
Abstract
Background
Recent evidence increasingly links schizophrenia to mitochondrial dysfunction, implicating altered mitochondrial respiratory and metabolic processes in its pathophysiology. Preclinical data suggest that atypical antipsychotics may inhibit mitochondrial respiratory chain complex I (MRCC I), raising concerns regarding mitochondrial impairment despite clinical efficacy. Hence, this study was conducted to evaluate mitochondrial dysfunction associated with schizophrenia and to compare changes during treatment with risperidone and aripiprazole.
Methods
In this randomized, open-label, clinical trial, 60 patients with schizophrenia were allocated to either risperidone or aripiprazole for 12 weeks. A healthy control group (n = 30) was recruited for baseline biochemical comparisons. The outcome measures were the change in platelet MRCC I, serum lactate, pyruvate, lactate:pyruvate (L:P) ratio, creatine kinase, Positive and Negative Syndrome Scale (PANSS), Newcastle Mitochondrial Disease Adult Scale (NMDAS), responder rate, and treatment-emergent adverse events (TEAEs).
Results
At baseline, schizophrenia patients exhibited significantly higher levels of MRCC I, lactate, pyruvate, and L:P ratio compared to healthy controls, consistent with underlying mitochondrial dysregulation. After 12 weeks of therapy, both risperidone and aripiprazole significantly reduced MRCC I concentration (−2.66 vs −3.13 nmol/108 platelets), with no inter-group difference (p = 0.508). Serum lactate and L:P ratio increased significantly, while serum pyruvate remained unchanged. PANSS scores improved significantly in both groups, with comparable responder rates. NMDAS scores showed no significant change. TEAEs were mostly mild and more frequent with risperidone, though not statistically significant.
Conclusion
Schizophrenia is associated with alterations in mitochondrial-related biochemical markers suggestive of disrupted cellular bioenergetics. Treatment with risperidone and aripiprazole was associated with changes in these markers, without differences between groups. These findings reflect altered bioenergetic status and warrant further investigation using functional assays.
Trial registration
ClinicalTrials.gov identifier: NCT06236451.
Keywords
Introduction
Schizophrenia is a complex psychiatric disorder that impacts approximately 1% of the adult population globally (Hany, 2023). Schizophrenia has been attributed to dysregulation of dopaminergic, glutamatergic, serotonergic, and GABAergic signaling; however, a growing body of evidence from transcriptomic, proteomic, and neuroimaging studies implicates mitochondrial dysfunction and impaired cellular bioenergetics in the pathophysiology of schizophrenia (Ben-Shachar, 2002; Prabakaran et al., 2004; Pruett and Meador-Woodruff, 2020; Zhou et al., 2025). Disruption of mitochondrial function may affect neural plasticity, potentially contributing to cognitive deficits, and abnormal behavioral manifestations (Ben-Shachar and Laifenfeld, 2004).
Recent cell-specific transcriptomic analyses of the prefrontal cortex suggest that neurons and glial cells in individuals with schizophrenia exhibit altered expression of genes related to mitochondrial function and ion regulation. Notably, these include genes involved in oxidative phosphorylation, such as NADH dehydrogenase subunits (e.g., NDUFS1 and NDUFV1), cytochrome c oxidase (COX family genes), and Adenosine Triphosphate (ATP) synthase components (e.g., ATP5F1A), indicating a widespread disruption of cellular energy metabolism and signaling in the disease (Bast et al., 2025). Multiple findings indicate impaired respiration, abnormal mitochondrial structure, altered mitochondrial DNA, reduced high-energy phosphates, and lower brain pH in schizophrenia (Clay et al., 2011; Fizikova et al., 2023). Notably, several studies have reported a paradoxical pattern in schizophrenia, wherein the expression of mitochondrial respiratory chain complex I (MRCC I) subunits is increased despite a concomitant reduction in enzymatic activity and oxidative phosphorylation efficiency (Akarsu et al., 2014; Dror et al., 2002; Mehler-Wex et al., 2006; Prabakaran et al., 2004; Taurines et al., 2010). This apparent compensatory upregulation is thought to reflect an adaptive response to impaired electron transport and ATP generation, rather than preserved mitochondrial function (Akarsu et al., 2014; Dror et al., 2002; Uittenbogaard and Chiaramello, 2014). Studies have shown that, although patients may exhibit increased expression of MRCC I subunits, the functional activity of the enzyme remains reduced, indicating a mismatch between quantity and bioenergetic efficiency (Mehler-Wex et al., 2006; Prabakaran et al., 2004). This paradoxical compensatory upregulation is thought to show the mitochondrial attempts to compensate for impaired electron transfer and ATP production (Uittenbogaard and Chiaramello, 2014).
The pharmacological management of schizophrenia primarily focuses on monoaminergic pathways, with atypical antipsychotics as the preferred first-line therapeutic agents (Meltzer, 2013). Antipsychotic therapy has been shown to influence brain energy metabolism and modify mitochondrial activity (De Simone et al., 2023). Previous preclinical investigations have shown that typical and atypical antipsychotic agents can decrease the activity of MRCC I (Burkhardt et al., 1993; Hardy et al., 2023; Luptak et al., 2021; Modica-Napolitano et al., 2003; Prince et al., 1997; Prince et al., 1998; Scaini et al., 2013). In a cross-sectional observational study, Casademont et al. (2007) observed markedly reduced MRCC I activity in patients treated with haloperidol or risperidone compared to drug-naive individuals. Meta-analysis of studies on postmortem brain tissue from individuals with schizophrenia has revealed consistent downregulation of several ATP synthase-related genes across both sexes, supporting the presence of impaired mitochondrial energy production in the disorder (Katz Shroitman et al., 2023). Dror et al. (2002) demonstrated phase-dependent alterations in MRCC I, with increased subunit expression during acute psychosis and reduced levels in residual schizophrenia. Furthermore, mitochondrial abnormalities linked to schizophrenia appear to persist despite antipsychotic treatment (Clay et al., 2011). Evidence from preclinical studies also suggests that antipsychotic-related extrapyramidal adverse effects may stem from disrupted mitochondrial energy metabolism, particularly through inhibition of MRCC I in the electron transport chain (Chan et al., 2020; Hardy et al., 2023; Maurer and Volz, 2001).
