levels differed between RGS12-null and wild-type mice. Dorsal and ventral striatal brain regions were rapidly dissected and frozen on dry ice. Tissue was then sonicated in 10 volumes of 0.3 N perchloric acid and homogenates were centrifuged at 12,000×g for 15 min. Supernatants were collected, passed through a 0.22 µm filter, and dopamine levels in the homogenates were determined by HPLC with dual-cell electrochemical detection. Cleared lysates underwent automated direct injection of 10 µL into a Dionex Ultimate 3000 HPLC system equipped with a pump (ISO-3100BM, Thermo Fisher Scientific, Waltham, Massachusetts, USA) linked to an autosampler (WPS-3000TBRS, Thermo Fisher Scientific, Waltham, MA, USA), ESA HPLC column (MD-150×3.2) and dual coulometric/amperometric electrochemical detectors (ECD-3000RS, Thermo Fisher Scientific, Waltham, Massachusetts, USA) set at −170 mV and +400 mV, respectively. Mobile phase (MD-TM; Thermo Fisher Scientific, Waltham, Massachusetts, USA) was pumped at a flow rate of 0.6 mL/min. Chromatograms were analyzed using Chromeleon software and dopamine peak height (concentration) was determined from a standard curve. The identity of the DA peak in the sample was confirmed by standard addition.
Statistics
All data are presented as means±standard error of the mean (SEM). Statistical significance was set at p<0.05 for all experiments, and data were analyzed by student’s t-test or two-way ANOVA followed by Sidak’s post-hoc test where appropriate.
Results
RGS12 protein is abundantly expressed in the ventral striatum of the mouse CNS
Our study was initiated by the generation of two independent strains of RGS12-null mice on the C57BL/6J background (Figure 1(a)–(c)). Rgs12 mRNA was detected within the brain by in situ hybridization in wild-type, but not in RGS12-null, mice (Figure 1(d), (e)). Rgs12 is expressed in the VTA and the substantia nigra pars compacta (SNc) (Figure 1(d)); there was also clear labeling of Rgs12 in the dentate gyrus and the cornu ammonis regions of the hippocampus (HPC). As shown in Figure 1(e), there was also diffuse labeling of Rgs12 in the striatum (STR) and cortex (CTX), as well as marked labeling of Rgs12 in the claustrum (CLA). RGS12 protein immunoreactivity was detected in the midbrain (the seat of the VTA and SNc) (Figure 1(f)); however, midbrain RGS12 protein content was low in comparison to the high abundance of Rgs12 mRNA. There was also detectable RGS12 immunoreactivity in the frontal CTX and dSTR (Figure 1(f)). RGS12 immunoreactivity was greater in the vSTR than in frontal cortex, dSTR, or midbrain (Figure 1(g)). Quantification of immunoblots is presented as the mean±SEM (n=6 per group), and statistical significance was assessed by one-way ANOVA with Tukey’s multiple comparisons post-hoc analysis: e.g. frontal CTX vs dSTR (p<0.05); dSTR vs vSTR (p<0.01); midbrain vs dSTR (not significant (ns)), p=0.72). Despite dense RGS12 immunoreactivity, Rgs12 mRNA labeling in the vSTR is quite sparse (Figure 1(e)).
RGS12 loss blunts hyperlocomotion to low doses of AMPH and cocaine
Given marked RGS12 expression in the vSTR, and an established role for the vSTR in psychostimulant actions, as a next step we evaluated RGS12-null mice in psychostimulant-induced behavioral assays. Both RGS12-null mouse strains exhibited a markedly reduced response to a standard hyperlocomotive-inducing dose (3 mg/kg) of AMPH compared with wild-type littermates (Figure 2(a)–(c)). Data indicating lack of a saline effect (Figure 2(a)) are displayed as means±SEM (n=7–8 per group) and analyzed by two-way ANOVA: genotype effect, F(1,13)=0.04, p=0.84; time, F(21,273)=5.4, p<0.0001; genotype×time, F(21,273)=1.1, p=0.40. Data indicating reduced hyperlocomotion of conditional RGS12-null mice to 3 mg/kg AMPH (Figure 2(b)) are displayed as means±SEM (n=7–8 per group) and analyzed by two-way ANOVA: genotype effect, F(1,12)=8.9, p=0.011; time, F(21,252)=11.3, p<0.0001; genotype×time, F(21,252)=3.2, p<0.0001. Data indicating reduced hyperlocomotion of constitutive RGS12-null mice (Figure 2(c)) are means±SEM (n=9–23 per group) and analyzed by two-way ANOVA: genotype effect, F(1,41)=18.6, p<0.0001; time, F(21,861)=42.3, p<0.0001; genotype×time, F(21,861)=9.6, p<0.0001.
