
Abstract
Background:
Oral S-ketamine (S-KETPO) is being explored as an alternative to intravenous maintenance treatment (S-KETIV) in treatment-resistant depression (TRD). However, the first-pass effect with S-KETPO significantly alters S-ketamine’s bioavailability and the systemic exposure of its active metabolites, raising potential safety and efficacy concerns.
Aims:
This study characterized the pharmacodynamic and safety profiles of S-KETPO related to its pharmacokinetics in healthy participants, compared head-to-head with an S-KETIV dose that had previously demonstrated antidepressant effects in TRD .
Methods:
In a randomized, double-blind, placebo-controlled, double-dummy, four-way cross-over study, 16 healthy participants received a single dose of S-KETPO 0.20 and 0.45 mg/kg, S-KETIV 0.40 mg/kg and placebo. Plasma concentrations of S-KET and its active metabolites norketamine (S-NOR) and S-hydroxynorketamine (S-HNK) were measured, safety assessments were conducted up to 24 hours post-dose, and central nervous system (CNS) effects were evaluated up to 6 hours post-dose.
Results:
Absolute bioavailability of S-KETPO was poor (9–12%). Peak plasma concentrations for S-KETPO 0.20 mg/kg, 0.45 mg/kg and S-KETIV were 9.81, 22.7 and 146 ng/mL (S-KET); 62.0, 127 and 55.2 ng/mL (S-NOR); and 29.5, 62.1 and 32.2 ng/mL (S-HNK), respectively. S-KETPO S-NOR:S-KET and S-HNK:S-KET metabolite-to-parent compound ratios were 7.52, 6.98 and 0.42, and 3.78, 3.71 and 0.23, for KETPO 0.20 mg/kg, S-KETPO 0.45 mg/kg, and S-KETIV, respectively. S-KETIV produced sedative, psychomotor and psychotomimetic effects coinciding with reductions in quantitative electroencephalography (qEEG) alpha, beta and delta power, whereas S-KETPO 0.45 mg/kg demonstrated inconsistent effects on vigilance and arousal but clear albeit relatively smaller reductions in qEEG alpha, beta and delta power coinciding with limited psychotomimetic effects, and finally, KETPO 0.20 mg/kg lacked effects altogether. Safety was comparable across treatments.
Discussion and Conclusion:
Oral administration of S-ketamine alters pharmacokinetic and pharmacodynamic profiles, resulting in poor bioavailability, increased pharmacologically active metabolite exposures, and limited CNS effects relative to intravenous administration. The current findings have implications for S-KETPO dose selection, which may impact safety and/or therapeutic outcomes in future TRD studies.
Trial Registration:
The study was registered in the ‘Overview of Medical Research in the Netherlands’ (OMON) under NL-OMON55261.
Keywords
Introduction
Major depressive disorder (MDD) is a common mental disorder that significantly contributes to the burden of disease worldwide (Proudman et al., 2021; Yan et al., 2024). Despite a broad range of effective antidepressant drugs (ADs) available on the market, up to one-third of patients do not achieve adequate symptomatic response or remission and, as a consequence, are considered treatment resistant (Matveychuk et al., 2020; Rush et al., 2006; Smith-Apeldoorn et al., 2022a). Treatment-resistant depression (TRD) is generally defined as insufficient response to at least two ADs with different mechanisms of action (MoA), administered at a therapeutic dose and for a duration of several weeks (Gaynes et al., 2020; Matveychuk et al., 2020; Nemeroff, 2007). Since the inefficacy of currently available ADs renders patients at risk of a chronic disease course and/or complications such as suicide, a significant need for more efficacious ADs currently exists. Since most conventional ADs modulate monoaminergic neurotransmitter pathways, novel ones acting via alternative MoAs may benefit TRD patients, and as a result, are currently a subject of interest to clinicians and drug developers alike.
The non-selective, non-competitive glutamate N-Methyl-
In a recent dose-ranging study in TRD, S-KET 0.20 and 0.40 mg/kg administered IV demonstrated similar antidepressant efficacy and time to depressive relapse following response (Singh et al., 2016). However, as 0.20 mg/kg was associated with superior tolerability, while maintaining similar efficacy, future S-KET maintenance treatment in the form of repeated IV dosing schemes following response to an initial IV dose seems a plausible approach. However, since repeated IV drug administrations are burdensome to patients and limit scalability due to logistical complexity, both wide-spread and patient-friendly access to S-KET will require an alternative, viable strategy. For that reason, several research groups are investigating the potential of orally administered S-KET as a patient-friendly and scalable alternative to IV maintenance treatment (Smith-Apeldoorn et al., 2019). This approach is arguably also supported by recent data on an immediate-release oral KET formulation showing no unexpected safety signals and key pharmacokinetic (PK) parameters for KET and its primary metabolites to be dose proportional (Qureshi et al., 2025). However, the application of orally administered S-KET as potential AD treatment is currently precluded by inadequate systematic characterization of its PK and pharmacodynamic (PD) characteristics in controlled clinical trials, and in addition, its ease of administration also necessitates careful evaluation of its safety profile, including abuse and dependence liability.