Given the systemic nature of mitochondrial dysfunction, peripheral tissues with high bioenergetic demand have been increasingly explored as accessible models to study disease-related mitochondrial alterations in schizophrenia (Clay et al., 2011; Fizikova et al., 2023; Moren et al., 2025). Platelets share key mitochondrial properties with neurons, including reliance on oxidative phosphorylation and sensitivity to MRCC I dysfunction, and have been validated as biologically relevant peripheral surrogates for systemic mitochondrial alterations in neuropsychiatric disorders (Dror et al., 2002; Mehler-Wex et al., 2006; Moren et al., 2025). Prior studies have demonstrated that platelet mitochondrial markers reflect disease state and treatment-related bioenergetic changes in schizophrenia, supporting their use in longitudinal clinical investigations (Bar-Yosef et al., 2020; Casademont et al., 2007; Dror et al., 2002).
Despite growing interest, the clinical relevance of antipsychotic-associated mitochondrial dysfunction remains poorly defined, with a lack of clinical trials or prospective observational studies directly evaluating its impact. Clarifying the mechanisms, magnitude, and clinical consequences of mitochondrial impairment is essential for understanding long-term treatment effects and for guiding the development of safer, mechanism-based therapies in schizophrenia. In this context, atypical antipsychotics may influence clinical outcomes and adverse effects in schizophrenia through their effects on mitochondrial function. The present randomized controlled trial was designed to evaluate mitochondrial dysfunction associated with schizophrenia and to compare biochemical and clinical changes during treatment with risperidone and aripiprazole.
Methods
Study design
The present study is a randomized, open-label, parallel-arm clinical trial. The study was approved by the Institutional Ethics Committee, All India Institute of Medical Sciences, Bhubaneswar, India, and the authors assert that all procedures contributing to this work comply with the ethical standards of the national ethical guidelines and with the Helsinki Declaration of 1975, as revised in 2013. Written informed consent was obtained from the legally authorized representatives of the patients, and confidentiality was ensured by restricted investigator access throughout the study. The study protocol was prospectively registered with ClinicalTrials.gov (NCT06236451) and has been published in a peer-reviewed journal, ensuring transparency, methodological rigor, and adherence to international standards for clinical research reporting (Shaju et al., 2025).
Study population
Patients with schizophrenia attending the outpatient Psychiatry department were screened and enrolled as per the predefined selection criteria. Patients diagnosed with schizophrenia according to the Diagnostic and Statistical Manual of Mental Disorders-5 (DSM-5) criteria, of either sex aged 18–60 years, who had not taken any antipsychotic drugs for at least 4 weeks before recruitment or were treatment naïve, were recruited (American Psychiatric Association, 2013). Patients diagnosed with other psychiatric diagnoses or patients with known mitochondrial disorders (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; Leber hereditary optic neuropathy; Leigh syndrome, Kearns–Sayre syndrome, Myoclonic Epilepsy with Ragged-Red Fibers, etc.) were excluded. Patients with medical comorbidities, patients with a history of substance abuse, pregnant, or lactating mothers were also excluded.
Randomization and allocation concealment
By employing computer-generated codes and block randomization (block size of 6), all the patients were divided into two groups with an allocation ratio of 1:1. The random allocation code was generated by the investigator who was not involved in recruitment. The sequentially numbered, opaque, sealed envelope technique was used for allocation concealment.
Study procedure and data collection
Baseline assessment included medical history, anthropometry, Positive and Negative Syndrome Scale (PANSS), Newcastle Mitochondrial Disease Adult Scale (NMDAS) scores, and mitochondrial biomarkers (platelet MRCC I, serum lactate, serum pyruvate, lactate:pyruvate (L:P) ratio and serum creatine kinase). Sixty patients were randomized to receive either risperidone (n = 30) or aripiprazole (n = 30) for 12 weeks. Risperidone was initiated at 2 mg/day and titrated to 6 mg/day over 2–3 weeks, while aripiprazole was started at 10 mg/day and titrated to 20 mg/day over 2–3 weeks. A healthy control group (n = 30) was recruited for baseline biochemical comparison. Treatment adherence was formally assessed at the 12-week follow-up using the pill-counting method, while telephonic follow-up at 6 weeks was conducted to monitor interim clinical status and adverse events. At 12 weeks, clinical scales and biochemical parameters were reassessed, and adverse events were documented and evaluated using the World Health Organization-Uppsala Monitoring Centre (WHO-UMC) causality system. A patient was considered compliant if they had ⩾80% pill intake. For all biochemical parameters, venous blood samples were collected under fasting conditions (after an overnight fast) to minimize variability in metabolic parameters.
Outcome measures
Biochemical parameters
The concentration of MRCC I in platelets was measured at baseline and at 12-week follow-up using a commercially available enzyme-linked immunosorbent assay kit. The concentration thus obtained was normalized to the platelet count for the patient at that visit.

Serum lactate, serum pyruvate, and the L:P ratio were measured at baseline and at the 12-week follow-up using spectrophotometric methods (Rosenberg and Rush, 1966).
Serum creatine kinase levels were measured at baseline and 12 weeks using an autoanalyzer.
Clinical parameters
NMDAS scores were measured at baseline and 12 weeks of follow-up. NMDAS is a validated clinical instrument that assesses mitochondrial disease burden across functional, systemic, and neurological domains and was included to provide a standardized clinical correlate of mitochondrial dysfunction alongside biochemical markers (Schaefer et al., 2006). The NMDAS comprises four sections: Section I (Current Function, reflecting functional status over the preceding 4 weeks; 4 items), Section II (System-Specific Involvement, capturing multi-organ manifestations including neurological, cardiac, respiratory, gastrointestinal, and endocrine systems; 20 items), Section III (Current Clinical Examination, based on objective clinical findings at assessment; 8 items), and Section IV (Quality of Life, assessed using the SF-12 v2 questionnaire and scored separately). Each item across Sections I–III is scored on a 0–5 ordinal scale, where 0 denotes normal function and 5 denotes the most severe impairment; section scores are obtained by summing item scores, with higher total scores reflecting greater disease burden. NMDAS was applied only to patients with schizophrenia to assess potential clinical manifestations of mitochondrial dysfunction and was not administered to healthy controls, as it is designed for use in individuals with suspected mitochondrial disease.