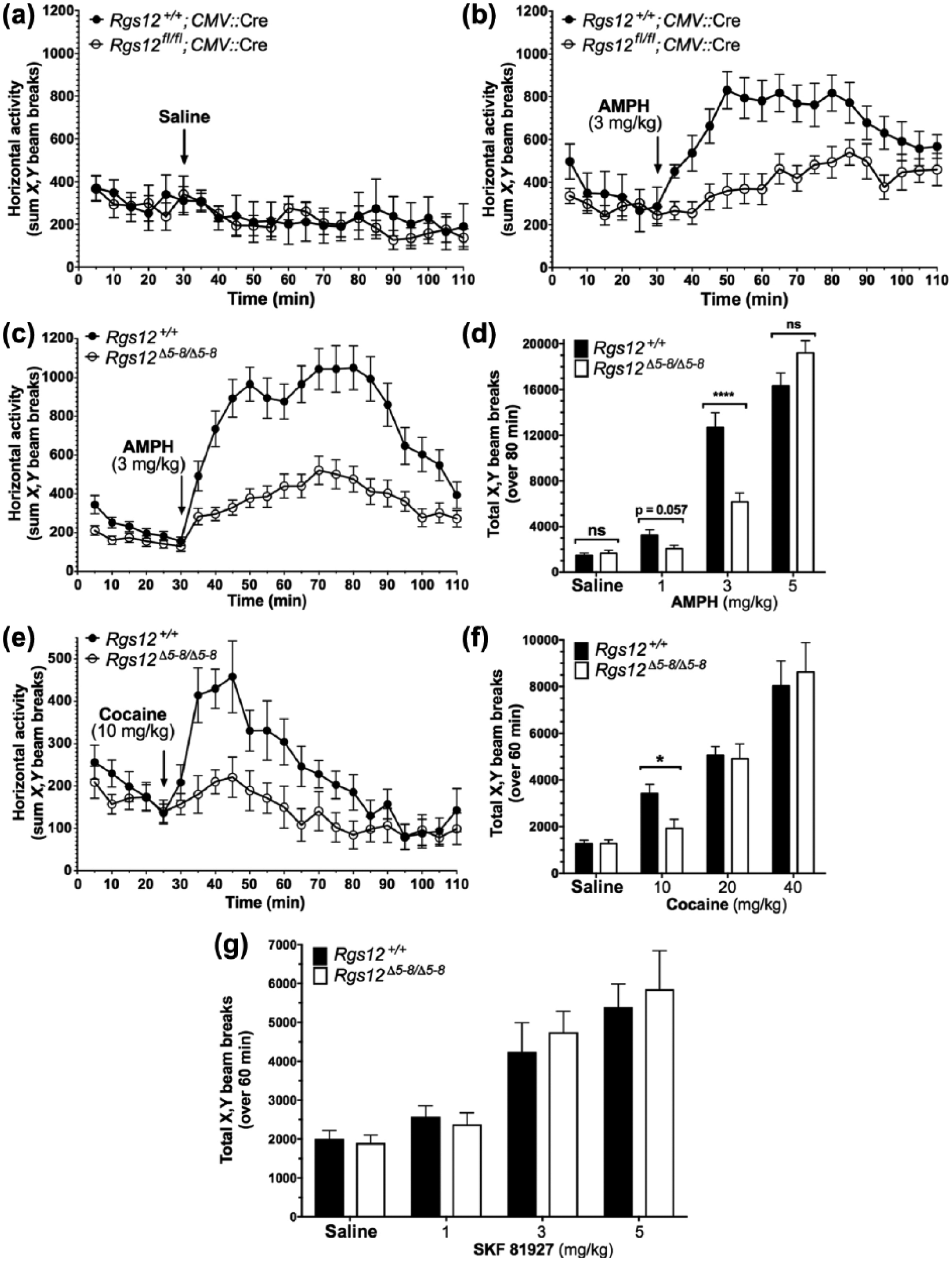
Acute psychostimulant-induced locomotion in Rgs12-null mice. (a) Locomotor activity in Rgs12 Δ5-6/Δ5-6 mice (i.e. conditional Rgs12fl/fl mice cross-bred with an ubiquitous CMV::Cre transgene) following 30 min acclimation to activity chambers and then saline (at a final volume of 10 mL/kg) administration. Data are means±standard error of the mean (SEM) (n=7–8 per group). (b) (+)-amphetamine (AMPH) (3 mg/kg)-induced hyperlocomotion in Rgs12 Δ5-6/Δ5-6 mice for 80 min following 30 min acclimation to activity chambers. Data are means±SEM (n=7–8 per group). (c) AMPH (3 mg/kg)-induced hyperlocomotion in constitutive Rgs12 Δ5-8/Δ5-8 mice for 80 min following 30 min acclimation to activity chambers. Data are means±SEM (n=9–23 per group). (d) Cocaine (10 mg/kg)-induced hyperlocomotion in constitutive Rgs12 Δ5-8/Δ5-8 mice for 60 min following 30 min acclimation to activity chambers. Data are means±SEM (n=9–10 per group). (e) Total locomotion by constitutive Rgs12 Δ5-8/Δ5-8 mice over 80 min following intraperitoneal (ip) injection of saline or various doses of AMPH after 30 min acclimation to activity chambers. Data are means±SEM (n=8–23 per group). (f) Total locomotion by constitutive Rgs12 Δ5-8/ Δ5- 8 mice over 60 min following ip injection of saline or various doses of cocaine after 30 min acclimation to activity chambers. Data are means±SEM (n=7–20 per group). (g) Total locomotion by constitutive Rgs12 Δ 5-8/Δ5-8 mice over 60 min following ip injection of saline or indicated doses of SKF 81927 after 30 min acclimation to activity chambers. Data are means±SEM (n=8 per group).
An initial dose of 3 mg/kg AMPH was selected because it has been extensively characterized as eliciting robust locomotor responses in wild-type C57BL/6 mice (Medvedev et al., 2013; Ramsey et al., 2008; Salahpour et al., 2008). We next assessed whether RGS12-null mice exhibit reduced locomotor responses to lower doses of AMPH. We found that AMPH at 1 mg/kg was sufficient to observe hyperlocomotion in wild-type mice relative to saline injection (Figure 2(d)); however, there was only a trend towards reduced AMPH-stimulated hyperlocomotion in RGS12-null mice at this lower dose (Figure 2(d)). At a high dose of AMPH (5 mg/kg), the hyperlocomotive responses of wild-type and Rgs12-null mice were identical (Figure 2(d)). Data from these additional AMPH doses are displayed as means±SEM (n=8–23 per group) and analyzed by multiple t-test analysis: saline, p=0.52; 1 mg/kg, p=0.057; 3 mg/kg, p<0.0001; 5 mg/kg: p=0.086.
