
Abstract
Background:
Previous studies suggested that Cg1 area of the cingulate cortex of rats controls glutamate-mediated fear-induced defensive behaviour and antinociception organised at the posterior hypothalamus. In turn, microinjection of the nitric oxide donor SIN-1 into the anterior hypothalamus of mice produced defensive behaviours and fear-induced antinociception. However, it remains unknown whether Cg1 also modulates the latter mechanisms in mice.
Aims:
The present study examined the influence of Cg1 on SIN1-evoked fear-induced defensive behaviour and antinociception organised at the anterior hypothalamus of mice.
Methods:
The fear-like behavioural and antinociceptive responses to the microinjection of SIN-1 (300 nmol) into the anterior hypothalamus were evaluated after the microinjection of either N-methyl-D-aspartic acid receptor agonist (0.1, 1 and 10 nmol) or physiological saline into the cingulate cortex of C57BL/6 male mice. In addition, neurotracing and immunohistochemistry were used to characterise Cg1-anterior hypothalamus glutamatergic pathways.
Results:
The data showed that activation of Cg1 N-methyl-D-aspartic acid receptors increased escape while reducing freezing and antinociceptive responses to SIN-1 microinjections into the anterior hypothalamus. Anterograde neural tract tracer co-localised with VGLUT2-labelled fibres suggests these responses are mediated by glutamatergic synapses at the anterior hypothalamus.
Conclusions:
In contrast with previous studies showing that Cg1 facilitates both escape and antinociception to chemical stimulation of the posterior hypothalamus in rats, the present data suggest that Cg1 facilitates escape while inhibiting defensive antinociception produced by the microinjection of SIN-1 in the anterior hypothalamus of mice. Accordingly, Cg1 may have opposite effects on antinociceptive responses organised in the anterior and posterior hypothalamus of mice and rats, respectively.
Keywords
Introduction
The cingulate cortex is a limbic structure comprising the anterior cingulate cortex (ACC), the midcingulate cortex (MCC) and the posterior cingulate cortex, among other areas of the medial surface of the brain (Vogt, 2005, 2009). In particular, the ACC and rostral MCC have been proposed as interfaces of pain and fear (Vogt, 2009). Indeed, evidence from tract-tracing and neuroimaging studies has shown the latter structures are both the target of pain-related afferents (from the dorsal horn, lateral parabrachial area and parafascicular nucleus) and the source of highly selective projections to fear-related areas of the amygdala, anterior hypothalamus (AH), and periaqueductal grey matter (PAG) (Craig et al., 1996; Fillinger et al., 2018; Floyd et al., 2000; Kong et al., 2010; Mobbs et al., 2007; Vogt, 2009). Although the ACC and the rostral part of the MCC of rodents were formerly named Cg1/Cg2, a recent study by Vogt and Paxinos (2014) assigned the rostral part of Cg1/Cg2 to the ACC dorsal and ventral areas 24b and 24a, respectively and the caudal part of Cg1/Cg2 to the MCC dorsal and ventral areas 24bʹ and 24aʹ, respectively. This terminology extended Brodmann’s cytoarchitectonics to rodents. Most notably, however, recent studies in mice showed that although the 24a projections target the posterior region of the AH and the dorsolateral PAG (dlPAG), 24b projections circumvent the AH and target the lateral PAG (Fillinger et al., 2018). Although the projections from the ‘dorsal AC’ (i.e., Cg1 or 24b) to the dlPAG have already been described in rats (Floyd et al., 2000), ACC projections to the hypothalamus appear to be less abundant (Falconi-Sobrinho et al., 2017a; Floyd et al., 2001). Interestingly, both the PAG and hypothalamus have been implicated in the modulation of fear-related unconditioned defensive behaviour (Anseloni et al., 1999; Fanselow, 1991; Krieger and Graeff, 1985; Shekhar, 1994; Ullah et al., 2015) and nociceptive responses in laboratory animals (Butler and Finn, 2009 for review; Coimbra et al., 2006). Cg1 has also been implicated in regulating contextual fear conditioning (Morgan and Ledoux, 1995; Zheng et al., 2008) and pain-related fear memory (Tang et al., 2005). In particular, evidence from rodent studies suggests that Cg1 controls fear-related responses via projections to hypothalamus and PAG defence areas (Coutinho and Menescal-de-Oliveira, 2010; Falconi-Sobrinho et al., 2017a, 2017b). Studies performed by Falconi-Sobrinho et al. (2017a) showed that defensive behaviour and antinociceptive responses to chemical stimulation of the posterior hypothalamus (PH) are attenuated by the microinjection of either lidocaine or glutamate antagonists in Cg1 and PH, respectively. The latter results were supported both by the demonstration of Cg1 glutamatergic projections to the PH (Falconi-Sobrinho et al., 2017a) and by the impairment of PH-evoked defensive responses and antinociception by the microinjection of an N-methyl-D-aspartic acid (NMDA) receptor antagonist (LY235959) into Cg1 (Falconi-Sobrinho et al., 2017b).
The fact that the ACC is crucial in human pain perception was shown by its selective activation by thermal painful stimuli, either real (5°C or 47°C) or illusory, produced by the spatial alternation of cool (20°C) and warm (40°C) innocuous stimuli (thermal grill illusion) (Craig et al., 1996). The cingulate cortex also seems to play a key role in expectation-induced pain modulation in humans (Shih et al., 2019).
In turn, fear-induced antinociception is has long been known as a defensive adaptive response (Bolles and Fanselow, 1980; Coimbra et al., 2017) mediated by spinally projecting pain inhibitory pathways (Basbaum and Fields, 1978; Fields and Basbaum, 1978) originating in the cerebral cortex (Falconi-Sobrinho et al., 2017a), hypothalamus (Biagioni et al., 2013), dlPAG (Coimbra et al., 2006) and ventrolateral PAG (Samineni et al., 2017; Watson et al., 2016). In particular, the limbic cortex appears to modulate nociceptive transmission via either the activation or the inhibition of PAG efferent connections to brainstem neurons that project to laminae I and V of the dorsal horn of the spinal cord (Butler and Finn, 2009 for review; Nathan, 1977; Ohara et al., 2005). Although PAG neurons also play an important role in pain expectation in humans (Fairhurst et al., 2007; Shih et al., 2019; Tracey et al., 2002) and receive inputs from hypothalamic neurons (Falconi-Sobrinho et al., 2017a; Ullah et al., 2017), it remains unknown whether the cortical modulation of nociception is mediated by hypothalamic projections to the PAG. In fact, the AH is part of the medial hypothalamus defensive system that is activated in animals exposed to a natural predator (Canteras, 2002; Canteras et al., 1997; Paschoalin-Maurin et al., 2018).
In turn, Falconi-Sobrinho and Coimbra (2018) showed that microinjections of the nitric oxide (NO) donor SIN-1 into the AH-induced freezing and escape reactions along with antinociception. Notably, the latter responses were blocked by prior microinjection of an antagonist (AP-7) of NMDA glutamate receptor into the AH. These results suggest the interaction of glutamatergic and nitrergic systems within the AH of mice. In fact, both glutamate and neuronal NO synthase (nNOS) were identified in the AH of rodents (Edelmann et al., 2007; Shioda et al., 2012; Vincent and Kimura, 1992).
There is a lack of studies demonstrating the top-down influence of AH-mediated fear-related behaviour and antinociception by cingulate cortex neurons. Thus, the aim of the present study was to investigate the role of the Cg1 area of the cingulate cortex on fear-related defensive behaviour and fear-induced antinociception mediated by AH. For this, the fear-like behaviours and the antinociceptive responses to the microinjection of SIN-1 into the AH were evaluated after the microinjection of either saline (0.9%) or NMDA (0.1, 1 and 10 nmol) into the cingulate cortex of C57BL/6 male mice. In addition, morphological techniques were performed to identify Cg1-AH glutamatergic pathways.
Methods
Animals
Male C57BL/6 mice (10–12 weeks, weighing 30–35 g) from the animal facility of Ribeirão Preto Medical School of the University of São Paulo (FMRP-USP) were kept in groups of four animals per cage, with water and food ad libitum, and habituated to the experimental room for at least 48 h before the onset of experiments. The cage was maintained under a light/dark cycle of 12/12 h (lights on from 7 am to 7 pm) and at a constant room temperature of 24°C ± 1°C. All efforts were made to minimise animal suffering. The experiments were performed in accordance with the recommendations of the Commission of Ethics in Animal Experimentation of the FMRP-USP (process 187/2015), which is consistent with the ethical principles in animal research adopted by the National Council for Animal Experimentation Control and were approved by the Commission of Ethics in Animal Research on 29 February 2016.