PANSS scores were measured at baseline and 12 weeks of follow-up (Kay et al., 1987).
Responder rate: A patient with a reduction of PANSS score by ⩾50% from baseline was considered a “responder” (Leucht et al., 2007).
Safety evaluation: During follow-up, patients were assessed using non-directive questioning and direct interaction to identify adverse events. All adverse events associated with risperidone or aripiprazole were recorded, irrespective of prior recognition, including details of causality, description, severity, duration, management, and outcome.
Evaluation of the severity of extrapyramidal adverse effects by the Modified Simpson–Angus Scale (Simpson and Angus, 1970).
Sample size calculation
A sample size of 30 participants per group (N = 60) was determined to be adequate to detect an intergroup difference of 4 nmol/min/mg in mitochondrial Complex I activity (SD 4.7) at a two-sided α of 0.05 with 80% power, based on prior data.(Casademont et al., 2007). Although previous studies reported activity, the primary outcome in the present study was the change in MRCC I concentration. Assuming a moderate-to-large effect size (~0.8), 26 participants per group were required; this was increased to 30 per group to account for a 10% attrition rate. An additional group of 30 healthy controls were included for baseline biochemical comparison.
Statistical analysis
Continuous variables were summarized as mean ± SD or median (interquartile range, IQR), and categorical variables as proportions. Analyses were performed using R software, following the intention-to-treat principle, with p ⩽ 0.05 considered statistically significant. Missing data were handled using multiple imputation under the assumption of missing at random (MAR), and pooled estimates were used for statistical inference. Categorical variables were compared using chi-square or Fisher’s exact test, as appropriate. Normally distributed data were analyzed using unpaired or paired t-tests, while non-parametric data were analyzed using the Wilcoxon signed-rank or Mann–Whitney U-test, with Hodges–Lehmann estimates and 95% confidence intervals reported. Associations were assessed using Spearman correlation, and regression analyses (linear and logistic) were performed to identify predictors of primary outcomes and treatment response. To test the hypothesis that antipsychotic treatment overall is associated with a reduction in platelet MRCC I, a linear mixed model was fitted with MRCC I as the outcome, time (baseline vs 12 weeks) and treatment group as fixed effects, their interaction as a test of between-drug difference, and subject as a random intercept to account for within-individual correlation across timepoints. Sensitivity analysis were done using complete case analysis and imputation using last observation carried forward (LOCF) to check for robustness of the primary findings. Biochemical outcomes were compared using paired Wilcoxon signed-rank or t-tests, as appropriate, with Benjamini–Hochberg False Discovery Rate (FDR) correction applied across all secondary comparisons within each domain, as reported in the Supplemental materials. All further analyses, including clinical scale scores, correlation analyses, and adverse event comparisons, are exploratory; uncorrected p-values are reported and clearly labelled as such throughout.
Results
Patients
Patient enrollment commenced on April 5, 2024, and follow-up for all participants was completed by November 3, 2025. Of 108 schizophrenia patients screened, 48 were excluded, and 60 were randomized to either risperidone or aripiprazole. Another 30 healthy volunteers were recruited to serve as baseline controls. Four participants in the risperidone group and five in the aripiprazole group did not attend their follow-up visits despite telephonic reminders (Figure 1). The loss of follow-up was due to geographical distance or financial limitations. At baseline, patients with schizophrenia differed significantly from healthy controls in MRCC I level, serum lactate, pyruvate, and the lactate-to-pyruvate ratio, while age, sex, BMI, and creatine kinase were comparable (Supplemental Table S1). To assess whether smoking confounded the baseline differences in mitochondrial markers, we performed sensitivity analyses using ANCOVA, with smoking status as a covariate.
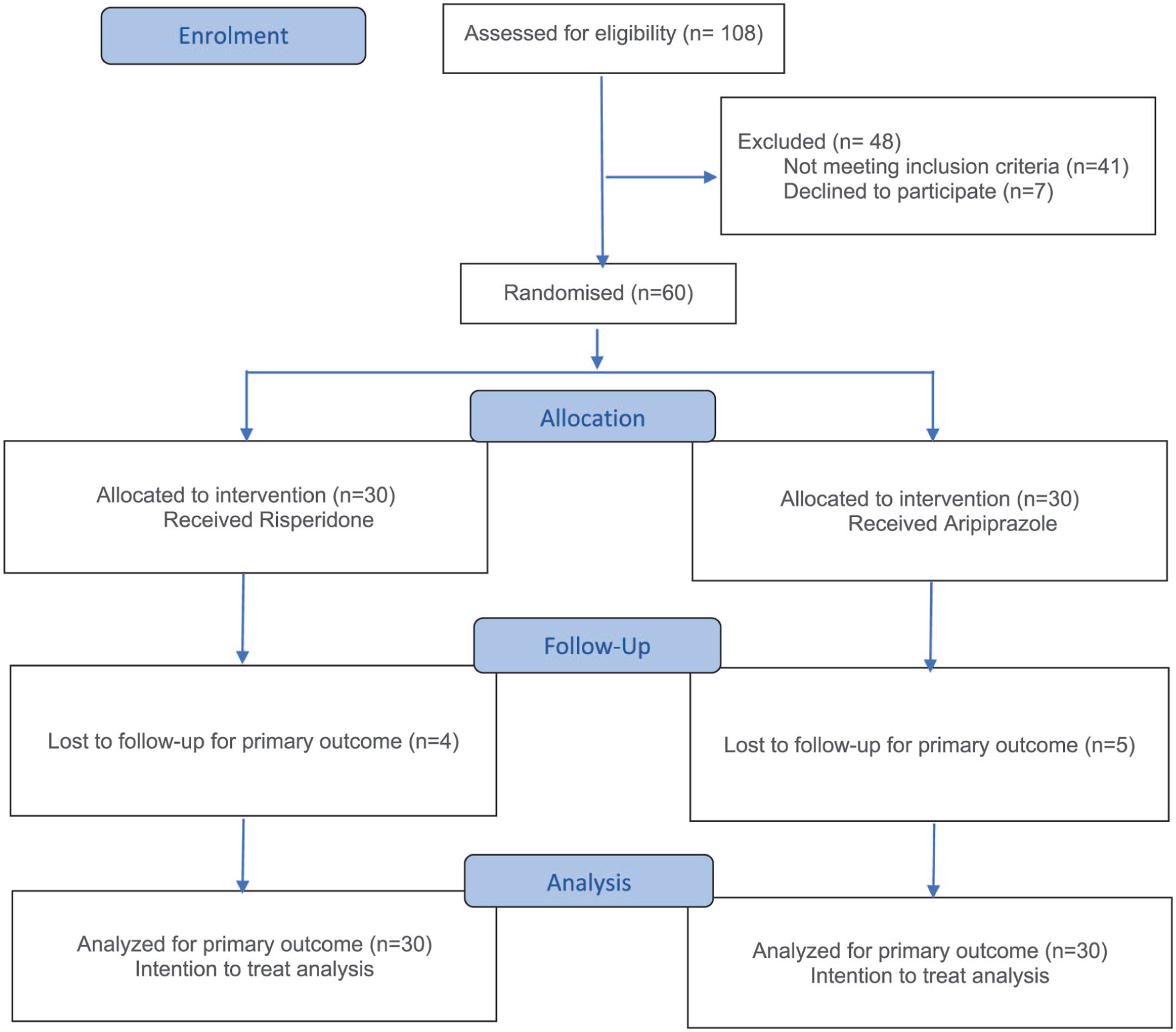
Consolidated Standards of Reporting Trials (CONSORT) flow diagram of the study.