Having obtained the described results with AMPH, we proceeded to determine whether the response to another dopaminergic psychostimulant, cocaine, was similarly affected upon RGS12 loss. As shown in Figure 2(e), 10 mg/kg cocaine elicited a hyperlocomotive response in wild-type mice that was markedly reduced in Rgs12-null mice. Data indicating reduced hyperlocomotion to cocaine in RGS12-null mice are displayed as means±SEM (n=9–10 per group) and analyzed by two-way ANOVA: genotype effect, F(1,17)=17.2, p=0.016; time, F(21,357)=8.8, p<0.0001; genotype×time, F(21,357)=2.4, p=0.0006. In follow-up studies with 20 mg/kg and 40 mg/kg of cocaine, doses established by others’ studies (Elliot, 2002; Ramsey et al., 2008; Reith et al., 1991; Salahpour et al., 2008), we observed no genotype-dependent differences in drug-induced hyperlocomotion (Figure 2(f)). Data from these additional cocaine doses are displayed as means±SEM (n=7–20 per group) and analyzed by multiple t-test analysis: saline, p=0.99; 10 mg/kg, p=0.02, p<0.01); 20 mg/kg, p=0.84; 40 mg/kg, p=0.72.
Both AMPH and cocaine are well known to exert their actions via the neurotransmitter DA (Ciccarone, 2011). One possible explanation for these observations of reduced hyperlocomotion is that, in RGS12-null mice, post-synaptic DA receptor sensitivity is decreased. SKF 81927 is a selective D1-like (D1/D5) DA receptor full agonist (Desai et al., 2005; Weed et al., 1993), which has been extensively characterized as eliciting robust horizontal locomotor activity in mice (Desai et al., 2005; Medvedev et al., 2013; Scott et al., 2005). However, we found no differences in hyperlocomotion between the two genotypes at each of the three different doses of SKF 81927 tested (Figure 2(g)), suggesting a pre-synaptic, rather than a post-synaptic, effect of RGS12 loss on dopaminergic psychostimulant action. Data on SKF 81927-mediated hyperlocomotion are displayed as means±SEM (n=8 per group) and analyzed by multiple t-test analysis: saline, p=0.73; 1 mg/kg, p=0.64; 3 mg/kg, p=0.59; 5 mg/kg, p=0.71.
RGS12 loss does not affect sensitization nor CPP
Having established a phenotype based on acute administration of AMPH or cocaine, we next tested the effects of repeated dosings of these psychostimulants on RGS12-null mice with doses known to elicit robust sensitization in wild-type C57BL/6 mice (Eisener-Dorman et al., 2011; Ramsey et al., 2008). Despite recapitulating the reduced acute locomotor responses to AMPH by RGS12-null mice (Figure 3(a)), sensitization to repeated administration of AMPH–a measure of neuronal plasticity of the mesolimbic dopamine system (Berg and Olsson, 2004; Steketee and Kalivas, 2011)–was intact in RGS12-null mice compared to wild-type littermates (Figure 3(b),(c)). Locomotion observations from the first day of acute 2 mg/kg AMPH administration (Figure 3(a)) were analyzed by two-way ANOVA: genotype effect, F(1,308)=10.3, p=0.002; time, F(21,308)=1.7, p=0.03; genotype×time, F(21,308)=7.6, p=0.75. Locomotion observations from the sixth day of acute 2 mg/kg AMPH administration (Figure 3(b)) were analyzed by two-way ANOVA: genotype effect, F(1,14)=0.45, p=0.52; time, F(21,294)=36.2, p<0.0001; genotype×time, F(21,294)=1.1, p=0.36. AMPH sensitization time-course data (Figure 3(c)) were analyzed by two-way, repeated-measures ANOVA: genotype effect, F(1,14)=2.7, p=0.6; time, F(6,84)=25.4, p<0.0001; genotype×time, F(6, 84)=25.4, p=0.95. Similarly, sensitization to cocaine was not affected in RGS12-null mice (Figure 3(d)–(f)). Locomotion observations from the first day of acute 20 mg/kg cocaine administration (Figure 3(d)) were analyzed by two-way ANOVA: genotype effect, F(1,10)=0.31, p=0.59; time, F(21,210)=14.2, p<0.0001; genotype×time, F(21,210)=3.6, p<0.0001. Locomotion observations from the sixth day of acute 20 mg/kg cocaine administration (Figure 3(e)) were analyzed by two-way ANOVA: genotype effect, F(1,10)=0.08, p=0.78; time, F(21,210)=17.7, p<0.0001; genotype×time, F(21,210)=0.61, p=0.91. Cocaine sensitization time-course data (Figure 3(f)) were analyzed by two-way, repeated-measures ANOVA: genotype effect, F(1,10)=1.0, p=0.33; time, F(21,210)=17.4, p<0.0001; genotype×time, F(21,210)=0.73, p=0.80.