Stereotaxic surgery
Animals were anaesthetised with intramuscular injections of 92 mg/kg ketamine (Ketamine Agener, União Química Farmacêutica Nacional, São Paulo, Brazil) and 9.2 mg/kg xylazine (Calmium, União Química Farmacêutica Nacional, São Paulo, Brazil) and fixed in a stereotaxic frame (David Kopf, Tujunga, California, USA). In the neuropharmacological experiment, stainless steel guide cannulae (outer diameter 0.6 mm, inner diameter 0.4 mm) were implanted in the cingulate cortex or diencephalon of mice, targeting 0.5 or 1 mm above the Cg1 region or AH, respectively. The following coordinates were used for Cg1: anteroposterior: 0.26 mm; mediolateral: −0.3 mm; and dorsoventral: −1.0 mm. The following coordinates were used for AH: anteroposterior: −0.70 mm; mediolateral: −0.5 mm; and dorsoventral: −4.0 mm. The guide cannulae were fixed to the skull using acrylic resin. At the end of the surgery, each guide cannula was sealed with a stainless-steel wire to protect it from obstruction. In mice subjected to the rotarod test, stainless steel guide cannulae were implanted only in the cingulate cortex area 24b. In neuroanatomical experiments, neural tract tracers were injected during stereotaxic surgery. A fluorescein-conjugated dextran (FCD) retrograde tract tracer was deposited into the AH through a dental needle directed to this hypothalamic nucleus according to the coordinates anteroposterior: 0.70 mm; mediolateral: −0.5 mm; and dorsoventral: −5.0 mm. In addition, a non-fluorescent biotinylated dextran amine (BDA) anterograde tract tracer was deposited into the Cg1 area of the cingulate cortex through a dental needle directed to this cortical region according to the coordinates anteroposterior: 0.26 mm; mediolateral: −0.3 mm; and dorsoventral: −1.5 mm (Paxinos and Franklin, 2001).
After stereotaxic surgery, each mouse was treated with an intramuscular injection of penicillin G-benzathine (120,000 UI; 0.1 mL) followed by subcutaneous injection of the non-steroidal analgesic and anti-inflammatory flunixin meglumine (2.5 mg/kg) (Schering-Plough, São Paulo, SP, Brazil). After surgery, the animals were allowed to recover for 4 (Falconi-Sobrinho and Coimbra, 2018) and 10 days before the behavioural tests and morphological study, respectively.
Behavioural tests
Fear-related behavioural defensive reactions were quantified by measuring the number and duration of freezing and escape behaviours. Freezing was characterised by defensive immobility for at least 6 s followed by an autonomic reaction, such as defecation, exophthalmia and/or micturition (Coimbra et al., 2017; Uribe-Mariño et al., 2012). Escape was defined as running and/or jumping responses and characterised as ‘oriented’ or ‘non-oriented’ based on whether they were directed towards elevated platforms or burrows (Almada and Coimbra, 2015; Almada et al., 2015; Falconi-Sobrinho and Coimbra, 2018). Additionally, the frequency of crossings (stepping with four legs within a defined rectangle in the polygonal arena floor after crossing the border of each section line) was recorded as a quantitative measure of escape behaviour expressed by running (dos Anjos-Garcia et al., 2017; Falconi-Sobrinho et al., 2017a, 2017b).
To perform the behavioural test, an enriched polygonal transparent acrylic arena was used (length 140 cm; width 62 cm; height 50 cm) with a black acrylic walls shelter box (10 × 7 × 5 pol) inside and two elevated platforms for escape (safe places) (Almada and Coimbra, 2015; Almada et al., 2015; dos Anjos-Garcia and Coimbra, 2020; Falconi-Sobrinho and Coimbra, 2018). The floor of the polygonal arena was made of a transparent acrylic platform. The floor of the polygonal arena was divided by red lines into 20 equal rectangles (4.2 mm width; Pritt mark-it).
Nociceptive test
The nociception thresholds of the experimental animals were recorded using the tail-flick test. Each mouse was placed in restraining apparatus (Insight, Ribeirão Preto, SP, Brazil) with acrylic walls, and its tail was placed on a heating wire coil of custom-built tail-flick equipment (FMRP-USP Precision Workshop facilities; Ribeirão Preto, Brazil). Beginning at room temperature (approximately 20°C), the current raised the coil temperature at a rate of 9°C per second until the mouse showed a tail flick response (Coimbra et al., 2006; da Silva-Soares et al., 2019; Falconi-Sobrinho and Coimbra, 2018; Prado and Roberts, 1985). Whenever necessary, the current was adjusted to obtain three baseline tail-flick latencies between 2.5 and 3.5 s. The cut-off latency was 6 s. Tail-flick latencies were recorded three times at 5 min intervals before defensive behaviours and at 10 min intervals for a period of 60 min after behavioural tests.
Rotarod test
The rotarod test has been used to evaluate the motor coordination and balance of rodents. It allows the investigation of the possible side effects of drugs on locomotor function (Coimbra et al., 2001; Dunham and Miya, 1957; Scheidt et al., 2002). The rotarod apparatus for mice (Ugo-Basile 47600, Germonio, Varese, Italy) consists of a revolving rod subdivided into five compartments by disks 25 cm in diameter.
Experimental groups
The following groups were performed in which the first and second abbreviations refer to area 24b and AH microinjections, respectively: (a) vehicle (10 min) + vehicle (n = 8) (Veh-Veh); (b) NMDA-Veh: NMDA (10 min) + Veh (n = 8 per dose); (c) Veh-SIN: Veh (10 min) + SIN (n = 8); and (d) NMDA-SIN: NMDA (10 min) + SIN (n = 8 per dose). The timeline of the experiments is shown in Figure 1.
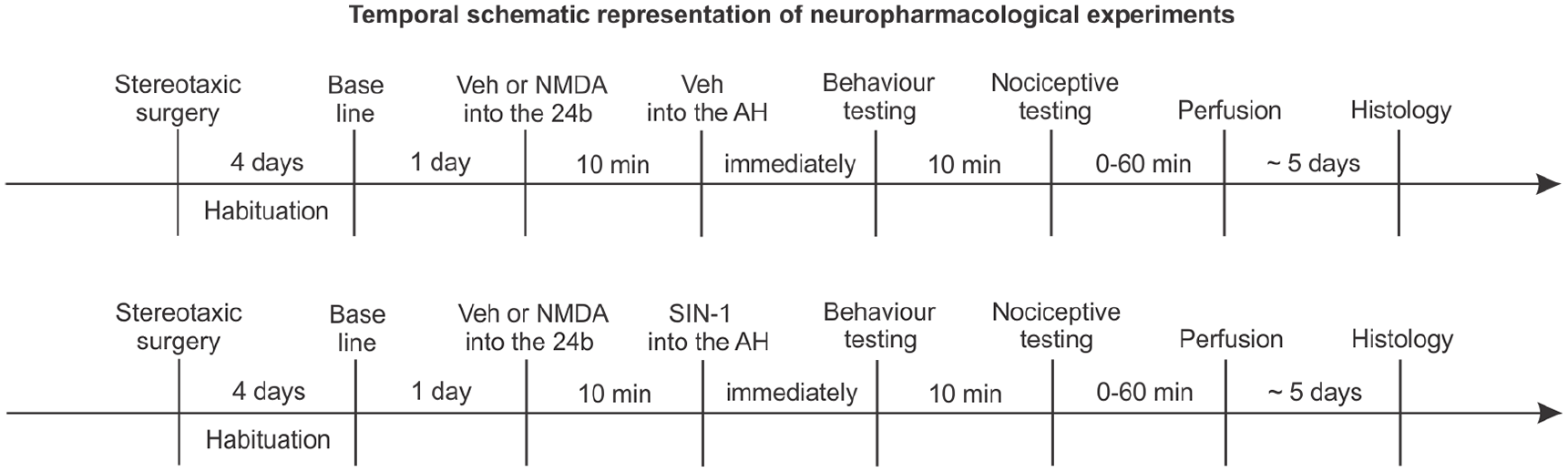
Timeline of neuropharmacological experiments.
An additional group was used to assess NMDA treatment motor effects. For this experiment, mice were only injected into area 24b with either Veh or NMDA (0.1, 1.0 or 10 nmol) (n = 6 per dose).