Additionally, Kruskal–Wallis analysis within the schizophrenia group confirmed that smoking status did not significantly differ across baseline biomarker levels (MRCC I: H = 1.674, p = 0.433; Lactate: H = 0.819, p = 0.664; L:P ratio: H = 3.515, p = 0.173), further supporting that the observed group differences reflect disease-related mitochondrial dysregulation rather than smoking behavior. After adjusting for smoking status as a covariate, the baseline differences in platelet MRCC I (F(1, 87) = 49.834, p < 0.001), serum lactate (F(1, 87) = 497.206, p < 0.001), and the L:P ratio (F(1, 87) = 67.102, p < 0.001) between schizophrenia patients and healthy controls remained statistically significant, supporting the conclusion that the observed biochemical differences reflect disease-related mitochondrial dysregulation rather than smoking behavior alone. Baseline characteristics were well matched between risperidone and aripiprazole groups (Table 1). BMI did not change significantly from baseline to 12 weeks in either treatment group, indicating stable body weight during the study period.
Baseline demographic, clinical, and laboratory characteristics of treatment groups.

BMI: body mass index; PANSS: Positive and Negative Syndrome Scale; NMDAS: New Castle Mitochondrial Disease Adult Scale; MRCC: mitochondrial respiratory chain complex I.
Median (interquartile range) and Mann–Whitney U-test.
Pearson’s chi-squared test.
Mean ± SD and unpaired t-test.
Missing follow-up data occurred in 9 of 60 schizophrenia patients (15.0%), balanced between treatment arms (risperidone: 4/30; aripiprazole: 5/30). Baseline comparison of completers versus non-completers revealed that patients lost to follow-up had significantly lower baseline MRCC I concentrations, suggesting an MAR mechanism. The primary analysis used multiple imputation under the MAR assumption with 20 imputed datasets; pooled estimates and standard errors were derived using Rubin’s rules. To assess robustness, two pre-specified sensitivity analyses were performed: (1) complete case analysis restricted to participants with observed follow-up data, and (2) a conservative intention-to-treat analysis using LOCF, in which missing post-treatment values were replaced by each participant’s own baseline value.
Primary outcome measure
Change in MRCC I level
To test the hypothesis that antipsychotic treatment overall is associated with a reduction in platelet MRCC I, a linear mixed model was fitted with MRCC I as the outcome, time (baseline vs 12 weeks) and treatment group (risperidone vs aripiprazole) as fixed effects, their interaction as a test of between-drug difference, and individual patient as a random effect. The main effect of time was highly significant (β = −2.227 nmol/108 platelets, 95% CI: −3.243 to −1.211, p < 0.001), confirming that antipsychotic treatment was associated with a significant overall reduction in MRCC I concentration across both arms combined. The time × group interaction was not significant (β = −1.008, 95% CI: −2.459 to 0.443, p = 0.173). The comparable reduction in platelet MRCC I concentration observed with both risperidone and aripiprazole is consistent with a shared pharmacological influence on peripheral mitochondrial-related biomarkers; however, whether this reflects a broader class effect of atypical antipsychotics on mitochondrial respiratory chain function cannot be determined from the present study. Post-treatment level of platelet MRCC I (Table 2) declined significantly in both study groups. In the risperidone group, levels declined from 9.02 ± 3.49 to 6.36 ± 3.82, with a mean reduction of 2.66 nmol/108 platelets (95% CI: −3.71 to −1.61). The aripiprazole group showed a comparable decrease from 8.61 ± 3.19 to 5.48 ± 2.66, with a mean reduction of 3.13 nmol/108 platelets (95% CI: −4.13 to −2.13). The mean change did not differ significantly between the groups (mean difference: 0.47 nmol/108 platelets; 95% CI: −0.94 to 1.89; p = 0.508).
Change in outcome measures from baseline to 12 weeks.