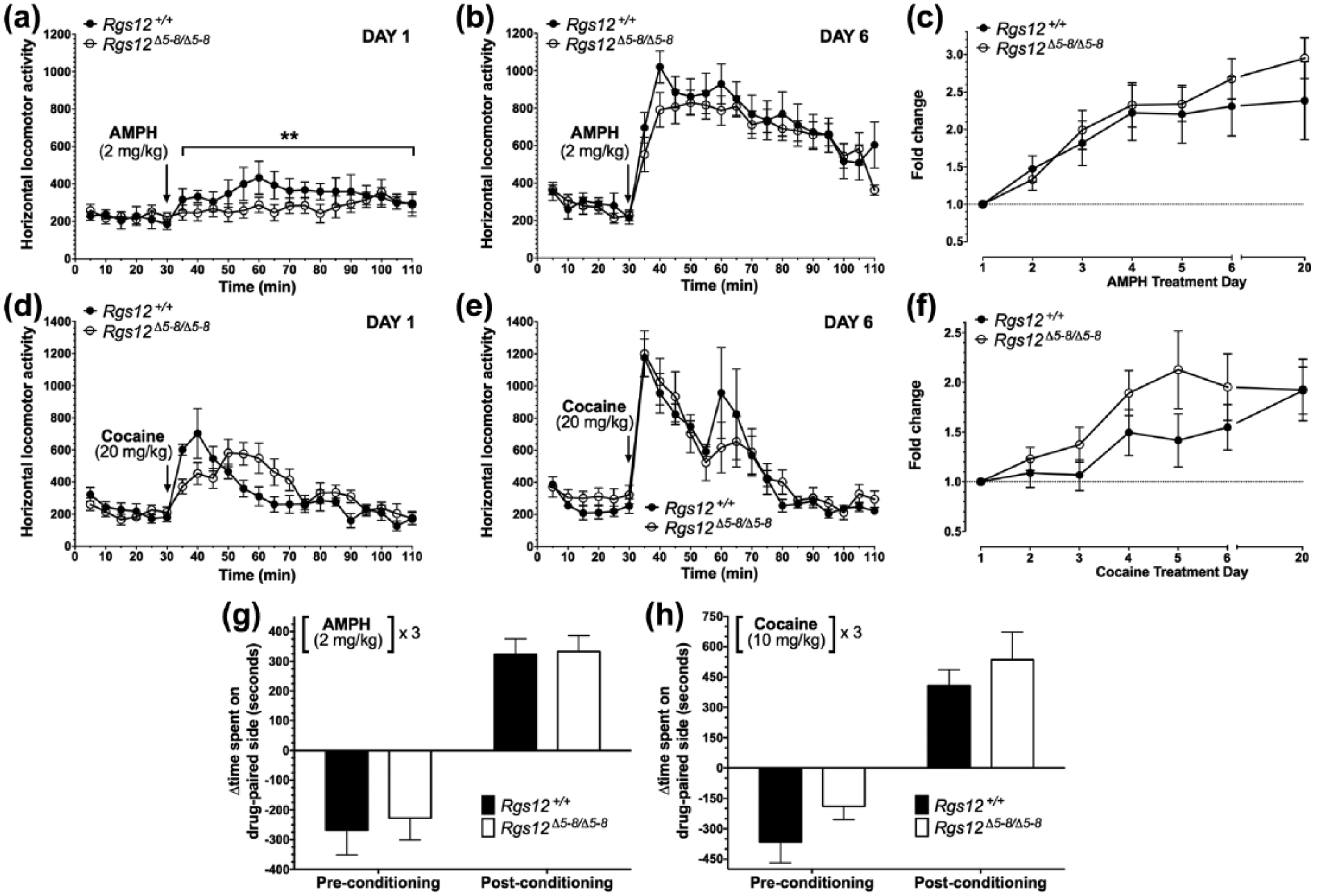
Psychostimulant-induced locomotor sensitization and conditioned place preference in regulators of G protein signaling (RGS)12-null mice. Total locomotion over 80 min following (+)-amphetamine (AMPH) (2 mg/kg; (a) and (b)) or 60 min following cocaine (20 mg/kg; (d) and (e)). Total locomotion normalized to Day 1 total locomotion plotted as fold change across drug treatment days ((c) and (f)). Data are means±standard error of the mean (SEM) (n=6–9 per group). Psychostimulant-induced conditioned place preference in Rgs12-null mice. Data shown are the difference in the time spent between AMPH (2 mg/kg) (panel (g))- or cocaine (10 mg/kg) (panel (h))-paired and saline-paired chamber for pre-conditioning (Day 2) and post-conditioning (Day 9). Data are means±SEM (n=7–8 per group).
In another behavioral paradigm elicited by repeated psychostimulant administration–CPP, which measures the ‘rewarding’ properties of drugs (Tzschentke, 2007)–we observed no differences in the effects of either AMPH or cocaine in RGS12-null vs wild-type mice (Figure 3(g), (h)). CPP as induced by 2 mg/kg doses of AMPH was analyzed by two-way, repeated-measures ANOVA: genotype effect, F(1,12)=0.23, p=0.64; time, F(1,12)=54.1, p<0.0001; genotype×time, F(1,12)=0.04, p=0.84. CPP as induced by 10 mg/kg doses of cocaine was analyzed by two-way, repeated-measures ANOVA: genotype effect, F(1,14)=3.43, p=0.09; time, F(1,14)=41.4, p<0.0001; genotype×time, F(1,14)=0.04, p=0.84. All CPP data are presented as means±SEM (n=6–9 per group).
AMPH-elicited neuronal excitation is reduced in the NAc upon RGS12 loss
In light of the locomotor data and RGS12 expression data, we suspected that RGS12 loss might affect psychostimulant-dependent neuronal activation in at least one locus within the vSTR: namely, the NAc. Therefore, we addressed this hypothesis by measuring c-Fos induction in AMPH-treated RGS12-null and wild-type mice. c-Fos immunoreactivity (measured as c-Fos-positive nuclei per field; Figure 4(a)) was pronounced in the NAc (i.e. ventro-lateral to the anterior commissure (ac)) after an acute dose of 3 mg/kg AMPH. Consistent with the reduced hyperlocomotion of RGS12-null mice to acute, low-dose AMPH, c-Fos immunoreactivity was significantly reduced in the NAc of AMPH-treated RGS12-null mice compared with wild-type littermates (Figure 4(b)). Conversely, in the dSTR, the number of c-Fos-positive nuclei was not different between AMPH-treated RGS12-null mice and wild-type littermates (Figure 4(b)). c-Fos immunoreactivity data are presented as means±SEM (n=3–4 mice per group) and were analyzed by two-way ANOVA with Sidak’s multiple comparisons: vSTR, p<0.0001; dSTR, p=0.85.