The clinical pharmacokinetics (PK) of IV administered S-KET has been extensively described elsewhere (Fanta et al., 2015; Zanos et al., 2018). In summary, IV administered S-KET is rapidly distributed as a result of its relatively high lipophilicity, and readily eliminated (Dinis-Oliveira, 2017; Zanos et al., 2018). Its metabolism primarily occurs via cytochrome P450 (CYP) 3A4 and, to a lesser extent via CYP2B6 and CYP2C9, converting S-KET into its active metabolites S-norketamine (S-NOR), S-dehydronorketamine, and S-hydroxynorketamine (S-HNK) (Hijazi and Boulieu, 2002). In addition, a small amount is excreted unchanged in urine (Dinis-Oliveira, 2017; Zanos et al., 2018). Following IV administered S-KET, S-KET and S-NOR demonstrate elimination half-lives (t1/2el) of 5–7 and 6–10 hours, respectively (Peltoniemi et al., 2012). Since S-KET’s pharmacologically active metabolites S-NOR and S-HNK seem to contribute to its antidepressant-like effects in preclinical models, alterations in PK characteristics impacting the metabolite-to-parent compound ratios may affect the efficacy of S-KET when being administered PO instead of IV in humans (Zanos et al., 2018). In fact, when administered PO in humans, S-KET undergoes extensive first-pass metabolism, resulting in S-NOR plasma exposures predicted to be up to 16.5 times higher than those of S-KET following multiple dosing (Fanta et al., 2015). This contrasts with IV administration, following which S-KET and S-NOR exposures are essentially inverted (Fanta et al., 2015). Moreover, considering the disparate NMDA receptor affinities and potencies for S-KET, S-NOR, and S-HNK (see Supplemental Table S1 for a summary of NMDA receptor affinities and potencies Moaddel et al., 2012; Zanos et al., 2018), changes to S-KET’s PK may alter its PD characteristics, and, as a consequence, potentially also its clinical safety and/or efficacy profiles in TRD. Together, these data suggest that orally administered S-KET may demonstrate divergent PD and/or safety profiles compared with IV administered S-KET, necessitating its detailed characterization in relation to its PK in humans prior to its broader application in TRD.
The PD profile of IV administered S-KET has previously been extensively characterized in healthy participants (Kleinloog et al., 2015) using the validated Neurocart central nervous system (CNS) test battery (Groeneveld et al., 2016). S-KET administered IV to achieve individualized pseudo-steady state plasma concentrations targeted at 120 and 240 ng/mL, demonstrated robust and predominantly exposure-dependent effects on saccadic peak velocity (SPV), smooth pursuit eye movements, adaptive tracking, body sway and visual analogue scales (VAS) for subjective drug effects (Kleinloog et al., 2015). Moreover, these PD effects aligned with plasma concentrations over time and resolved within maximally 2 hours after discontinuation of the infusion. Since Neurocart has been shown to be sensitive to the CNS effects of S-KET in healthy humans, it represents a reliable, non-invasive tool to characterize its PD in relation to its PK. In addition, resting-state quantitative electroencephalography (qEEG) has been proposed as a pharmacodynamic biomarker sensitive to the effects of S-KET (De la Salle et al., 2022).
The current study characterized orally administered S-KET’s PD and safety profiles in relation to its PK in healthy participants, and compared these directly to an established efficacious antidepressant dose of IV administered S-KET in TRD. As the first-pass effect was anticipated to significantly impact orally administered S-KET’s PK, systematic pharmacological characterization is considered a crucial basis for adequate design and interpretation of future studies with orally administered S-KET in TRD.
Methods
Study design and participants
Sixteen healthy male and female participants, 18–45 years of age, were recruited via media advertisements or from the participants’ database of the Centre for Human Drug Research (CHDR). Participants were selected following a medical screening to be included in a randomized, double-blind, double-dummy, placebo-controlled, four-way cross-over study receiving S-KET per os (i.e., orally administered; S-KETPO), 0.20 mg/kg, S-KETPO 0.45 mg/kg, S-KETIV (i.e., IV administered S-KET) 0.40 mg/kg, or placebo. No prescription medication and over-the-counter medication were permitted within 14 days prior to study drug administration, or less than 5 half-lives (whichever was longer), and during the course of the study. Participants were excluded if they had a positive urine test for drugs of abuse and/or pregnancy at screening or pre-dose, a (family) history or symptoms of any significant medical disease or psychiatric disorder, or regular recreational use of illicit drugs within 12 months of screening.
Each participant provided written informed consent before any screening procedures were performed. The study was conducted from August 2021 to March 2022 at CHDR in Leiden, The Netherlands. The study was approved by the BEBO Foundation for the Assessment of Ethics of Biomedical Research (Assen, The Netherlands), was conducted according to the Dutch Act on Medical Research Involving Human Subjects (WMO) and in compliance with all International Conference on Harmonisation-Good Clinical Practice (ICH-GCP) guidelines and the Declaration of Helsinki. This study was registered in the ‘Overview of Medical Research in the Netherlands’ (OMON) under NL-OMON55261.