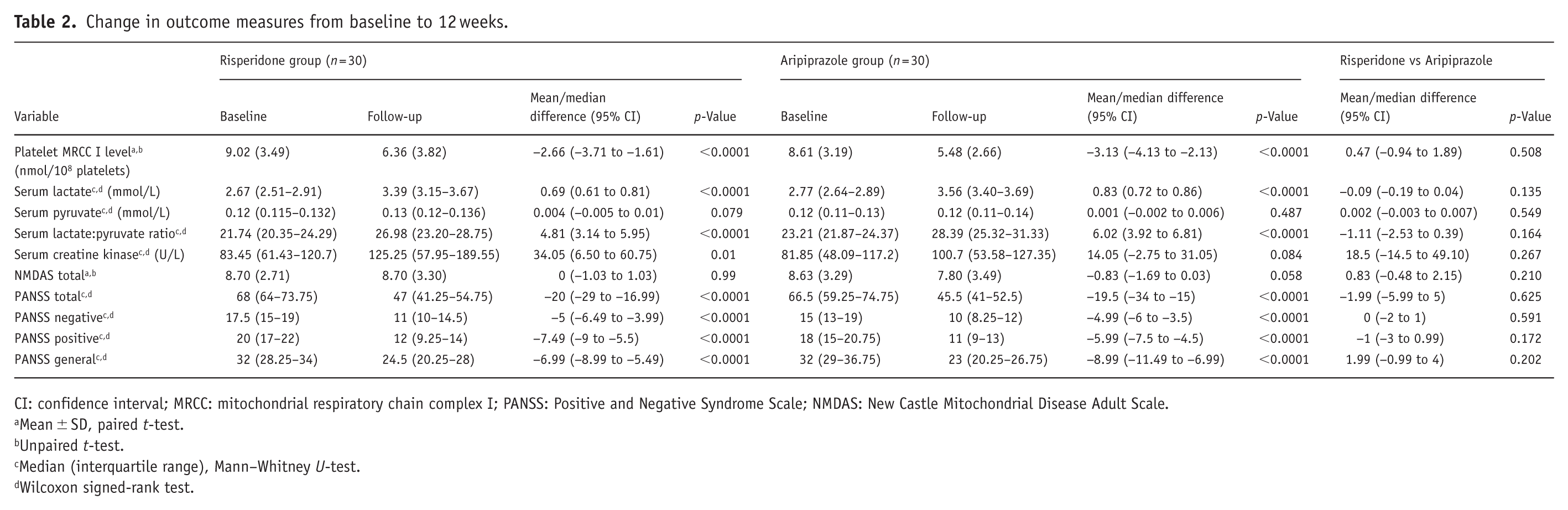
CI: confidence interval; MRCC: mitochondrial respiratory chain complex I; PANSS: Positive and Negative Syndrome Scale; NMDAS: New Castle Mitochondrial Disease Adult Scale.
Mean ± SD, paired t-test.
Unpaired t-test.
Median (interquartile range), Mann–Whitney U-test.
Wilcoxon signed-rank test.
Complete case analysis: Restricting the analysis to the 51 patients with complete data (risperidone n = 26, aripiprazole n = 25), the mean MRCC I reduction was 2.23 nmol/108 platelets in the risperidone arm (p = 0.0003) and 3.24 nmol/108 platelets in the aripiprazole arm (p < 0.0001).
LOCF analysis: As a conservative intention-to-treat analysis, missing 12-week values were imputed using each participant’s own baseline value that is, equivalent to assuming no change in MRCC I among non-completers. Under this approach, the mean MRCC I reduction was 1.93 nmol/108 platelets in the risperidone arm (p = 0.0004) and 2.70 nmol/108 platelets in the aripiprazole arm (p < 0.0001), representing a full intention-to-treat cohort of 60 patients. All three approaches yielded consistent conclusions.
Secondary outcome measures
Change in serum lactate
As shown in Table 2, serum lactate increased significantly over 12 weeks in both study groups. In the risperidone group, the median (IQR) of serum lactate increased from 2.67 (2.51–2.91) to 3.39 (3.15–3.67), with a median change of 0.69 (95% CI: 0.61–0.81). The aripiprazole group showed an increase from 2.77 (2.64–2.89) to 3.56 (3.40–3.69), with a median change of 0.83 (95% CI: 0.72–0.86). The change did not differ significantly between groups (median difference: −0.09; 95% CI: −0.19 to 0.04; p = 0.135).
Change in serum pyruvate
Serum pyruvate levels showed no significant change in either of the groups, risperidone [0.12 (0.115–0.132) to 0.13 (0.120–0.136)] or aripiprazole [0.12 (0.11–0.13) to 0.12 (0.11–0.14)]. The between-group difference was also not significant (median difference: 0.002; 95% CI: −0.003 to 0.007; p = 0.549; Table 2).
Change in serum L:P ratio
The serum L:P ratio increased significantly in both study groups. In the risperidone group, the ratio increased from 21.74 (20.35–24.29) to 26.98 (23.20–28.75), with a median change of 4.81 (95% CI: 3.14–5.95). The aripiprazole group showed a similar increase from 23.21 (21.87–24.37) to 28.39 (25.32–31.33), with a median change of 6.02 (95% CI: 3.92–6.81). Between-group comparison revealed no significant difference (median difference: −1.11; 95% CI: −2.53 to 0.39; p = 0.164; Table 2).
Change in serum creatine kinase
Serum creatine kinase increased significantly in the risperidone group from 83.45 to 125.25 U/L (median change: 34.05; 95% CI: 6.50–60.75). In the aripiprazole group, the level increased from 81.85 to 100.7 U/L, but the change was not significant (median change: 14.05 U/L; 95% CI: −2.75 to 31.05). The between-group difference was also not significant (median difference: −18.5; 95% CI: −14.5 to 49.10; p = 0.267; Table 2).
Change in NMDAS total score
No significant change in the NMDAS total score was observed in either group (Table 2). In the risperidone group, scores remained unchanged from baseline to follow-up (8.70 ± 2.71 vs 8.70 ± 3.30; mean difference: 0; 95% CI: −1.03 to 1.03). The aripiprazole group showed a statistically nonsignificant reduction from 8.63 ± 3.29 to 7.80 ± 3.49 (mean difference: −0.83; 95% CI: −1.69 to 0.03). Between-group comparison of change in scores was also not significant (mean difference: 0.83; 95% CI: −0.48 to 2.15; p = 0.210).