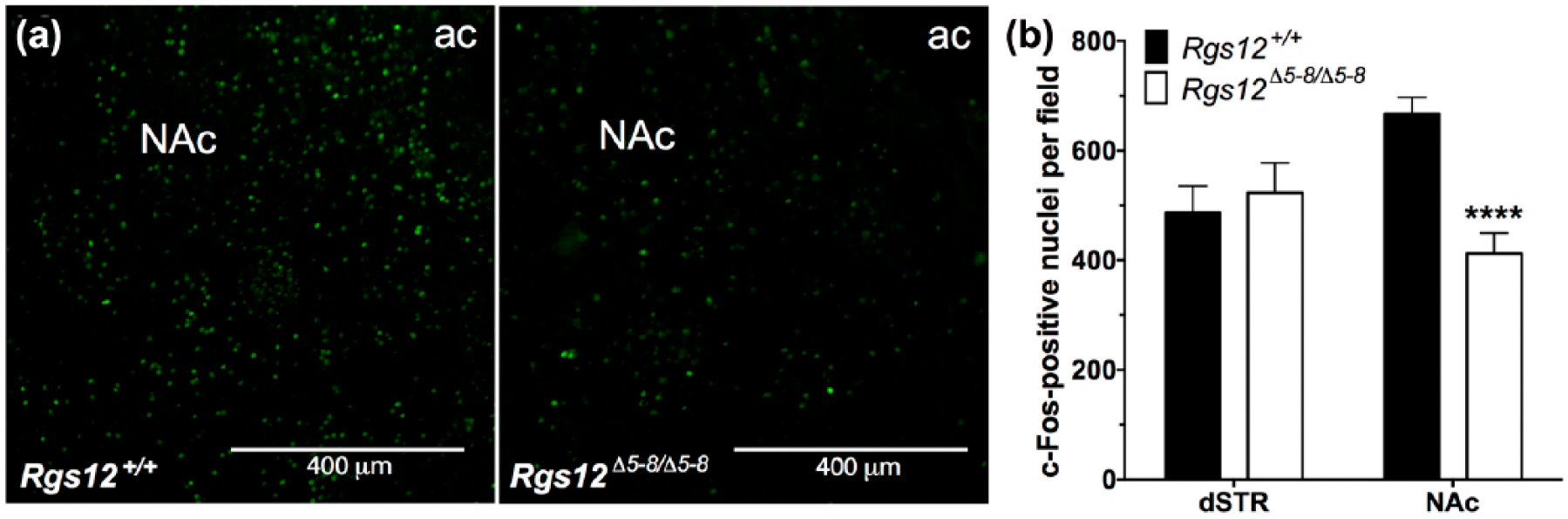
(+)-Amphetamine (AMPH)-stimulated c-Fos induction in regulators of G protein signaling (RGS)12-null mice. (a) Immunohistochemical analysis of c-Fos in the nucleus accumbens (NAc) of RGS12-null and wild-type (WT) mice injected with AMPH (3 mg/kg). (b) Quantification of the number of c-Fos-positive neurons per field in RGS12-null vs WT dorsal striatum (dSTR) and ventral striatum (vSTR) (3–4 matched sections per genotype). Data are means±standard error of the mean (SEM) (n=3–4 mice per group).
Dopamine uptake is enhanced in ventral striatal synaptosomes from RGS12-null mice
The reduced effects of psychostimulants on RGS12-null mice could be explained by increased DAT-mediated uptake in the vSTR of DA, the major neurotransmitter affected by AMPH and cocaine (Ciccarone, 2011). Thus, to test this hypothesis, we next prepared ventral and dorsal striatal synaptosomes from RGS12-null mice to measure [3H]dopamine uptake. A significant increase in the Vmax (maximal rate of [3H]dopamine uptake) of RGS12-null ventral striatal synaptosomes was observed as compared with wild-type ventral striatal synaptosomes (Figure 5(a) and (b) inset); however, no genotype-specific differences in Vmax were observed for dorsal striatal synaptosomes (Figure 5(b)). Km values were equivalent between genotypes in both ventral and dorsal striatal synaptosomes (Figure 5(a) and (b)). [3H]Dopamine uptake Vmax data are presented in Figure 5(b) as means±SEM (n=9–12 per group) and were analyzed by genotype using two-way ANOVA with Sidak’s multiple comparisons: vSTR, p=0.0054; dSTR, p=0.91 (ns). [3H]Dopamine uptake Km values were as follows: vSTR, wild-type mice=0.36±0.16 µM, RGS12-null mice=0.33±0.16 µM; dSTR, wild-type mice mice=0.86±0.32 µM, RGS12-null mice=0.80±0.32 µM; two-way ANOVA with Sidak’s multiple comparisons: vSTR, p=0.99; dSTR: p=0.49.