Neuroanatomical procedures were also performed in independent groups: (a) FCD tract tracer deposits in AH (n = 2); (b) non-fluorescent BDA tract tracer deposits in cingulate cortex area 24b (n = 3) + VGLUT2 immunohistochemistry.
Experimental procedure
Neuropharmacological experiments
Microinjection of the NMDA into the cingulate cortex area 24b
At the end of the habituation period, each mouse was submitted to three measures of control tail-flick latencies to determine the baseline nociceptive threshold. Thereafter, independent groups of mice were randomly assigned to receive microinjections of Veh (0.9% NaCl/0.1 μL) or 0.1, 1 or 10 nmol/0.1 μL NMDA into the cingulate cortex area 24b. Then 10 minutes later, Veh (0.9% NaCl/0.1 μL) was microinjected into the AH. The microinjections were performed through a dental needle (0.3 mm in outer diameter) attached to a polyethylene tube (PE-10) connected to a 5 μL syringe (Hamilton, Reno, Nevada, USA) propelled by an infusion pump (Stoelting, Kiel, Wisconsin, USA). Drugs were injected in a volume of 0.1 μL over 30 s. To prevent reflux, the dental needle was left in place for 15 s after the end of each injection.
After the intra-cingulate cortex area 24b microinjections of NMDA or veh, mice were placed in a polygonal arena and filmed for 10 min with a video camera (Handycam, Sony Corporation, Osaki, Shinagawa-ku, Tokyo, Japan). Immediately after the end of the behavioural tests, tail-flick latencies were measured at 10 min intervals for 60 min. The videos were analysed offline.
Microinjection of the NMDA into cingulate cortex area 24b followed by intra-AH microinjection of SIN
At the end of the habituation period, each mouse was submitted to three measures of control tail-flick latencies to determine the baseline nociceptive threshold. On the day of the experiment, each mouse was pre-treated with microinjections of Veh (0.9% NaCl/0.1 μL) or 0.1, 1 or 10 nmol/0.1 µL NMDA into the cingulate cortex area 24b. Then 10 minutes later, 300 nmol of the NO donor SIN was microinjected into the AH as described in the previous experiment. After the intra-AH administration of the NO donor, the defensive responses displayed by mice in the polygonal arena were quantitatively analysed for 10 min, and immediately after the behavioural tests, the tail-flick latencies were measured at 10 min intervals for 60 min.
Effects of microinjections of NMDA into cingulate cortex area 24b on motor function
Before stereotaxic surgery, the animals received three consecutive training sessions on the rotarod test (Falconi-Sobrinho et al., 2017a; Scheidt et al., 2002). Then 5 days after the surgery, an independent group of mice received microinjections of different doses of NMDA (0.1, 1.0 or 10 nmol) or Veh into the 24b area of the cingulate cortex and were submitted to the rotarod test. The motor test was conducted at a constant speed of 22 revolutions per min and the latency to fall from the apparatus was recorded for each animal, with a maximum latency of 120 s (cut-off).
Neuroanatomical experiments
Labelling the cingulate cortex area 24b-AH pathways using anterograde and retrograde neural tract tracers
An FCD retrograde tract tracer (deposited into the AH; n = 2) or the non-fluorescent BDA tract tracer (deposited into the cingulate cortex area 24b; n = 3) was microinjected in independent groups of mice in a volume of 0.2 or 0.1 μL, respectively, over the course of 5 min. Infusions were delivered using an infusion pump (Stoelting, Kiel, Wisconsin, USA) through a polyethylene tube (PE10) attached to a dental needle, targeting the hypothalamic nucleus. The dental needle was left in place for 2 min after the end of each microinjection to allow local drug diffusion. After completing the microinjection procedure, the dental needle was removed and the skin was sutured. Then 10 days after the neural tract tracer microinjections, mice were deeply anaesthetised with intramuscular injections of 92 mg/kg ketamine (Ketamina®) and 9.2 mg/kg xylazine (Dopaser®) and perfused intracardially with physiological saline followed by 4% paraformaldehyde (PFA, Sigma) dissolved in 0.1 M phosphate buffer (pH 7.4). The encephalon was removed, post-fixed in PFA for 4 h then transferred to 10% and 20% sucrose dissolved in 0.1 M sodium phosphate buffer, pH 7.3, at 4°C for at least 12 h in each solution. The nervous tissue was immersed in 2-methylbutane (Sigma), frozen in dry ice (30 s), embedded in Tissue-Tek, sectioned (20 mm thick) using a cryostat (CM 1950, Leica, Wetzlar, Germany) at 20°C and mounted with Fluoromount with DAPI (Electron Microscopy Sciences, Hatfield, Pennsylvania, USA) on silanised slides. After this procedure, the forebrain slices with FCD retrograde tract tracer were mounted on glass slides and cover-slipped with mineral oil, and both the AH and cingulate cortex area 24b were observed under fluorescence microscopy (AxioImager Z1 with APOTOME II, Zeiss, Oberkochen, Germany).
During the processing of non-fluorescent BDA-labelled cortical neural tissues, endogenous peroxidase activity was blocked by pre-incubation in 3% hydrogen peroxide. BDA labelling was visualised using the avidin-biotin method (ABC standard Elite kit; Vector Laboratories, Burlingame, California, USA). Then, the AH sections were incubated with anti-vesicular glutamate monoclonal transporter 2 (VGLUT2, 1: 300; Merck/Sigma/Aldrich, St. Louis, Missouri, USA) primary antibody (procedure omitted in control groups). Following an overnight incubation period with the primary antibody, slides were washed three times for 5 min each wash and subsequently incubated with secondary antibodies for a period of 60 min. The slides were then washed again, incubated with AB solution and stained with nickel-enhanced chromogen. Finally, slides were mounted and histological sections were analysed under light microscopy (AxioObserver Z1, Zeiss, Oberkochen, Germany).
Drugs
Neuropharmacological experiments: (a) NMDA (Sigma/Aldrich, St. Louis, Missouri, USA) at 0.1, 1 (Ullah et al., 2017) and 10 nmol/0.1 μL or Veh (physiological saline; 0.9% NaCl/0.1 μL) into the cingulate cortex area 24b; (b) NO donor SIN-1 (SIN) (3-morpholinosydnonimine hydrochloride; Tocris Biosciences, Bristol, United Kingdom) at 300 nmol/0.1 μL (de Oliveira et al., 2000; Falconi-Sobrinho and Coimbra, 2018) or Veh (physiological saline; 0.9% NaCl/0.1 μL) into the AH. Both SIN and NMDA were dissolved in physiological saline shortly before each central microinjection.
Neuroanatomical experiments: (a) FCD retrograde tract tracer (10% FCD, 3000 MW; Molecular Probes, Eugene, Oregon, USA); (b) BDA anterograde tract tracer (10% DBA, 10,000 MW; Molecular Probes, Eugene, Oregon, USA).
Histology
Upon completion of the experiments, the animals were anaesthetised with ketamine at 92 mg/kg and xylazine at 9.2 mg/kg and perfused through the left cardiac ventricle using an infusion pump (Master Flex® L/S TM, Vernon Hills, IL, USA). The thoracic descending aortic artery was clamped, perfusion was performed through the left cardiac ventricle and blood was washed out with Tyrode buffer (40 mL at 4°C). The animal was then perfused with 200 mL of ice-cold 4% (w/v) PFA in 0.1 M sodium phosphate buffer (pH 7.3) for 15 min. The brain was quickly removed and maintained in 4% PFA for at least 4 h and was then immersed in a 10% and 20% sucrose solution for at least 12 h in each solution. Tissue pieces were immersed in 2-methylbutane (Sigma-Aldrich, St. Louis, Missouri, USA), frozen on dry ice (30 s), embedded in Tissue-Tek and cut on a cryostat (CM 1950, Leica, Wetzlar, Germany). Slices were then mounted on glass slides that were coated with chrome alum gelatine and stained in a robotic autostainer (CV 5030 Leica Autostainer, Wetzlar, Germany) with haematoxylin-eosin. The sections were viewed under a motorised photomicroscope (AxioImager Z1, Zeiss, Oberkochen, Germany) and the positions of the tips of the guide cannulae were localised according to Paxinos and Franklin’s mouse brain in the stereotaxic coordinate atlas (2001).