Change in PANSS score
All PANSS domains showed significant improvement in both groups (Table 2). In the risperidone group, PANSS total scores decreased from 68 (64–73.75) to 47 (41.25–54.75), with a median change of −20 points (95% CI: −29 to −16.99; p < 0.0001). The aripiprazole group showed a similar reduction from 66.5 (59.25–74.75) to 45.5 (41–52.5), with a median change of −19.5 points (95% CI: −34 to −15; p < 0.0001). The magnitude of improvement did not differ significantly between groups (median difference: −1.99; 95% CI: −5.99 to 5; p = 0.625). A comparable pattern of significant improvement was also observed across PANSS subscales (positive, negative, and general psychopathology) in both treatment groups, with no statistically significant differences in the magnitude of change between risperidone and aripiprazole.
Responder rates
Responder rates (⩾50% PANSS reduction) did not differ between groups: 13/26 (50%) with risperidone and 9/25 (36%) with aripiprazole, with comparable non-response rates (50% vs 64%). Pearson’s chi-square test with Yates’ correction showed no significant between-group difference (χ2 = 0.53, df = 1, p = 0.468).
Correlation analyses
Baseline MRCC I correlated inversely with the L:P ratio (ρ = −0.26, p = 0.045), with no other biochemical or clinical correlations (Supplemental Figure S1) The analysis showed a borderline negative association between ΔMRCC I and ΔPANSS (ρ = −0.27, p = 0.05). Baseline PANSS strongly predicted symptom improvement (ΔPANSS: ρ = −0.51, p < 0.001; Supplemental Figure S2). Baseline MRCC I predicted both greater post-treatment decline in MRCC I (ρ = −0.42, p = 0.002; Supplemental Figure S3) and reduced clinical improvement (ΔPANSS: ρ = 0.36, p = 0.004; Supplemental Figure S4), suggesting a potential ceiling or compensatory effect rather than causality. No significant correlations were observed between changes in MRCC I and changes in lactate, pyruvate, or L:P ratio. Baseline NMDAS correlated with Δcreatine kinase (ρ = 0.34, p = 0.02; Supplemental Figure S5). No other baseline measures were predictive (p > 0.05). Change in BMI over 12 weeks was not significantly associated with change in MRCC I (ρ = 0.133, p = 0.351), serum lactate (ρ = −0.049, p = 0.733), serum pyruvate (ρ = 0.168, p = 0.239), the L:P ratio (ρ = −0.093, p = 0.515), or creatine kinase (ρ = −0.115, p = 0.420), indicating that the observed alterations in mitochondrial-related biomarkers were independent of changes in body weight.
Regression analyses
Baseline MRCC I was the only independent predictor of change in MRCC I level (β = −0.36, p = 0.0013); treatment group showed a nonsignificant trend favoring greater reduction with aripiprazole (β = −1.00, p = 0.152). The final model was significant (F(2, 48) = 6.90, p = 0.0023) and explained 22.3% of the variance (adjusted R2 = 0.19; Supplemental Table S2). For treatment response, longer duration of illness reduced the likelihood of response (OR = 0.76 per year, p = 0.039), while no other baseline variables were predictive (Supplemental Table S3).
Safety evaluation
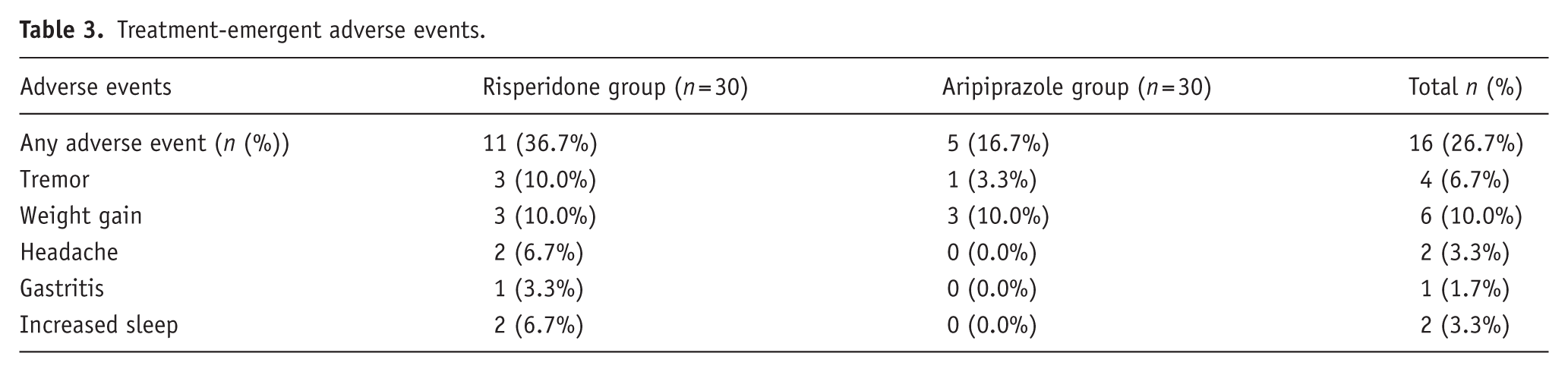
Treatment-emergent adverse events occurred in 11/30 (36.7%) risperidone and 5/30 (16.7%) aripiprazole patients, with no significant between-group difference (Fisher’s exact p = 0.143; OR = 0.35, 95% CI: 0.08–1.33). Events were mostly mild, more frequent with risperidone, and commonly included weight gain, increased sleep, tremors, headaches, and gastritis (Table 3). Tremor occurred in three risperidone-treated patients and one aripiprazole-treated patient. Median SAS score was 1.0 (IQR: 1.0–2.0; range 1–2) with risperidone and 1 in the aripiprazole case. All tremors were mild and self-limiting. SAS severity did not differ between groups (p = 0.99), though interpretation is limited by the small number of cases.
Treatment-emergent adverse events.