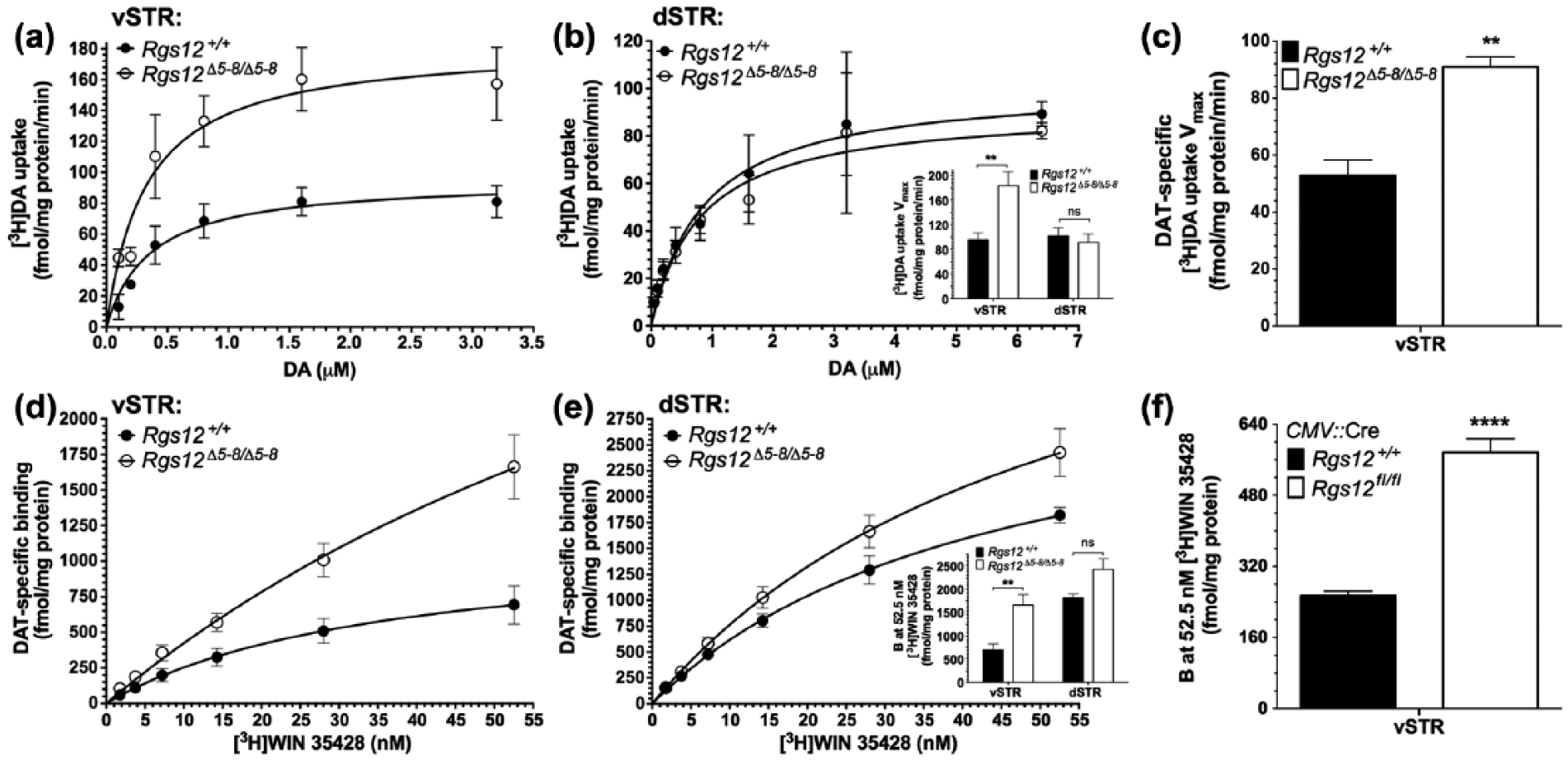
Analysis of dopamine transporter (DAT) function and expression in regulators of G protein signaling (RGS)12-null mice. [3H]Dopamine uptake in ventral (a) and dorsal (b) striatal synaptosomes prepared from RGS12-null and wild-type mice. ((b, inset) Uptake rates (Vmax) of [3H]dopamine in ventral and dorsal striatal synaptosomes derived from data in panels (a) and (b); non-specific uptake determined in the presence of 10 µM cocaine. Data are means±standard error of the mean (SEM) (n=9–12 per group). (c) Uptake rates (Vmax) by DAT of [3H]dopamine in ventral striatal synaptosomes derived from data in Supplementary Material, Figure S4; non-specific uptake determined in the presence of 10 µM GBR12935 (a selective dopamine reuptake inhibitor). Data are means±SEM (n=6 per group). (d) and (e) Levels of DAT determined from [3H]WIN 35428 saturation binding analysis of ventral (d) and dorsal (e) striatal crude membrane fractions. Non-specific binding was determined in the presence of 10 µM GBR12935. ((e) inset) Maximal DAT binding sites at 52.5 nM [3H]WIN 35428 in ventral and dorsal striatal membranes derived from data in (d) and (e). Data are means±SEM (n=6–9 per group). (f) Maximal DAT binding sites at 52.5 nM [3H]WIN 35428 in ventral striatal membranes derived from data in Supplementary Material, Figure S4. Data are means±SEM (n=6 per group). dSTR: dorsal striatum; vSTR: ventral striatum.
To evaluate whether the observed increase in [3H]dopamine uptake in the ventral striata of RGS12-null mice was mediated specifically by DAT, we also performed [3H]dopamine uptake studies using RGS12-null ventral striatal synaptosomes in the presence of the DAT-selective re-uptake inhibitor GBR12935 in order to quantify (and subtract out) non-specific uptake. A significant increase in the Vmax of DAT-specific [3H]dopamine uptake was observed in RGS12-null vSTR synaptosomes as compared with those derived from wild-type littermates (Figure 5(c)). DAT-specific [3H]dopamine uptake data are presented as means±SEM (n=6 per group) and were analyzed by unpaired t-test: p=0.001.
DAT binding sites are significantly elevated in the ventral striatum
Data indicating increased DA uptake by ventral striatal synaptosomes prompted us next to probe the expression levels of DAT in striatal tissue from RGS12-null mice. Radioligand saturation binding assays were performed with [3H]WIN 35428 (a DAT-selective radioligand) to determine the maximal number of DAT binding sites in wild-type and Rgs12-null crude membrane fractions of ventral and dorsal striatal tissue. A marked increase was seen in [3H]WIN 35428 binding to ventral striatal membrane fractions from Rgs12-null mice compared with wild-type mice (Figure 5(d) and (e) inset). Although unable to accurately determine Bmax given the absence of saturation binding, we were able to establish relative expression levels of DAT by comparing the binding of wild-type and RGS12-null ventral and dorsal striatal crude membrane fractions at the maximal concentration of [3H]WIN 35428 tested (52.5 nM). As shown in the bar graph inset within Figure 5(e), [3H]WIN 35428 binding to RGS12-null ventral striatal crude membrane fractions was markedly greater than radioligand binding to wild-type ventral striatal crude membrane fractions; however, there was no statistically significant, genotype-specific effect on [3H]WIN 35428 binding to dorsal striatal crude membrane fractions. Data for [3H]WIN 35428 binding in Figure 5(e inset) are presented as means±SEM (n=6–9 per group) and analyzed by genotype using two-way ANOVA with Sidak’s multiple comparisons: vSTR, p=0.0051; dSTR, p=0.11 (ns).