Study procedures and randomization
All participants underwent screening to ensure eligibility criteria, followed by CNS battery training and four in-clinic periods of approximately 3 days, separated by a wash-out period of 14–21 days, during which participants returned for a visit on day 7 (+3 days) post-dose during which PD-measurements were performed. A final follow-up visit took place at least 14 days after the last dose. Medical screening was performed maximally 42 days before the first dose, consisting of medical and psychiatric history, physical examination, blood chemistry and haematology, urinalysis and electrocardiogram (ECG). Upon admission for each in-clinic period (day 1), an alcohol breath, urine pregnancy and drug test were performed, and eligibility was reconfirmed. At regular intervals, PD and safety assessments and plasma PK sampling were performed. After inclusion participants were randomized in a consecutive order starting with the lowest number using SAS 9.4 for Windows or newer (SAS Institute Inc., Cary, NC, USA) by an unblended, study-independent statistician. The sequence of drug administration was randomized for each participant via a William’s square design using a block size of 4.
Investigational drugs
S-KET was sourced as Ketanest-S from Pfizer B.V. (Capelle a/d IJssel, The Netherlands). S-KETPO and S-KETIV were administered as single doses of a 0.20 and 0.45 mg/kg oral solution, and 0.40 mg/kg IV over 40 minutes, respectively. Participants drank the S-KETPO solution or the placebo saline solution, which was swallowed in a single go. To mask the difference in taste between S-KETPO and the oral placebo condition, a strong-tasting mint strip was administered prior and after oral dosing. A double-dummy placebo was administered as a saline infusion over 40 minutes or a matching oral solution. In order to align the expected time to maximum concentration (Tmax) for S-KETPO and S-KETIV, S-KETPO administrations were performed 15 minutes after the start of IV administration.
Dose rationale
S-NOR is S-KET’s primary pharmacologically active metabolite and has been demonstrated to contribute to antidepressant-like effects in preclinical studies (Swainson et al., 2019; Yang et al., 2018; Yokoyama et al., 2020; Zanos et al., 2018). While both S-KET and S-NOR have previously been demonstrated to antagonize NMDAR, the precise cellular target for S-HNK still remains elusive, although converging evidence supports enhanced signalling of the α7 nicotinic acetylcholine receptor (α7nAChR; Guhathakurta et al., 2024). Changes in S-KET, S-NOR, and/or S-HNK exposure ratios for S-KETPO in comparison to S-KETIV as a result of first-pass metabolism may result in relevant differences in terms of antidepressant efficacy and/or safety in humans (Fanta et al., 2015; Smith-Apeldoorn et al., 2019). S-KETIV 0.40 mg/kg administered over 40 minutes was selected since it demonstrated antidepressant efficacy in TRD, and furthermore, was expected to reach exposures comparable to the 120 ng/mL targeted infusion administration in a previous clinical study that demonstrated robust PD effects. S-KETPO 0.45 mg/kg was predicted to reach maximum S-NOR plasma exposures (Cmax) similar to a multiple dose S-KETPO study in TRD, which was being conducted elsewhere, while 0.20 mg/kg ensured S-NOR Cmax exposures approximately similar to those expected to be attained with 0.40 mg/kg S-KETIV (Fanta et al., 2015; Smith-Apeldoorn et al., 2019).
Dose selection was based on simulated S-KET and S-NOR concentrations of 1000 virtual participants (assumed weight distribution: mean 70 kg, standard deviation (SD): 10) using two population PK models, the development of which was based on in-house data following IV administration (Otto et al., 2021) and, as no PO data were available, from the literature (Fanta et al., 2015). A single 0.40 mg/kg IV dose, a single 0.45 mg/kg PO-dose and a single 0.20 mg/kg PO-dose were simulated, resulting in a factor ~27 higher mean ± SD S-KET Cmax (126 ± 23.7 ng/mL) for S-KETIV compared to S-KETPO 0.20 mg/kg (4.63 ± 1.92 ng/mL), whereas mean S-NOR Cmax was comparable for both treatments (60.7 ± 13.7 and 60.5 ± 17.9 ng/mL, respectively) as illustrated in Supplemental Figure S2 (Fanta et al., 2015; Otto et al., 2021). In addition, mean S-NOR Cmax for S-KETPO 0.45 mg/kg (136 ± 40.2 ng/mL) was up to a factor ~2 higher in comparison to both S-KETIV and S-KETPO 0.20 mg/kg with more than doubled S-KET Cmax (10.4 ± 4.33 ng/mL) as compared to S-KETPO 0.20 mg/kg (Fanta et al., 2015; Otto et al., 2021). Hence, comparing S-KETIV 0.40 mg/kg with both S-KETPO 0.20 mg/kg and 0.45 mg/kg was expected to shed light on the differential PD effects of S-KET and S-NOR.