Discussion
This randomized, parallel-arm trial demonstrates that schizophrenia is associated with alterations in mitochondrial-related biochemical markers suggestive of disrupted cellular bioenergetics, and a metabolic shift toward anaerobic energy production. Compared with healthy controls, patients exhibited elevated platelet MRCC I levels and increased lactate-based indices, consistent with baseline bioenergetic dysregulation. Following 12 weeks of treatment with risperidone or aripiprazole, MRCC I levels declined, accompanied by increases in serum lactate and the lactate-to-pyruvate ratio, without significant changes in pyruvate or creatine kinase levels. Both treatments yielded comparable clinical improvements, stable NMDAS scores, and similar response rates. The absence of significant change in NMDAS scores despite alterations in biochemical markers highlights an important distinction between subclinical mitochondrial dysfunction and overt mitochondrial disease. NMDAS, originally developed to assess multisystem involvement in primary mitochondrial disorders, may have limited sensitivity in detecting subtle bioenergetic disturbances in schizophrenia. The low baseline scores observed in our cohort suggest minimal overt clinical burden, resulting in a potential floor effect with restricted variability, which may limit the ability to detect treatment-related changes over a relatively short follow-up period. In this context, the elevated biochemical markers likely reflect subclinical mitochondrial dysregulation that does not translate into measurable clinical impairment within 12 weeks. NMDAS was included to provide a standardized clinical framework to explore whether biochemical alterations are associated with clinically detectable mitochondrial manifestations in this population, and its findings should therefore be interpreted as exploratory. Correlation and regression analyses identified baseline MRCC I and psychopathology severity as key predictors of biochemical and clinical change, while both drugs were generally well tolerated. The absence of significant change in BMI suggests that the observed alterations in mitochondrial and metabolic parameters are unlikely to be confounded by changes in body weight.
Baseline findings align with converging evidence that schizophrenia involves disrupted mitochondrial bioenergetics (Clay et al., 2011; Fizikova et al., 2023; Moren et al., 2025; Prabakaran et al., 2004). Postmortem, transcriptomic, and proteomic studies consistently demonstrate impaired MRCC I function and oxidative phosphorylation, often accompanied by increased expression of electron transport chain components (Akarsu et al., 2014; Katz Shroitman et al., 2023; Mehler-Wex et al., 2006; Prabakaran et al., 2004; Taurines et al., 2010). This paradoxical upregulation is widely interpreted as a compensatory response to inefficient electron transfer and reduced ATP generation, rather than a preservation of mitochondrial capacity (Akarsu et al., 2014; Mehler-Wex et al., 2006; Taurines et al., 2010; Uittenbogaard and Chiaramello, 2014). Dysregulated MRCC I activity may also promote redox imbalance through impaired NADH oxidation, contributing to oxidative stress and altered cellular signaling (Kim et al., 2017; Moren et al., 2025; Prabakaran et al., 2004). Given the central role of MRCC I in initiating mitochondrial respiration via NADH oxidation and proton pumping, its dysfunction disproportionately affects high-energy-demand neurons, particularly cortical pyramidal cells that rely heavily on oxidative metabolism (Moren et al., 2025; Trigo et al., 2022). Regionally divergent alterations in MRCC I subunit expression observed in schizophrenia, with reductions in prefrontal regions and relative increases in posterior cortical areas, further support ongoing, region-specific compensatory adaptations to chronic bioenergetic insufficiency within vulnerable neural networks (Dror et al., 2002; Karry et al., 2004).
The observed elevation in lactate and the L:P ratio suggests a shift toward aerobic glycolysis under conditions of mitochondrial stress, consistent with a Warburg-like metabolic phenotype, in which cells preferentially generate energy through glycolysis, leading to increased lactate production despite adequate oxygen availability, typically due to impaired oxidative phosphorylation (Fizikova et al., 2023; Pruett and Meador-Woodruff, 2020; Rowland et al., 2016). Impaired NADH oxidation by Complex I promotes pyruvate reduction to lactate via lactate dehydrogenase, resulting in lactate accumulation despite oxygen availability (Fizikova et al., 2023; Kim et al., 2017; Pruett and Meador-Woodruff, 2020). In vivo magnetic resonance spectroscopy studies demonstrating elevated brain lactate and altered NAD⁺/NADH redox balance in schizophrenia support this interpretation and indicate compromised mitochondrial oxidative capacity (Kim et al., 2017; Rowland et al., 2016).
Accumulating evidence indicates that both typical and atypical antipsychotics can inhibit MRCC I, thereby impairing mitochondrial respiration independent of their dopaminergic effects (Anglin et al., 2012; Burkhardt et al., 1993; Chan et al., 2020; Hardy et al., 2023; Luptak et al., 2021; Modica-Napolitano et al., 2003; Prince et al., 1997; Prince et al., 1998; Scaini et al., 2013). Risperidone has been shown to reduce ATP-linked oxygen consumption, while aripiprazole inhibits electron transfer at the ubiquinone site of Complex I, disrupting proton pumping and ATP synthesis (Bar-Yosef et al., 2020; Hardy et al., 2023). Despite this bioenergetic suppression, antipsychotic efficacy is maintained, suggesting dissociable dopaminergic and mitochondrial mechanisms (De Simone et al., 2023). However, sustained mitochondrial inhibition may contribute to oxidative stress, metabolic vulnerability, and neuroprogressive processes, highlighting mitochondria as a critical interface between therapeutic benefit and long-term risk (Chan et al., 2020; De Simone et al., 2023; Moren et al., 2025).
Although the absence of a placebo arm precludes definitive causal attribution, several lines of evidence argue against spontaneous temporal decline of MRCC I as the primary explanation for the post-treatment reduction observed in this study. First, cross-sectional studies across multiple illness phases, including chronic and residual schizophrenia, have consistently documented elevated MRCC I subunit expression, suggesting that the compensatory upregulation characteristic of this disorder reflects a relatively stable bioenergetic adaptation rather than an acuity-dependent fluctuation that would be expected to normalize spontaneously over a 12-week period (Akarsu et al., 2014; Dror et al., 2002; Mehler-Wex et al., 2006). Second, the preclinical literature provides a direct pharmacodynamic basis for the observed decline, with multiple independent studies demonstrating that risperidone reduces ATP-linked oxygen consumption while aripiprazole inhibits electron transfer at the ubiquinone binding site of Complex I, both suppressing MRCC I-mediated activity (Burkhardt et al., 1993; Hardy et al., 2023; Luptak et al., 2021; Prince et al., 1997). This pharmacological mechanism is further supported by the clinical observation of Casademont et al. (2007), who reported significantly lower MRCC-I activity in risperidone-treated patients than in drug-naïve individuals. Taken together, the post-treatment decline in MRCC I is most parsimoniously interpreted as a treatment-associated longitudinal change; however, a placebo-controlled design remains necessary to establish this relationship with certainty, and the findings should continue to be interpreted accordingly.