To assess whether the observed increase in vSTR DAT binding sites in Rgs12Δ5-8/Δ5-8 mice was recapitulated in an independent mouse strain lacking RGS12 expression, we next performed [3H]WIN 35428 binding assays on crude membrane fractions from Cre-recombinase dependent RGS12-null (Rgs12Δ5-6/Δ5-6) mice (Figure 1(c) and Supplementary Material, Figure S1(e); i.e. Rgs12fl/fl mice expressing an ubiquitously-expressed, cytomegalovirus (CMV)-driven Cre transgene). We found that maximal DAT binding sites are also elevated in this Rgs12 Δ5-6/Δ5-6 mouse strain relative to wild-type (Rgs12+/+; CMV::Cre) littermate controls (Figure 5(f)). Data for [3H]WIN 35428 binding in Figure 5(f) are presented as means±SEM (n=6 per group) and were analyzed by genotype using unpaired t-test: p<0.0001.
Discussion
Our major finding from these analyses is that the loss of RGS12 engenders reduced acute responses (hyperlocomotion and c-Fos induction) in response to the psychoactive drugs AMPH or cocaine. This reduction is likely caused by increased DA uptake in the vSTR–in particular, the NAc–due to elevated levels of surface-exposed (and, hence, functional) DAT. The presence of RGS12, in one (or more) brain region(s), must in some way negatively regulate DAT expression levels and/or activity, given that RGS12 loss has been observed in this study to increase DAT expression and activity, at least in the vSTR.
The expression pattern of Rgs12 mRNA compared with the distribution of RGS12 protein renders it tempting to speculate that RGS12 is being synthesized in the dopaminergic projections from the midbrain (i.e. the VTA and SNc; e.g. Figure 1(d)) and trafficked to striatal terminals (e.g. Figure 1(f)) Put another way, RGS12 appears to be a pre-synaptic protein in the STR. Furthermore, the lack of genotype-dependent differences in locomotor responses to a post-synaptically active dopaminergic agonist (i.e. SKF 81927; Figure 2(g)) suggests RGS12 acts pre-synaptically. Biochemical data are also in line with this notion: the expression and function of DAT, a pre-synaptic protein (Torres and Amara, 2007), are both increased upon RGS12 loss (Figure 5). However, baseline DA levels in the vSTR and dSTR of wild-type and RGS12-null mice are identical, as measured by HPLC with dual-cell electrochemical detection (Supplementary Material, Figure S2). In addition, no changes were observed in the levels of tyrosine hydroxylase, the rate-limiting enzyme for DA synthesis (Daubner et al., 2011) (Supplementary Material, Figure S2). Thus, the observed changes in DAT expression levels and function are likely a direct result of RGS12 loss, and not necessarily caused indirectly by a compensatory upregulation (Jakowec et al., 2004; Kimmel et al., 2001; Salvatore et al., 2016) in response to altered DA synthesis or basal levels upon RGS12 loss per se.
Despite reduced locomotor responses to acute administration of AMPH and cocaine, RGS12-null mice developed locomotor sensitization and CPP to each drug in a manner similar to wild-type mice. Although these latter behaviors are mediated by mesolimbic DA (Baik, 2013; Henry et al., 1989; Steketee and Kalivas, 2011), their expression is found to be dependent on neuroplastic changes (i.e. increased VTA excitability and firing rate) to postsynaptic NMDA glutamate and DA receptor signaling/expression levels, as well as to somatodendritic DA autoreceptors located on VTA neurons (Henry et al., 1989; Neisewander et al., 1998; Ramsey et al., 2008; Tanabe et al., 2004; White and Wang, 1984). These findings also support the notion that RGS12 is acting pre-synaptically and thus why only acute behavioral responses to AMPH and cocaine are affected.
Biochemical assays revealed no significant differences in DAT levels or activity in the dSTR. Thus, the phenomenon of RGS12 regulation of DAT appears to be region specific; i.e. RGS12 affects DAT in the mesolimbic system (i.e. VTA to the NAc–a major effector of locomotor behavior). Notably, the levels of RGS12 are greater in the ventral striatum than in the dorsal striatum (Figure 1).
One possible explanation for this enrichment could be a functional and/or physical interaction with pre-synaptic GPCR(s) in the vSTR that is not expressed or relevant in the dSTR. For example, striatal DAT expression and function are potently regulated by two Gαi-coupled GPCRs expressed on striatal presynaptic membranes: D2 receptors and κ-opioid receptors (KORs). Activation of these receptors serves as a negative feedback mechanism upon elevated striatal DA tone, thus augmenting DA uptake and increasing surface DAT expression (Bolan et al., 2007; Kivell et al., 2014) in a region-specific manner. Acute administration of KOR agonists increases DA uptake in vSTR, but not dSTR (Thompson et al., 2000). Additionally, D2 receptor agonists decrease DAT expression in the dSTR, but increase DAT expression in the vSTR (Kimmel et al., 2001). Thus, removal of RGS12 as a negative regulator of G protein-mediated signaling downstream of either GPCR may feasibly lead to augmented pre-synaptic D2 receptor and/or KOR-mediated signaling, resulting in elevated DAT surface expression and DA uptake.