Pharmacokinetic assessments and analysis
PK samples were collected pre-dose and at approximately 15 minutes, 45 minutes, 1 hour, 1 hour 30 minutes, 2 hours, 2 hour 30 minutes, 3 hours, 4 hours, 6 hours, 9 hours, and 24 hours after start of the IV infusion. As oral administration occurred 15 minutes later, these sampling time points correspond to −15 minutes relative to the oral dosing time. Approximately 2 mL blood per sample were collected via an IV catheter placed in the contralateral antecubital vein in K2EDTA tubes for assessment of S-KET, S-NOR and S-HNK PK parameters in plasma. The indwelling catheter was kept patent by saline flush after each blood sampling. Within 30 minutes of collection, the tubes were centrifuged at approximately 2000g for 10 minutes at 2°C–8°C. The plasma was transferred into two labelled polypropylene tubes. All samples were stored in an upright position at −20°C or lower. Samples were shipped on dry ice to Ardena, Assen, the Netherlands in a single shipment and stored at a temperature ⩽ −18°C. A detailed description of the LC–MS/MS assay, including sample preparation, instrumentation, and validation results (within- and between-run precision and accuracy), is provided in the Supplemental Tables S2 and S3.
Pharmacodynamic assessments
Neurocart
The Neurocart is an integrated test battery consisting of assessments sensitive to the PD effects of CNS-active drugs for a wide range of CNS domains. It was performed pre-dose and approximately 20 minutes, 50 minutes, 1 hour 20 minutes, 2 hours, 4 hours, 6 hours, 24 hours and 7 days after start of the IV infusion and oral administration. The results related to S-KET’s potential sustained PD effects up to 24 hours and 7 days following a single administration, will be reported elsewhere since this report focusses on the effects of direct exposure, up to 6 hours. Assessed Neurocart parameters were SPV, smooth pursuit eye movements, adaptive tracking, body sway, VAS Bond and Lader and VAS Bowdle, based on their sensitivity for measuring drug-induced CNS depression (Van Steveninck et al., 1991, 1999), distinct drug-induced changes in the eye movement system, postural instability and subjective effects of CNS-active drugs including subjective alertness, mood, calmness, and psychotomimetic effects, respectively (Kleinloog et al., 2015). The symbol-digit substitution test, a computerized version of the digital symbol substitution test (DSST), was used to assess effects on processing speed, executive function and working memory (Jaeger, 2018).
Resting-state qEEG recording
Resting-state qEEG was recorded in accordance with IPEG guidelines, using 5-minute alternating periods of eyes closed and eyes open every 64 seconds (Jobert et al., 2012). Frequency bands of interest included delta, theta, alpha, beta and gamma bands.
Mystical experiences questionnaire (MEQ30)
The MEQ30 was developed during an online survey on psilocybin-containing mushrooms and validated using data from experimental studies with controlled doses of psilocybin (Barrett et al., 2015). The revised version contains 30 items regarding subjective drug effects and is completed retrospectively. The questionnaire was translated into Dutch using standard CHDR operating procedures. Effects were scored in total and on four subdomains (mystical, positive mood, space/time, ineffability) pre-dose and approximately 8 and 24 hours after start of the IV infusion.
Safety evaluations
Clinical laboratory values (haematology, biochemistry and urinalysis), vital signs, and 12-lead ECGs were assessed at regularly predefined intervals up to 24 hours after the start of the IV infusion. Physical examinations and the Columbia-Suicide Severity Rating Scale (C-SSRS) were performed. All treatment-emergent adverse events (TEAEs) including serious adverse events (SAEs) were recorded.
Statistical methodology
A sample size of 16 participants was considered sufficient to characterize the PD effects of S-KET using Neurocart (Kleinloog et al., 2015). Participants who completed at least the first three out of four treatment periods were considered completers. Additional participants could be enrolled if a participant missed one or more of the four treatment periods. To establish significant treatment effects for the repeatedly measured PD parameters, each parameter was analyzed with a mixed model analysis of covariance (ANCOVA), using SAS 9.4 for Windows or newer (SAS Institute Inc.). In the ANCOVA model, treatment, time, period and treatment by time were fixed factors, participant, participant by treatment and participant by time were random factors and the (average) baseline measurement was included as covariate. Single-measured PD parameters were analyzed with a mixed model analysis of variance with treatment and period as fixed factors and participant as a random factor. In addition, the Kenward-Roger approximation was used to estimate denominator degrees of freedom and model parameters were estimated using the restricted maximum likelihood method. The general treatment effect and specific contrasts using measurements up to 6 hours post-dose on day 1 (S-KETPO 0.20 mg/kg vs placebo, S-KETPO 0.45 mg/kg vs placebo and S-KETIV 0.40 mg/kg vs placebo) were reported with the estimated difference and the 95% confidence interval (CI), the least square mean (LSM) estimates and the p-value.
Non-compartmental PK analysis was performed for S-KET, S-NOR, and S-HNK up to 24 hours post-dose using the PKNCA package in R(V4.0.3; Buckeridge et al., 2015; R Core Team, 2023). Calculations were based on the actual time relative to administration, that is, time to maximum concentration (Tmax) did not include the 15-minute time difference between administration of S-KETIV and S-KETPO. The trapezoidal linear-up and log-down method was used for calculation of the area under the curve. Elimination half-life was determined by linear regression of the terminal part of the log-concentration time curve, with a minimal r2 of 0.85 and covering a time interval of at least 1.5 times the estimated half-life, with a minimum of three datapoints after the maximum concentration had been reached. Absolute clearance (CL) and apparent clearance (CL/F) were calculated by dividing the administered dose by the area under the curve from time zero extrapolated to infinity, if available (i.e., terminal half-life regression was successful). CL/F was calculated for S-KET after S-KETPO dosing, where F represents the unknown fraction absorbed, that is, absolute bioavailability. CL/F was also calculated for S-NOR and S-HNK after S-KETIV administration, where F represents the unknown fraction metabolized. Volume of distribution during the terminal elimination phase was calculated by dividing the (apparent) clearance by the terminal first-order elimination rate constant. Volume of distribution at steady state for intravenous administration was determined by multiplying the mean residence time with absolute clearance. PK parameters were summarized by arithmic and geometric mean, SD, and coefficient of variation (CV), as well as median and range, with the exception of Tmax and Tlag, for which only the median and range were determined as they are dependent on scheduled sampling timepoints and thus considered non-continuous variables.