This randomized trial has several strengths. It employed prospective randomization and included healthy controls, enabling the separation of disease-related mitochondrial changes from drug-induced effects. A comprehensive biomarker panel assessed both mitochondrial concentration (MRCC I) and metabolic indices (lactate, pyruvate, L:P ratio, and creatine kinase). Biochemical measures were evaluated alongside clinical outcomes (PANSS and NMDAS), thereby improving clinical relevance. The use of standardized laboratory methods also reduced analytical bias. Despite these strengths, certain limitations should be acknowledged. First, an important limitation of the present study is that the biochemical markers used (serum lactate, pyruvate, and L:P ratio) are indirect and nonspecific indicators of mitochondrial function and may be influenced by multiple metabolic pathways beyond oxidative phosphorylation. Similarly, platelet MRCC I concentration reflects protein abundance rather than enzymatic activity and does not directly measure mitochondrial respiratory function. The absence of significant correlations between changes in MRCC I and metabolic parameters further suggests that these measures may capture related but distinct aspects of cellular bioenergetics. Therefore, the findings should be interpreted as indicative of altered bioenergetic status rather than definitive evidence of impaired mitochondrial respiratory chain function. Second, the follow-up duration was limited to 12 weeks, which may not fully capture the long-term trajectory of mitochondrial changes with continued antipsychotic exposure; however, this time frame is adequate to detect early biochemical adaptations associated with treatment initiation. Third, platelet-based measures were used as peripheral surrogates of mitochondrial function and may not directly reflect central nervous system bioenergetics. Platelets share key bioenergetic features with neurons, including reliance on oxidative phosphorylation and expression of MRCCs and have been used as accessible indicators of systemic mitochondrial alterations in schizophrenia.(Casademont et al., 2007; Dror et al., 2002). However, unlike neurons, platelets lack synaptic activity, regional specialization, and the complex metabolic demands of neural circuits. Moreover, mitochondrial dysfunction in schizophrenia may exhibit region- and cell-type-specific variability within the brain, which cannot be captured by peripheral measures. Therefore, platelet MRCC I should be interpreted as a surrogate marker of systemic bioenergetic status rather than a direct proxy for central mitochondrial function. Future studies integrating peripheral biomarkers with neuroimaging modalities, such as magnetic resonance spectroscopy or PET-based metabolic assessments, may help bridge this gap. Fourth, direct assessments of MRCC I enzymatic activity, oxidative stress markers, mitochondrial DNA parameters, and high-resolution respiratory flux analyses were not performed; nevertheless, the combined evaluation of MRCC I concentration with metabolic indices, such as lactate and the L:P ratio, provides convergent evidence of altered bioenergetic status, although these measures do not directly assess mitochondrial respiratory function. Fifth, treatment adherence was assessed using the pill-counting method at follow-up, which, although more objective than self-report, may still overestimate adherence due to factors such as pill dumping or non-ingestion despite removal from packaging. Sixth, healthy controls were not followed longitudinally; therefore, potential nonspecific temporal effects such as changes related to time, diet, or lifestyle factors could not be fully accounted for. Seventh, pharmacogenomic, lifestyle, and metabolic modifiers were not examined, which may influence individual variability in mitochondrial responses. However, randomization and comparable baseline characteristics between groups help mitigate potential confounding effects. These findings should therefore be interpreted as reflecting alterations in bioenergetic status rather than direct evidence of impaired mitochondrial respiratory function. Finally, we acknowledge that normalization to total protein or citrate synthase activity, a surrogate for mitochondrial mass, would have allowed more precise per-mitochondrion quantification and could not be done due to resource constraints. This remains a recommendation for future studies. Additionally, multiple secondary and exploratory analyses were performed without pre-registration of a formal correction hierarchy; findings from these analyses should be considered hypothesis-generating and interpreted in the context of uncorrected p-values.
Future studies should include more extended follow-up periods to clarify the progression of mitochondrial changes with sustained antipsychotic treatment. The use of advanced neuroimaging techniques, such as 31P-MRS, lactate spectroscopy, and PET-based metabolic measures, may help link peripheral biomarkers with central nervous system bioenergetics. Evaluating a broader range of antipsychotic agents and investigating mitochondria-protective adjunctive strategies, including pharmacological and lifestyle interventions, could help reduce bioenergetic stress. Together, these approaches may advance bioenergetics-informed, precision psychiatry that balances effective symptom control with preservation of neuronal energy metabolism.
Conclusion
In conclusion, this randomized trial demonstrates that schizophrenia is associated with alterations in peripheral mitochondrial-related biomarkers suggestive of disrupted cellular bioenergetics, and treatment with risperidone and aripiprazole is associated with further changes in these markers. Risperidone and aripiprazole produced comparable clinical improvement but were associated with biochemical shifts consistent with altered cellular bioenergetics and increased reliance on anaerobic metabolism. Baseline psychopathology and MRCC I predicted treatment-related changes, highlighting a potential bioenergetic trade-off in antipsychotic therapy and supporting the need for mitochondria-informed treatment strategies in schizophrenia. Future studies incorporating direct functional assays of mitochondrial activity, such as enzymatic measurements of complex I activity, high-resolution respirometry, or metabolic flux analyses, are required to validate and extend these findings.
Supplemental Material
sj-docx-1-jop-10.1177_02698811261453937 – Supplemental material for Mitochondrial dysfunction in schizophrenia and its modulation by atypical antipsychotic drugs: A randomized controlled trial
Supplemental material, sj-docx-1-jop-10.1177_02698811261453937 for Mitochondrial dysfunction in schizophrenia and its modulation by atypical antipsychotic drugs: A randomized controlled trial by Amiya Shaju, Biswa Ranjan Mishra, Debadatta Mohapatra, Archana Mishra, Anand Srinivasan and Rituparna Maiti in Journal of Psychopharmacology
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The first author received funding from the Department of Health Research, Ministry of Health and Family Welfare, Government of India vide grant number HRD/DHR-ICMR/PG-2024/0495.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.