Statistical analysis
One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was used to analyse data related to behavioural and motor function integrity studies. Data from the nociceptive threshold experiments collected immediately after the end of the defensive behaviour were submitted to a repeated measures two-way ANOVA followed by Tukey’s post hoc test. All behavioural data and tail-flick latencies are expressed as the mean ± S.E.M. for n = 8 or 6 mice per group. A p value < 0.05 was considered statistically significant. Considering that NMDA-Veh treatments did not produce any behavioural effects (zero mean and variance), these data were not included in the statistical analysis. The software used for statistical analysis and graph plotting was GraphPad Prism version 7.0.
Results
Effects of the activation of glutamatergic projections from cingulate cortex area 24b to the AH on fear-induced defensive behaviours
Microinjections of SIN in AH elicited defensive behaviour characterised by freezing and both oriented and non-oriented escape behaviour, as shown in Figure 2. NMDA-Veh treatment did not produce any defensive behaviour (data not shown). In contrast, NMDA-SIN mice showed a dose-dependent robust increase in the number of crossings (F3,28 = 40.29; p < 0.001) relative to the Veh-SIN group (Figure 2(g)). Except for a small but significant increase in the number crossings (F3,28 = 4.412; p < 0.05, Table 1), behaviours did not change in NMDA-Veh mice (data not shown). In contrast, NMDA-SIN groups were significantly different regarding the durations of both freezing (F3,28 = 5.607; p < 0.01) and non-oriented escape (F1,56 = 4.327; p < 0.01) and for the number of crossings (F3,28 =40.29; p < 0.001) (Figure 2(a)–(g)). However, whereas the duration of freezing was dose dependently decreased in the NMDA-SIN groups, the number of crossings showed a dose-dependent increase (Figure 2(b), (g)). Compared to the Veh-SIN group, the duration of non-oriented escape of NMDA-SIN mice was also significantly increased for the highest dose of NMDA (10 nmol) (Figure 2(f)).
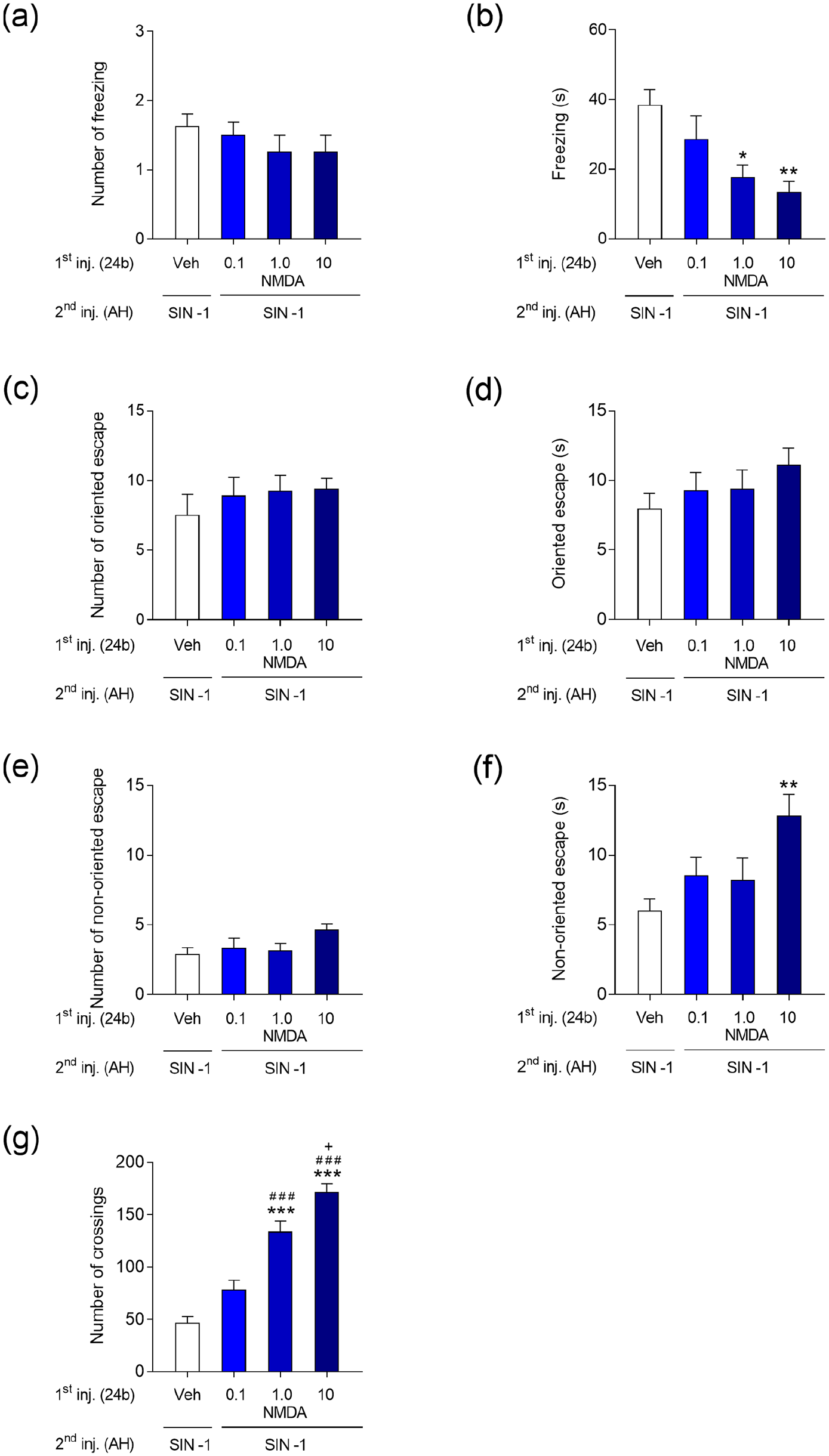
Effect of activation of the cingulate cortex, area 24b, with N-methyl-D-aspartic acid receptor agonist (NMDA) (0.1, 1 and 10 nmol/0.1 µL) or vehicle (NaCl 0.9%/0.1 µL) on the number and duration of freezing (a, b), number and duration of oriented escape (c, d), number and duration of non-oriented escape (e, f) and number of crossings (g) elicited by microinjection of SIN-1 (300 nmol/0.1 µL) into the anterior hypothalamus (AH) of mice. Data are presented as the mean ± standard error of the mean (S.E.M.); n = 8 per group; ***p < 0.001, **p < 0.001, *p < 0.001 compared with the Veh (24b) + SIN-1 (AH)-treated group; ###p < 0.001 compared with the 0.1 nmol NMDA (24b) + SIN-1 (AH); +p < 0.01 compared with the 1 nmol NMDA (24b) + SIN-1 (AH)-treated group, according to one-way analysis of variance (ANOVA) followed by Tukey’s post hoc tests.
Motor behaviour elicited by C57/BL6 mice in the control groups. The crossings were analysed and expressed as the mean ± S.E.M.

p < 0.05 in relation to the vehicle (Veh)-Veh-treated group by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc tests.
Intra-cingulate cortex area 24b microinjections of Veh or NMDA at different doses did not produce any significant effect on the motor function (F3,20 = 0.6923, p > 0.05) of mice subject to the rotarod test. All animals remained on the rotarod apparatus bar from 114 seconds to 120 min (cut-off time).
Effects of the activation of glutamatergic projections from cingulate cortex area 24b to the AH on tail-flick latencies
Tail-flick latencies of the NMDA-Veh groups were significantly different with respect to treatment (F3,28 = 3.524, p < 0.05), time (F9,252 = 2.274, p < 0.01) and the treatment-by-time interaction (F27,252 = 3.783, p < 0.001). Tail-flick latencies displayed by NMDA (1 and 10 nmol)-Veh mice were dose dependently decreased compared to those of the Veh-Veh group (Figure 3(a)).