There are several possible mechanisms by which RGS12 could impact DAT trafficking and/or function, given the multiple protein-protein interaction domains present within RGS12 (Kimple et al., 2002; Snow et al., 1998; Willard et al., 2007). One simple explanation for involvement of RGS12 in curtailing DAT function evokes the C-terminal GoLoco motif of RGS12– a guanine nucleotide dissociation inhibitor (GDI) activity that results in the sequestration of inactive Gα subunits (Kimple et al., 2002), which normally re-associate with Gβγ subunits. This re-association blocks any signaling that is mediated by Gβγ. Thus, by sequestering inactive Gα subunits, the GoLoco motif of RGS12 may extend the lifetime of free Gβγ subunits, which are known to, among other things, inhibit DAT activity (Garcia-Olivares et al., 2013).
Another plausible explanation for RGS12 affecting DAT function in the vSTR lies in the ability of RGS12 to interact with components of the mitogen activated protein kinase MAPK/MEK/ERK cascade, the activity of which supports DAT trafficking to the cell surface (Moron et al., 2003; Willard et al., 2007). We speculate that RGS12, as a (MAPK)/MEK/ERK scaffold, diverts this signaling cascade from facilitating the trafficking of DAT to the synaptic plasma membrane in the vSTR. Another possibility that cannot be excluded is that RGS12, through its established interactions with Ras, Raf, MEK, and/or other as-yet unidentified proteins, regulates transcription factors that negatively modulate the transcription of DAT. To explore this possibility, we performed immunoblot analyses on whole lysates of vSTR using a validated DAT antibody (Chen et al., 2006; Foster et al., 2012; Garcia-Olivares et al., 2013; Steinkellner et al., 2012); total DAT levels were seen to be comparable in wild-type and RGS12-null ventral striatum (Supplementary Material, Figure S2), arguing against any transcriptional effect (e.g. in VTA-to-NAc projection neurons). Thus, we are currently assessing whether RGS12 regulates the trafficking of DAT to the synaptic membrane in the vSTR.
There is a clear dose-dependence to the differences seen in acute responses to AMPH and cocaine between RGS12-null and wild-type mice (Figure 2). We suspect that this is due to a saturation of DAT by the uptake inhibitors AMPH and cocaine at higher concentrations. We propose that, at lower doses of DAT inhibitor (1 and 3 mg/kg AMPH; 10 mg/kg cocaine), the excess DAT that is expressed in RGS12-null ventral striatum is available to clear more synaptic DA, reducing the acute locomotor responses of RGS12-null mice vs those of wild-type mice. However, at higher doses of DAT inhibitor (5 mg/kg AMPH; 20 mg/kg and 40 mg/kg cocaine), even the extra DAT expressed in RGS12-null mice becomes saturated by drug, so all DAT molecules in the vSTR of both genotypes are blocked, thereby eliminating any difference in acute locomotor responses between RGS12-null and wild-type mice.
This hypothesis of extra, unsaturated DAT in RGS12-null mice would suggest that DAT levels would become saturated at lower doses in wild-type mice. Yet, in both genotypes, locomotor response is seen to increase with 20 mg/kg cocaine over that of 10 mg/kg, and again with 40 mg/kg cocaine over that of 20 mg/kg (Figure 2(f)). Therefore, an additional possibility is that RGS12 loss affects the regulation of autoreceptors on DAT-positive terminals, thereby lowering the threshold for pre-synaptic feedback by synaptic DA and resulting in curtailed DA release at low drug concentrations. On the other hand, it has been reported that 20 mg/kg cocaine increases locomotor response even in mice lacking vesicular monoamine transporter 2 (VMAT2), critical for DA transport into synaptic vesicular stores (Wang et al., 1997). Thus, another possibility to consider is a role for RGS12 in VMAT2 function affecting DA release–a role absent in RGS12-null mice but dependent on DAT inhibitor dose for full elaboration and observation in behavioral outcomes.
It is also notable that AMPH acts not only as an antagonist of DAT, but also as a releaser of DA. This dual effect explains the increased locomotor activity elicited by AMPH compared with cocaine, which is solely a DAT antagonist (Figure 2(d) vs 2(f), note the difference in y-axis scales). We propose that, at higher doses of AMPH (5 mg/kg), due to saturation of DAT, the maximum quantity possible of DA is released (i.e. at 5 mg/kg, dopamine is completely depleted from the pre-synaptic terminals of both wild-type and RGS12-null mice), potentially explaining why there is no difference between the genotypes. Moreover, in ventral striatal synaptosomal preparations, the ability of AMPH to enhance the release of [3H]dopamine is not markedly different between wild-type and RGS12-null mice (Supplementary Material, Figure S3). This latter observation suggest that RGS12 somehow modulates the uptake functionality–but not the release functionality–of DAT. Molecular studies to address this issue are currently underway.
In summary, our studies have uncovered a novel regulator of ventral striatal DAT expression and activity: namely, RGS12. The neuroanatomical site(s) of RGS12 action and the mechanism(s) by which it regulates ventral striatal DAT expression and function remain to be identified. However, the observed functional interaction between RGS12 and DAT clearly impacts acute mesolimbic-dependent responses to dopaminergic psychostimulants. Future work is required to establish the molecular determinants within both proteins that underlie this regulation, be it direct (e.g. via possible RGS12/DAT interactions) or indirect via the presynaptic regulation of G protein subunits or other intermediates.
Footnotes
Declaration of conflicting interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for these studies was supported in part by the WVU EJ Van Liere Medicine Professorship (to DPS). JDG acknowledges early predoctoral support from the WVU Behavioral and Biomedical Sciences T32 training grant (NIH 5T32GM081741) and current support from a NIDA predoctoral fellowship (NIH 1F31DA043331). The monoclonal antibody UNC60-80.4.1, originally developed by DPS, was re-obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.