Results
Demographics
Sixteen participants (eight males and eight females) completed the study. Mean (min-max) age and body mass index were 26.9 (21–35) years and 22.5 (19.4–27.2) kg/m2, respectively. Supplemental Figure S1 depicts the CONSORT flow diagram. One participant discontinued after the second treatment period and was replaced according to the study protocol, resulting in data being available for minimally 16 and maximally 17 participants.
Pharmacokinetics
The average time-concentration profiles up to 24 hours post-dose for S-KET, S-NOR, and S-HNK following S-KETPO and S-KETIV are presented in Figure 1. Table 1 summarizes key PK parameters for S-KET, S-NOR, and S-HNK following S-KETPO 0.20 mg/kg, S-KETPO 0.45 mg/kg, and S-KETIV. Following oral administration, S-KET concentrations did not exceed the lower limit of quantification during the terminal phase of the PK profile. Consequently, calculation of absolute bioavailability (F) was only possible based on the area under the curve from the time of dosing to the last measurable concentration (AUClast), and no S-KET terminal half-life (t1/2) or t1/2 derived parameters (e,g., Vz, CL) could be determined for either S-KETPO 0.20 or 0.45 mg/kg, resulting in missing PK parameters.
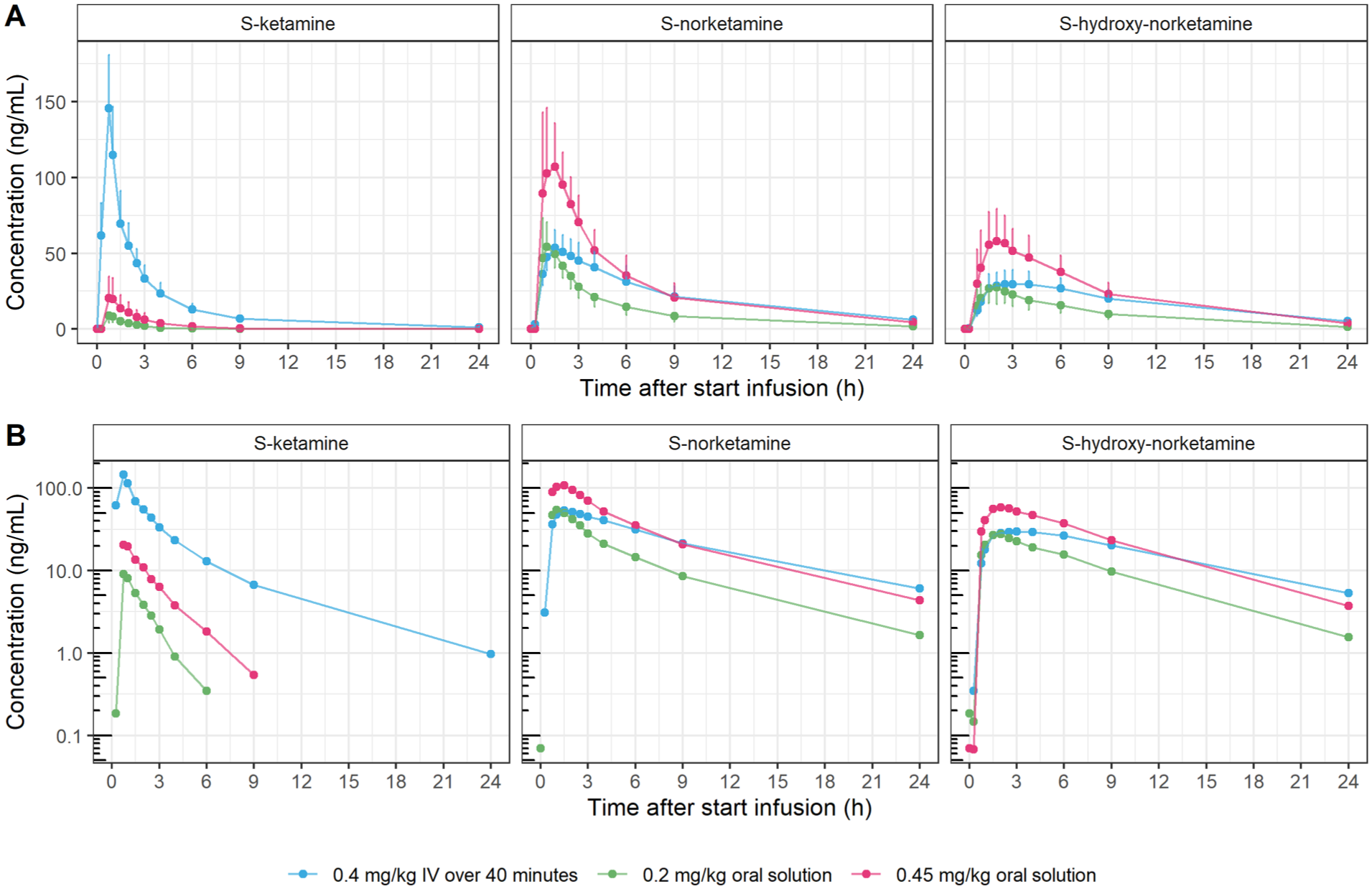
Mean S-KET, S-NOR and S-HNK plasma time-concentration profiles up to 24 hours post-dose following administration of S-KETPO 0.20 mg/kg, S-KETPO 0.45 mg/kg, and S-KETIV 0.4 mg/kg. Mean S-KET, S-NOR, and S-HNK plasma time-concentration profiles up to 24 hours post-dose for oral S-ketamine 0.20 mg/kg, 0.45 mg/kg and intravenous S-ketamine. Concentrations are presented (a) on a linear scale (with uni-directional error bars representing standard deviation); and (b) on a logarithmic scale. Error bars = standard deviation, blue dot = S-KETIV 0.40 mg/kg; pink dot = S-KETPO 0.45 mg/kg; green dot = S-KETPO 0.20 mg/kg.