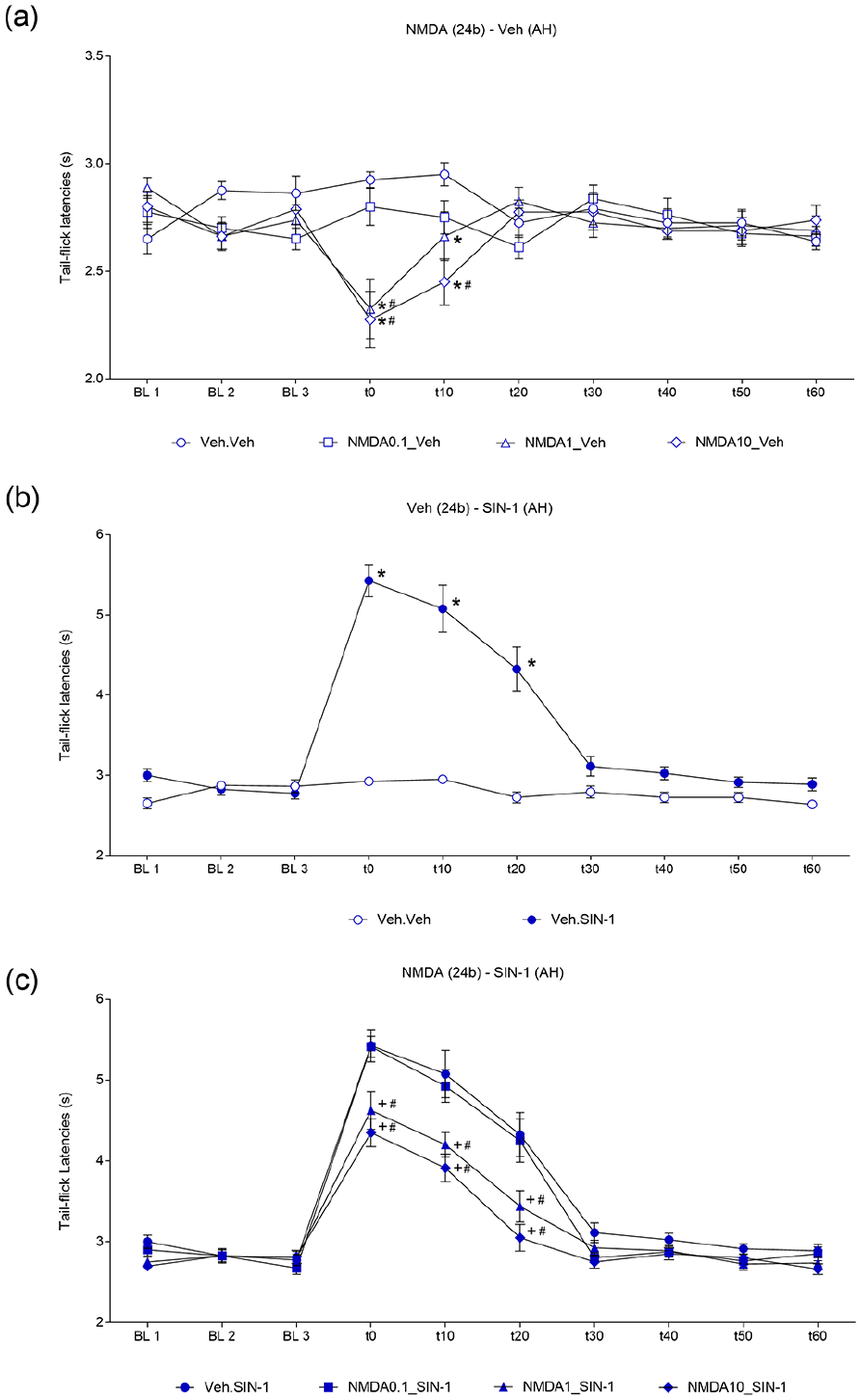
(a) Effect of activation of the cingulate cortex, 24b area, with N-methyl-D-aspartic acid receptor agonist (NMDA) (0.1, 1 and 10 nmol/0.1 µL) or vehicle (NaCl 0.9%/0.1 µL) followed by microinjection of vehicle into the anterior hypothalamus (AH) on tail-flick latencies. Data are presented as the mean ± standard error of the mean (S.E.M.); n = 8 per group; *p < 0.05 compared with the Veh (24b) + Veh (AH)-treated group; #p < 0.05 compared with the 0.1 nmol NMDA (24b) + Veh (AH)-treated group, according to repeated measures two-way analysis of variance (RM-ANOVA) followed by Tukey’s post hoc tests. (b) Effect of intra-AH microinjections of SIN-1 (300 nmol/0.1 µL) preceded by administration of vehicle into area 24b. Data are presented as the mean ± S.E.M.; n = 8 per group; *p < 0.05 compared with the Veh (Cg1) + Veh (AH)-treated group, according to two-way RM-ANOVA followed by Tukey’s post hoc tests. (c) Effect of the activation of the cingulate cortex, area 24b, with NMDA (0.1, 1 and 10 nmol/0.1 µL) followed by microinjection of vehicle into the AH on tail-flick latency. Data are presented as the mean ± S.E.M.; n = 8 per group; +p < 0.05 compared with the Veh (24b) + SIN-1 (AH)-treated group; #p < 0.05 compared with the 0.1 nmol NMDA (24b) + Veh (AH)-treated group, according to two-way RM-ANOVA followed by Tukey’s post hoc tests. BL: Baseline nociceptive threshold.
Tail-flick latencies of the Veh-SIN groups were significantly different with respect to treatment (F1,14 = 105.7, p < 0.001), time (F9,126 = 44.88, p < 0.001) and the treatment-by-time interaction (F9,126 = 34.89, p < 0.001). In particular, the Veh-SIN group showed tail-flick latencies that were significantly longer than those of the controls (Figure 3(b)).
Tail-flick latencies of NMDA-SIN groups were significantly different with respect to treatment (F3,28 = 13.1, p < 0.001), time (F9,252 = 172.9, p < 0.001) and the treatment-by-time interaction (F27,252 = 4.485, p < 0.001). NMDA (1 and 10 nmol)-Veh mice showed a decrease in tail-flick latencies from 0 to 20 min. The latter differences were due to shorter tail-flick latencies in comparison to Veh-SIN and NMDA (1 nmol)-SIN mice (Figure 3(c)).
Sites of drug microinjections into the cingulate cortex area 24b and AH
Histologically confirmed sites of NMDA (0.1, 1 and 10 nmol) or Veh microinjections into the cingulate cortex area 24b and of SIN (300 nmol) or Veh administration into the AH are shown in schematic drawings of sections of the C57BL/6 mouse brain. The number of points illustrated in the figure is apparently less than the total number of mice used in the present work because of overlapping microinjection sites (Figure 4).
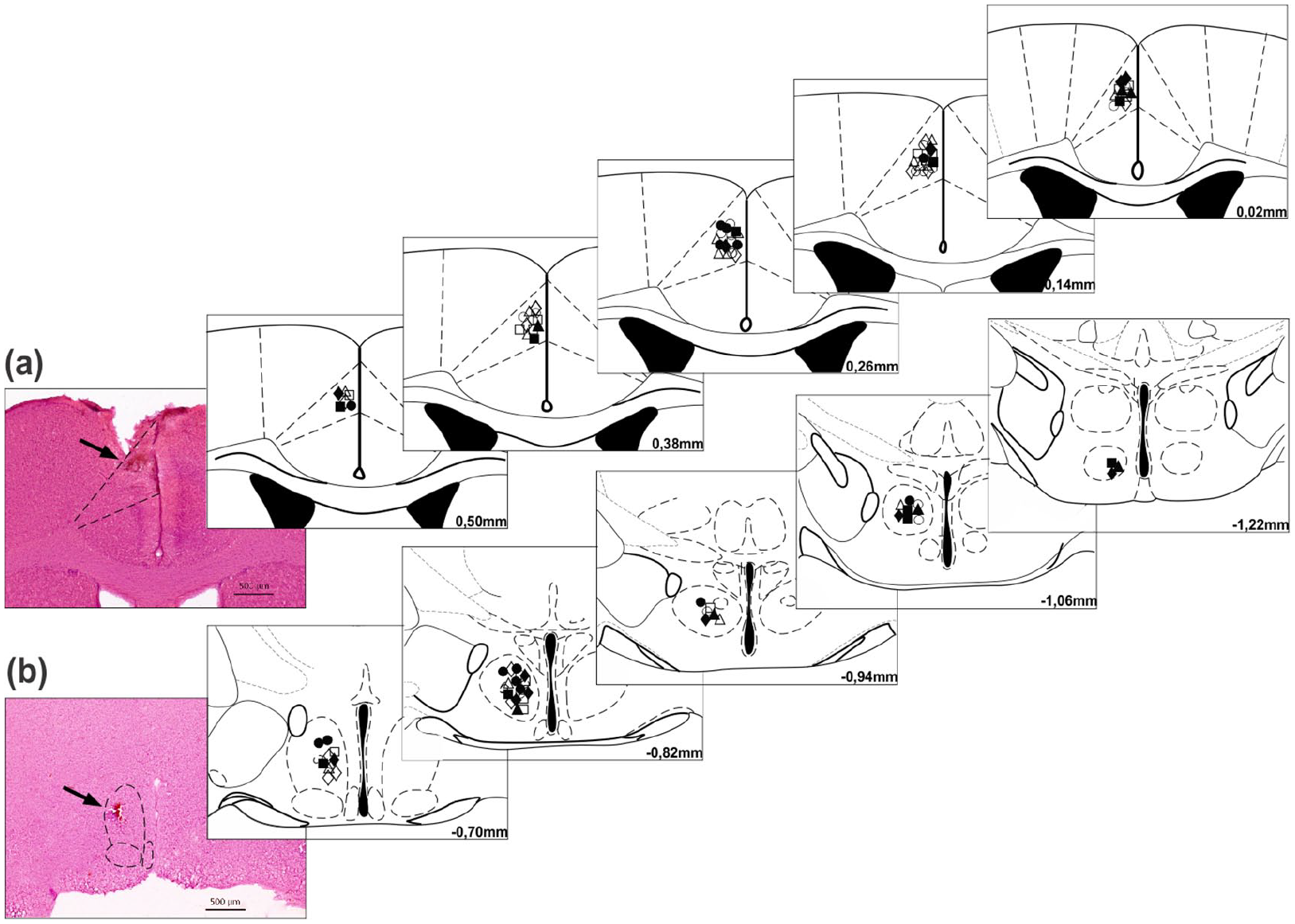
Diagrammatic representation of coronal sections of the C57BL/6 mouse brain showing the sites of central administration of N-methyl-D-aspartic acid receptor agonist (NMDA) or vehicle microinjection into cingulate cortex area 24b (a) and of SIN-1 or vehicle microinjection into the anterior hypothalamus (b). Photomicrographs of cortical and diencephalic (in the left) coronal sections of the brain at Bregma 0.26 mm and −0.70 mm showing representative sites (black arrows) of the central microinjections of drugs in cingulate cortex area 24b (a) and the anterior hypothalamus (b), respectively. Histological staining: haematoxylin and eosin.