Pharmacokinetic parameters for S-KET, S-NOR, and S-HNK in plasma up to 24 hours post-dose following administration of S-KETPO 0.20 mg/kg, S-KETPO 0.45 mg/kg, and S-KETIV 0.4 mg/kg.
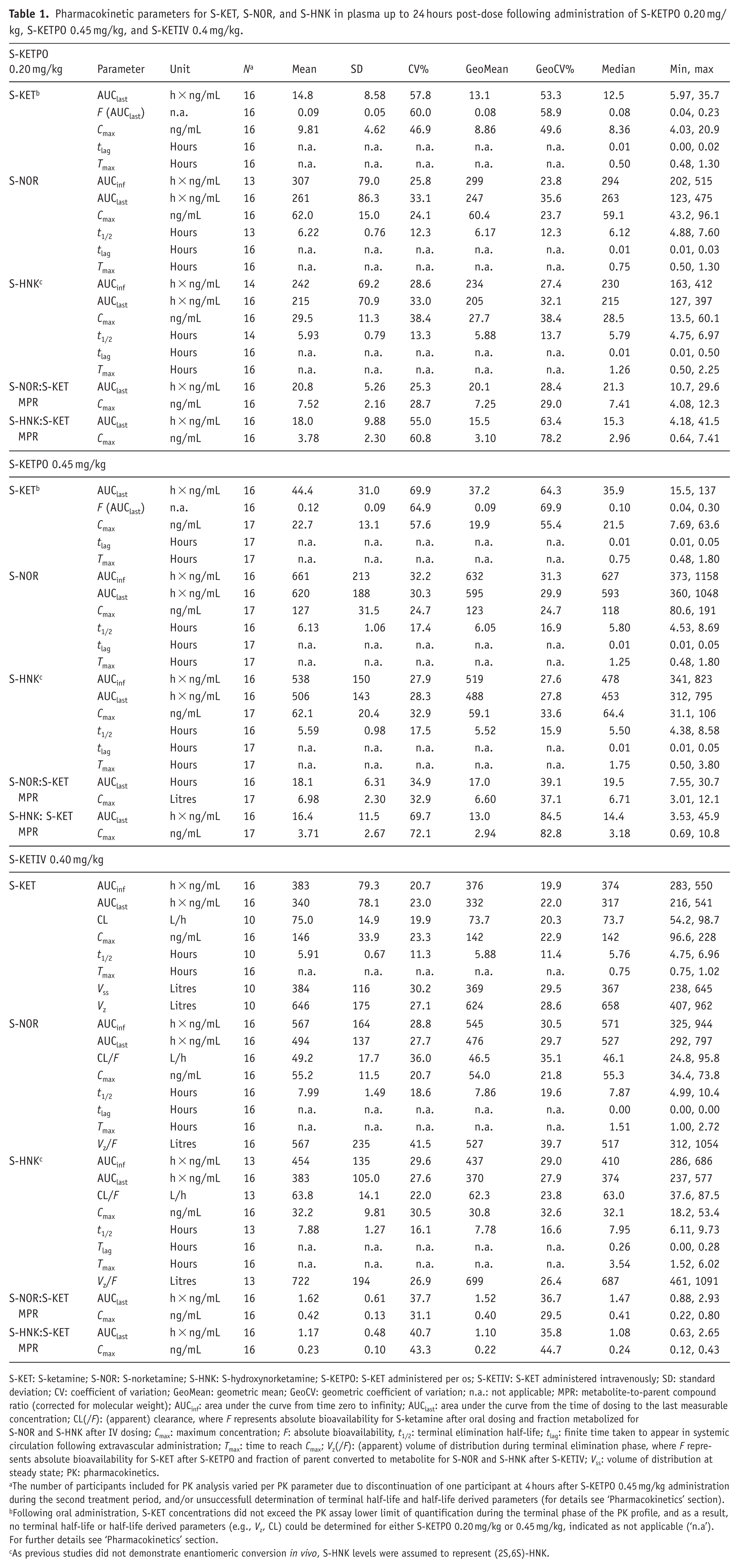
S-KET: S-ketamine; S-NOR: S-norketamine; S-HNK: S-hydroxynorketamine; S-KETPO: S-KET administered per os; S-KETIV: S-KET administered intravenously; SD: standard deviation; CV: coefficient of variation; GeoMean: geometric mean; GeoCV: geometric coefficient of variation; n.a.: not applicable; MPR: metabolite-to-parent compound ratio (corrected for molecular weight); AUCinf: area under the curve from time zero to infinity; AUClast: area under the curve from the time of dosing to the last measurable concentration; CL(/F): (apparent) clearance, where F represents absolute bioavailability for S-ketamine after oral dosing and fraction metabolized for S-NOR and S-HNK after IV dosing; Cmax: maximum concentration; F: absolute bioavailability, t1/2: terminal elimination half-life; tlag: finite time taken to appear in systemic circulation following extravascular administration; Tmax: time to reach Cmax; Vz(/F): (apparent) volume of distribution during terminal elimination phase, where F represents absolute bioavailability for S-KET after S-KETPO and fraction of parent converted to metabolite for S-NOR and S-HNK after S-KETIV; Vss: volume of distribution at steady state; PK: pharmacokinetics.
The number of participants included for PK analysis varied per PK parameter due to discontinuation of one participant at 4 hours after S-KETPO 0.45 mg/kg administration during the second treatment period, and/or unsuccessfull determination of terminal half-life and half-life derived parameters (for details see ‘Pharmacokinetics’ section).
Following oral administration, S-KET concentrations did not exceed the PK assay lower limit of quantification during the terminal phase of the PK profile, and as a result, no terminal half-life or half-life derived parameters (e.g., Vz, CL) could be determined for either S-KETPO 0.20 mg/kg or 0.45 mg/kg, indicated as not applicable (‘n.a’). For further details see ‘Pharmacokinetics’ section.
As previous studies did not demonstrate enantiomeric conversion in vivo, S-HNK levels were assumed to represent (2S,6S)-HNK.
As expected, S-KET absorption was poor following oral administration, with a limited absolute oral bioavailability of 9–12% that was also highly variable (range 4–30%). Consistent with this, S-KET mean maximal plasma concentrations (Cmax) were higher for S-KETIV compared with S-KETPO (Cmax 146 ng/mL vs 9.81and 22.7 ng/mL for S-KETIV, S-KETPO 0.20 mg/kg, and KETPO 0.45 mg/kg, respectively). In contrast, the opposite was observed (ie. Cmax was relatively higher for S-KETPO compared with S-KETIV) for S-NOR and S-HNK (Cmax 62.0 and 29.5, 127 and 62.1, and 55.2 and 32.2 ng/mL, for S-KETPO 0.20 mg/kg, KETPO 0.45 mg/kg and S-KETIV, respectively), which resulted in significantly altered mean S-NOR to S-KET (S-NOR:S-KET) and S-HNK to S-KET (S-HNK:S-KET) metabolite-to-parent compound ratios (MPRs) of 7.52, 6.98 and 0.42, and 3.78, 3.71 and 0.23, for KETPO 0.20 mg/kg, S-KETPO 0.45 mg/kg, and S-KETIV, respectively. Time-to-maximal plasma concentration (Tmax) showed rapid absorption for S-KET following S-KETPO administration, with a median of 0.5 (range 0.48–1.20) and 0.75 hours (0.48–1.80 hours), largely overlapping with Tmax of S-KETIV at median 0.75 (0.75–1.02). Peak S-NOR levels closely followed S-KET, with median Tmax of 0.75, 1.25, and 1.51 hours, for S-KETPO 0.20 mg/kg, S-KETPO 0.45 mg/kg, and S-KETIV, respectively, while S-HNK maximum concentrations were observed later, at 1.26, 1.75, and 3.54 hours, respectively. t1/2 for S-KET, S-NOR and S-HNK were similar and ranged from 4.75 to 6.96, 4.99 to 10.4 and 6.11 to 8.73 hours, for S-KETIV, and for S-NOR and S-HNK from 4.88 to 7.60 and 4.65 to 6.97 hours; and 4.53 to 8.69 and 4.38 to 8.58 hours for S-KETPO 0.20 mg/kg and S-KETPO 0.45 mg/kg, respectively.
Pharmacodynamic effects
S-KETPO 0.20 mg/kg did not demonstrate PD effects. Compared with placebo, S-KETPO 0.45 mg/kg statistically significantly reduced smooth pursuit eye movements, qEEG alpha, beta and delta power (Pz-O1), and increased VAS ‘feeling high’. In contrast compared with placebo, S-KETIV statistically significantly decreased SPV, smooth pursuit eye movements, adaptive tracking, VAS ‘alertness’, DSST total number of correct responses and qEEG alpha, beta, and delta power (Pz-O1), and increased body sway, VAS ‘feeling high’, VAS ‘mood’, VAS ‘external perception’, DSST average response time and MEQ30 total score. For VAS ‘feeling high’, smooth pursuit eye movements and qEEG, which were affected by both S-KETPO 0.45 mg/kg and S-KETIV. However, effects for S-KETPO 0.45 mg/kg were 31% and 35% that of KETIV for smooth pursuit eye movements and VAS ‘feeling high’, respectively, and 70%, 78%, and 79% that of KETIV for qEEG alpha, beta and delta power, respectively. qEEG theta and gamma power remained unaffected for both S-KETPO and S-KETIV. A summary of PD results is presented in Table 2 and Figure 2.