Neural tract-tracing of pathways connecting cingulate cortex area 24b to the AH
Microinjections of the FCD retrograde tract tracer were performed in independent mice (n = 2) aimed at the AH (Figure 6(a), (b)), showing connections between this hypothalamic nucleus and the cingulate cortex area 24b. FCD retrograde tract tracer-labelled pyramidal neurons were found in the internal pyramidal layer of cingulate cortex area 24b ipsilateral to the site in which the neural tract tracer was microinjected (Figure 5(c)–(f)).
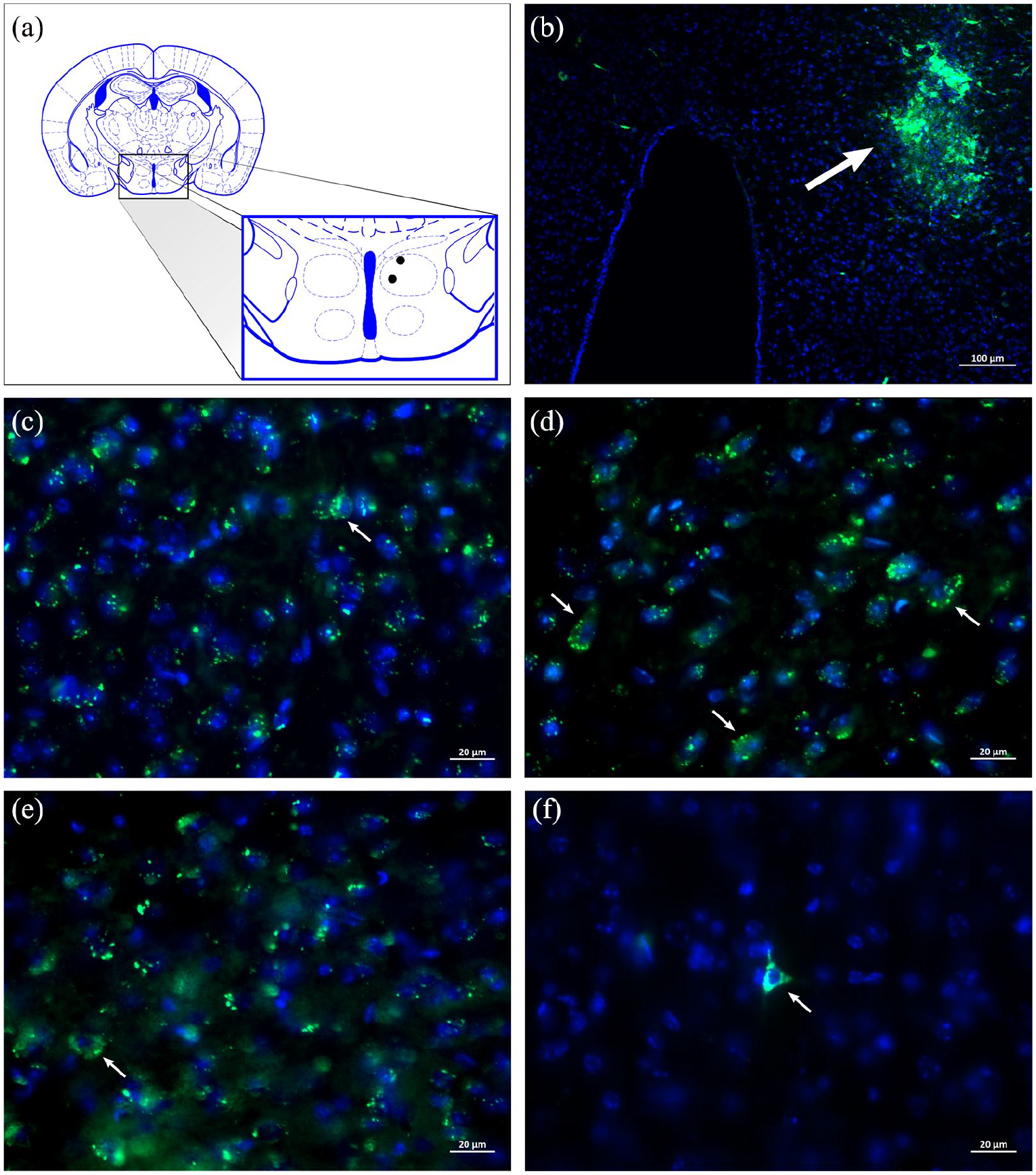
Diagrammatic representation of a transverse section of the diencephalon showing (a) the histologically confirmed microinjection sites (black circles) of a retrograde neural tract tracer in the anterior hypothalamus (AH) (Bregma −1.22 mm) depicted in a modified drawing from Paxinos and Franklin’s stereotaxic atlas (2001). (b) Photomicrograph of a transverse section of the diencephalon of a C57BL/6 mouse showing a representative microinjection site (white arrow) of the fluorescein-conjugated retrograde dextran (3000 MW) neural tract tracer in the AH. (c) to (f): Photomicrographs of transverse sections of the cingulate cortex of C57BL/6 mice showing fluorescein-conjugated dextran (FCD) retrograde tract tracer-labelled pyramidal cortical neurons (white arrows) located in cingulate cortex area 24b ipsilateral to the site in which the neural tract tracer was microinjected.
Microinjections of the BDA anterograde neural tract tracer were performed in independent mice (n = 3) aimed at cingulate cortex area 24b (Figure 6(a)–(e)), showing connections between this limbic cortical region and the AH. Cingulate cortex efferent pathways seem to originate from the internal pyramidal layer of cingulate cortex area 24b (Figure 6(d), (e)). Glutamatergic vesicle-labelled axonal fibres from cingulate cortex area 24b were found in the AH surrounding the perikarya with terminal buttons, suggesting synaptic contacts (Figure 7(a)–(f)). Considering the BDA-labelled projections from cingulate cortex area 24b to AH neurons, we identified that approximately 29% of axonal fibres were labelled as glutamatergic vesicles.
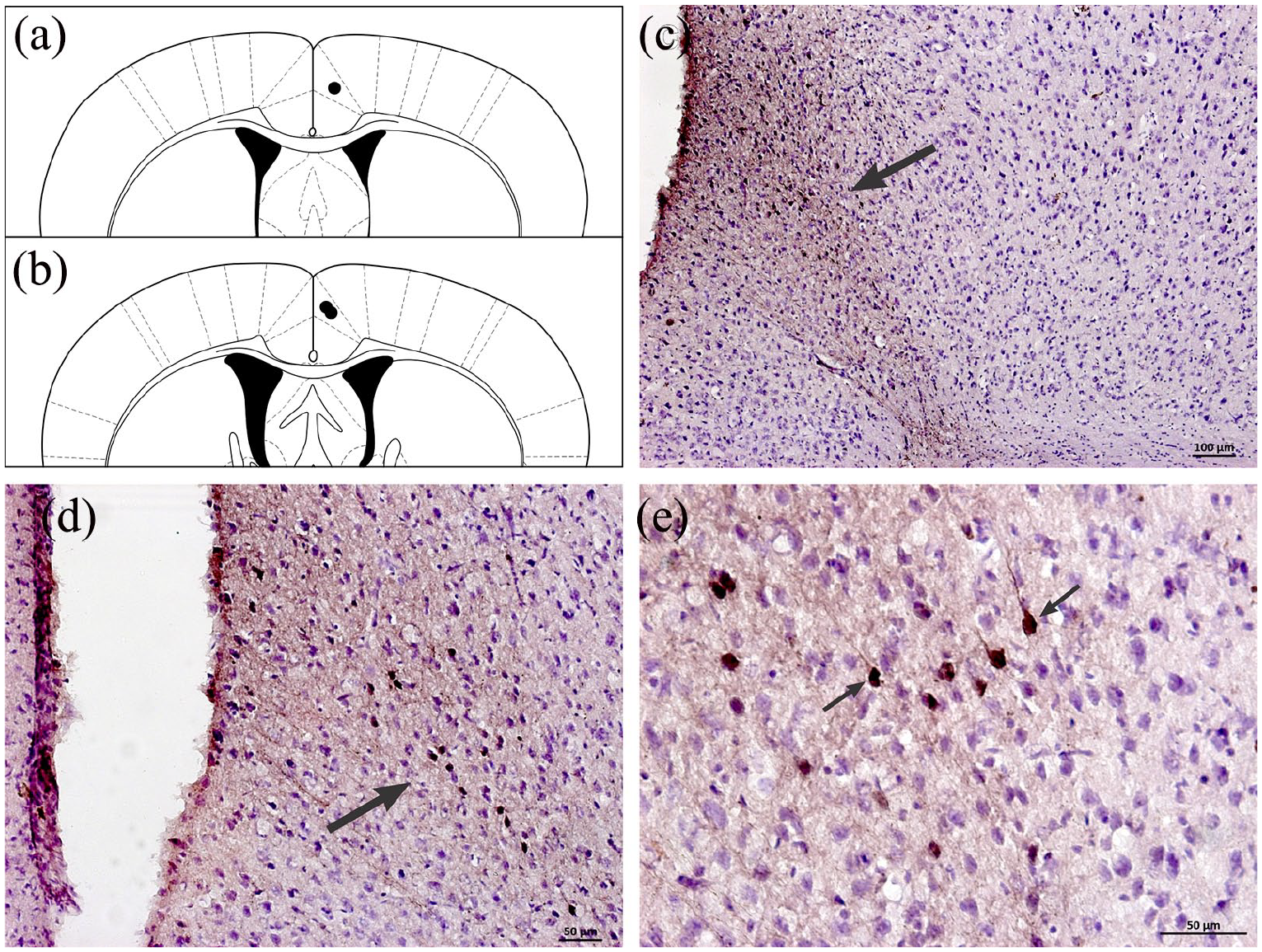
Diagrammatic representations of transverse sections of the cingulate cortex, showing (a) and (b) histologically confirmed microinjection sites (black circles) of the biotinylated dextran amine (BDA) (10,000 MW) anterograde tract tracer in cingulate cortex area 24b (Bregma 0.26 mm and 0.02 mm, respectively) depicted in a modified drawing from Paxinos and Franklin’s stereotaxic atlas (2001). (b) to (d): Photomicrographs of transverse sections of the cingulate cortex of C57BL/6 mice showing representative microinjection sites (black arrows) of the neural tract tracer in cingulate cortex area 24b. Note in (e) that the cortical fugal pathways originate from neurons situated in the internal pyramidal layer of the cingulate cortex area 24b.
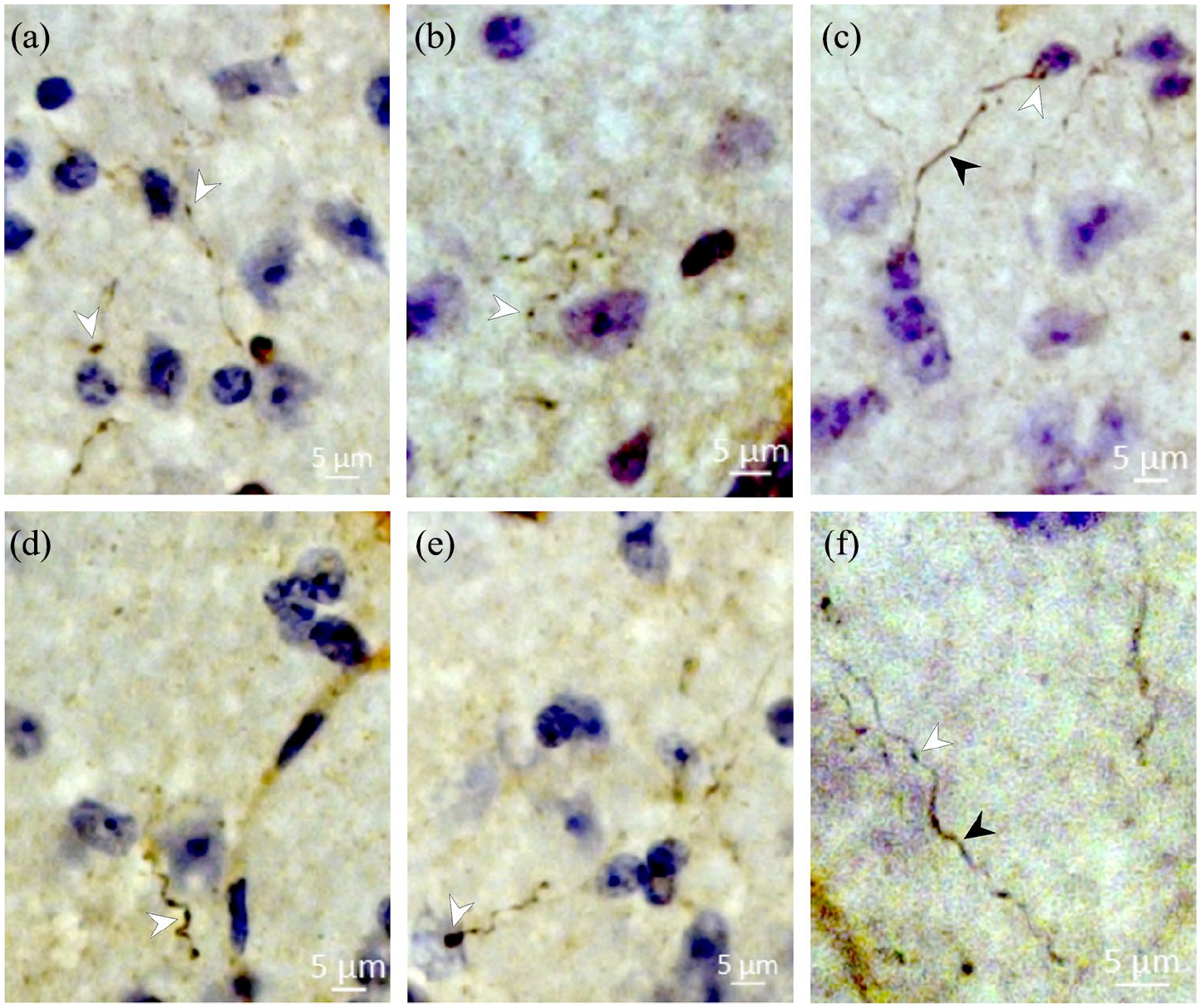
Photomicrographs of transverse sections of the anterior hypothalamus (AH) of C57BL/6 mice, showing from (a) to (f), cortical-hypothalamic axonal fibres (black arrowheads) with glutamatergic synaptic vesicle-labelled terminal buttons (white arrowheads) surrounding perikarya in the AH, suggesting glutamatergic synaptic contacts.
Discussion
The present study sought to characterise the cingulate cortex area 24b projections to the AH of mice. The data showed that activation of cingulate cortex area 24b NMDA receptors facilitated escape while reducing freezing and antinociception produced by SIN-1 microinjections into the AH. In addition, neural tract tracing and immunocytochemical data suggested these responses are mediated by glutamatergic synapses at AH. These data corroborated previous studies showing that SIN-1 injections into the AH of mice produce defensive behaviours and antinociceptive responses that were markedly attenuated by local pre-treatment with an NMDA receptor antagonist (Falconi-Sobrinho and Coimbra, 2018). Fear-related behaviours started within 1 min after the microinjection of NO donor into the AH. This delay could be the result of the slow release of NO through the formation of the intermediate compound SIN-1C (Feelisch et al., 1989; Southam and Garthwaite, 1991). In contrast, previous studies in rats showed Cg1 facilitates both escape and fear-induced antinociceptive responses produced by blockade of GABA receptors of the PH (Falconi-Sobrinho et al., 2017a, 2017b). Altogether, these studies suggest Cg1 has differential effects on antinociception evoked by SIN-1 microinjections into the AH and PH of mice and rats, respectively. In any event, NO could act on several targets. In particular, it is long known that NO can act as a retrograde transmitter, causing further increases in the pre-synaptic release of glutamate (Garthwaite et al., 1989).
Glutamate binding to NMDA receptors promotes an influx of calcium that activates nNOS, an enzyme responsible for NO synthesis. Once synthesised at the post-synaptic neuron, NO acts as a retrograde transmitter because it is synthesised in post-synaptic neurons (mostly soma) and diffuses to the pre-synaptic terminal, where it increases glutamate release (Garthwaite et al., 1988, 1989 for review).
More recently, Eguchi et al. (2012) presented evidence that NO increases glutamate release through the facilitation of cellular exocytosis (i.e., increased vesicular cycling) regulated by cGMP-dependent protein kinase (PKG). This mechanism would maintain the exoendocytic coupling that is critical during sustained synaptic transmission at a high rate.
Considering that NO signalling within the AH is increased by NMDA injections into Cg1, NMDA microinjections are expected to increase both fear and escape behaviour. Several studies have shown the hypothalamus predominantly organises defensive escape behaviours. Chemical stimulation of most nuclei of the medial zone of the hypothalamus elicits escape and jumps interspersed with exploratory behaviours (Biagioni et al., 2012, 2013; dos Anjos-Garcia et al., 2017; Falconi-Sobrinho and Coimbra, 2018). In addition, previous studies performed by Falconi-Sobrinho et al. (2017a, 2017b) showed that although the GABAergic disinhibition of PH neurons did not cause freezing behaviour, it induced vigorous escape reactions. Here, we show that the activation of AH neurons provoked by intra-AH administration of SIN-1 caused both escape and freezing behaviours.
To our knowledge, the present study is the first demonstration that escape behaviours evoked by the microinjection of SIN-1 into the AH are facilitated by the pre-injection of NMDA into the 24b area of the cingulate cortex. As shown by neuroanatomical evidence reported herein, these effects were most likely due to the increased activity of 24b area glutamatergic projections to the AH. NMDA injections in area 24b were also able to produce a significant increase in the distance travelled (crossings) by these animals. These data suggest that 24b glutamatergic projections modulate NO-mediated fear-related escape behaviours organised in the AH.
Pharmacological data were supported by the identification of glutamate-labelled fibres, terminal buttons and vesicles in the AH of mice injected with BDA into the 24b area of the cingulate cortex.
As expected, the facilitation of SIN-evoked escape following NMDA microinjection into the 24b area was paralleled by a decrease in freezing behaviour. The glutamatergic neurons of area 24b may also project to limbic structures that influence the hypothalamus defence mechanisms. In particular, the amygdaloid complex is both the target of the cingulate cortex (Buchanan et al., 1994; Jhang et al., 2018) and a source of dense projections to the AH (Petrovich et al., 1996). Moreover, studies by Jhang et al. (2018) showed that optogenetic activation of Cg1 glutamatergic projections to the amygdaloid complex inhibits freezing reactions displayed by mice exposed to a predator odour. Conversely, the optogenetic inactivation of Cg1 increased the freezing of mice submitted to the same test. In fact, neuroimaging studies in humans presented evidence that the rostral cingulate cortex is implicated in complex ‘decisions’ on the defensive strategy to a distal threat (Mobbs et al., 2007). However, these authors linked the escape response to deactivation of the rostral cingulate cortex.
Although NMDA receptors are predominantly located on excitatory neurons, we cannot rule out the possibility that the anti-aversive effects reported herein were due to the activation of NMDA somato-dendritic receptors of GABAergic interneurons, as already reported elsewhere (Homayoun and Moghaddam, 2007; Zanos and Gould, 2018). As a result, NMDA activation of GABAergic interneurons of the cingulate cortex would facilitate escape at the expense of freezing. Although this hypothesis is in line with the findings of Mobbs et al. (2007), it contradicts the present findings suggesting the NMDA direct activation of hypothalamus-projecting glutamatergic neurons. The present study also showed the microinjection of SIN-1 into the AH increases nociceptive thresholds measured by the tail-flick test. Recent findings have suggested the chemical stimulation of both the medial hypothalamus (MH) (de Freitas et al., 2014) and PH (Falconi-Sobrinho et al., 2017a, 2017b) activates pain-inhibitory descending pathways that mediate the antinociceptive response that follows defensive behaviour. In addition, these authors have demonstrated that MH fear-induced antinociception organised in both the MH and PH can be mediated by different regions of the limbic cortex, such as the prelimbic and cingulate cortex. Neurons located in the medial prefrontal cortex, including the Cg1 region of the cingulate cortex, are involved in both the encoding of nociceptive stimulus intensity (Zhang et al., 2004) and in emotion suppression of pain (Butler and Finn, 2009 for review; Falconi-Sobrinho et al., 2017a, 2017b). Falconi-Sobrinho et al. (2017a, 2017b) showed that Cg1 inhibition by local microinjections of either lidocaine or an NMDA receptor antagonist attenuated both the defensive behaviours and the antinociception in response to chemical stimulation of the PH. Recently, Coimbra et al. (2017) presented evidence that rats exposed to different anxiety (elevated plus maze) and fear experimental models (elevated T-maze and confrontation with rattlesnakes in polygonal arenas for snakes) trigger different levels of antinociception. Indeed, the exposure of prey to natural predators elicits defensive antinociception (Lexter and Fanselow, 1985; Mendes-Gomes et al., 2020). However, dissociation between unconditioned fear and defensive antinociception was also reported (Biagioni et al., 2016; da Silva et al., 2015).
Conversely, however, the present study showed that NMDA injections into area 24b of the cingulate cortex facilitate both defensive behaviours and nociception. A previous study in rats (Falconi-Sobrinho et al., 2017a, 2017b) found that injections of an antagonist of NMDA receptor into area 24b impair defensive behaviours while facilitating nociceptive responses to chemical stimulation of the PH. The results of the present and previous studies suggest that AH and PH play opposite roles in stress modulation of pain perception, activating descending pathways that either facilitate or inhibit nociception, respectively. It remains unknown whether the opposite results of these studies are related to the differences in the hypothalamic area or the species.
Footnotes
Author contributions
LLF-S performed the experiments, analysed and interpreted the data, and wrote the manuscript. TdA-G worked on the morphological approaches. NCC designed the experiments, analysed and interpreted the data, wrote the manuscript and approved the final manuscript. All authors approved the final version of the manuscript and are entirely responsible for the scientific content of this paper.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (grant numbers 2007/01174-1, 2012/03798-0 and 2017/11855-8) and Conselho Nacional de Pesquisa e Desenvolvimento Tecnológico (CNPq) (grant numbers 483763/2010-1, 474853/2013-6 and 427397/2018-9). LLF-S was supported by FAPESP (Scientiae Magister grant 2013/10984-8; post-doctoral grant 2019/05255-3) and CNPq (ScM fellowship grant 134267/2013-3; Scientiae Doctor fellowship grant 145258/2015-7). TdA-G was financially supported by CNPq (ScM fellowship grant 130124/2012-5; Sc.D. fellowship grant 141124/2014-8) and FAPESP (Postdoctoral grant 2017/22647-7). NCC is a researcher (level 1A) at CNPq (processes 301905/2010-0 and 301341/215-0). The authors also thank Daoud Hibrahim Elias-Filho for his expert technical assistance. DHE-F received technician scholarships from FAPESP (TT-2, process 02/01497-1) and CNPq (processes 501858/2005-9, 372654/2006-1, 372810/2008-0, 372877/2010-9, 474853/2013-6, and 372838/2018-9).