Average S-KET effects for pharmacodynamic assessments up to 6 hours post-dose following administration of S-KETPO 0.20 mg/kg, S-KETPO 0.45 mg/kg, and S-KETIV 0.4 mg/kg up to 6 hours post-dose, represented as LSM change from baseline and compared with placebo, with 95% CI.
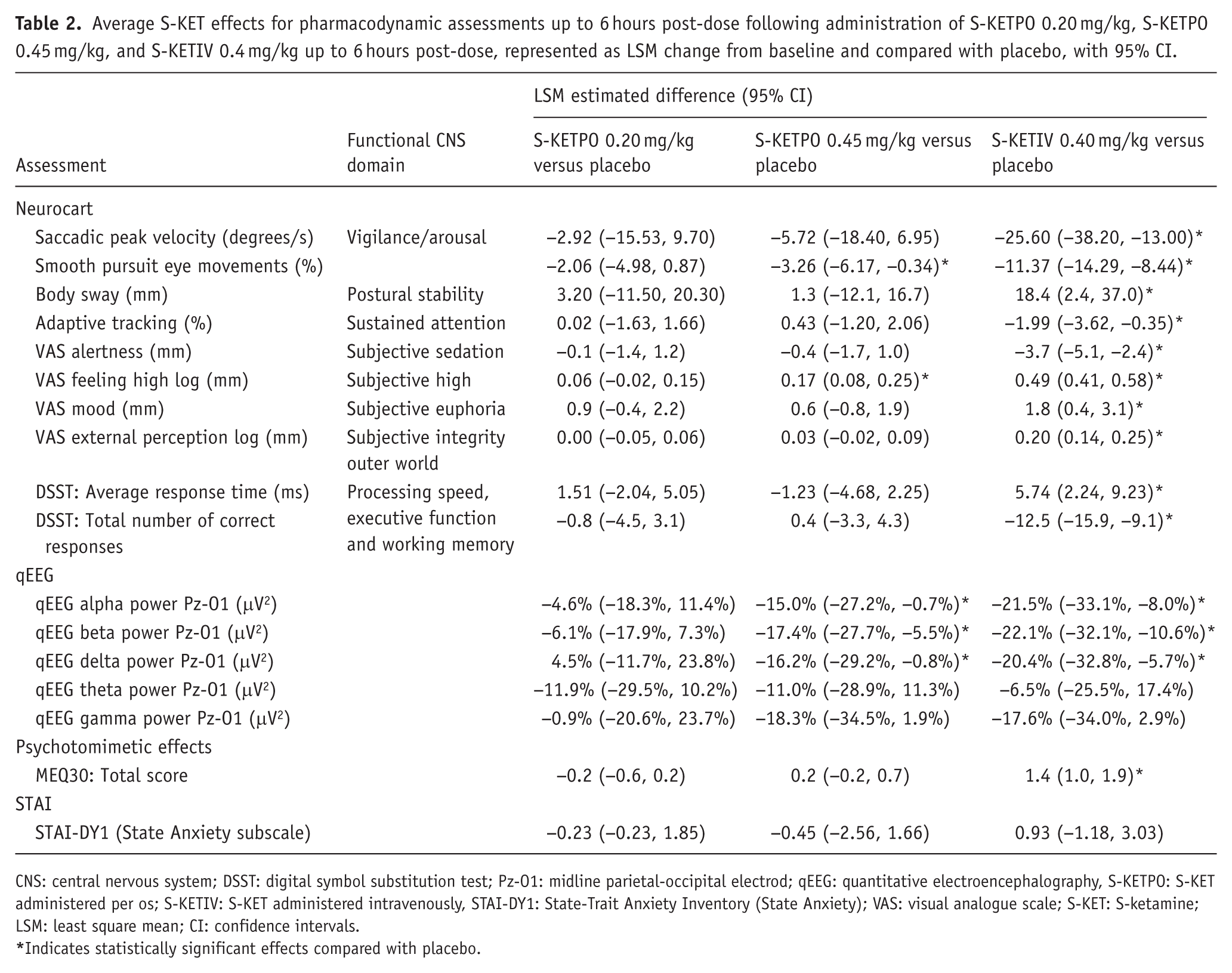
CNS: central nervous system; DSST: digital symbol substitution test; Pz-O1: midline parietal-occipital electrod; qEEG: quantitative electroencephalography, S-KETPO: S-KET administered per os; S-KETIV: S-KET administered intravenously, STAI-DY1: State-Trait Anxiety Inventory (State Anxiety); VAS: visual analogue scale; S-KET: S-ketamine; LSM: least square mean; CI: confidence intervals.
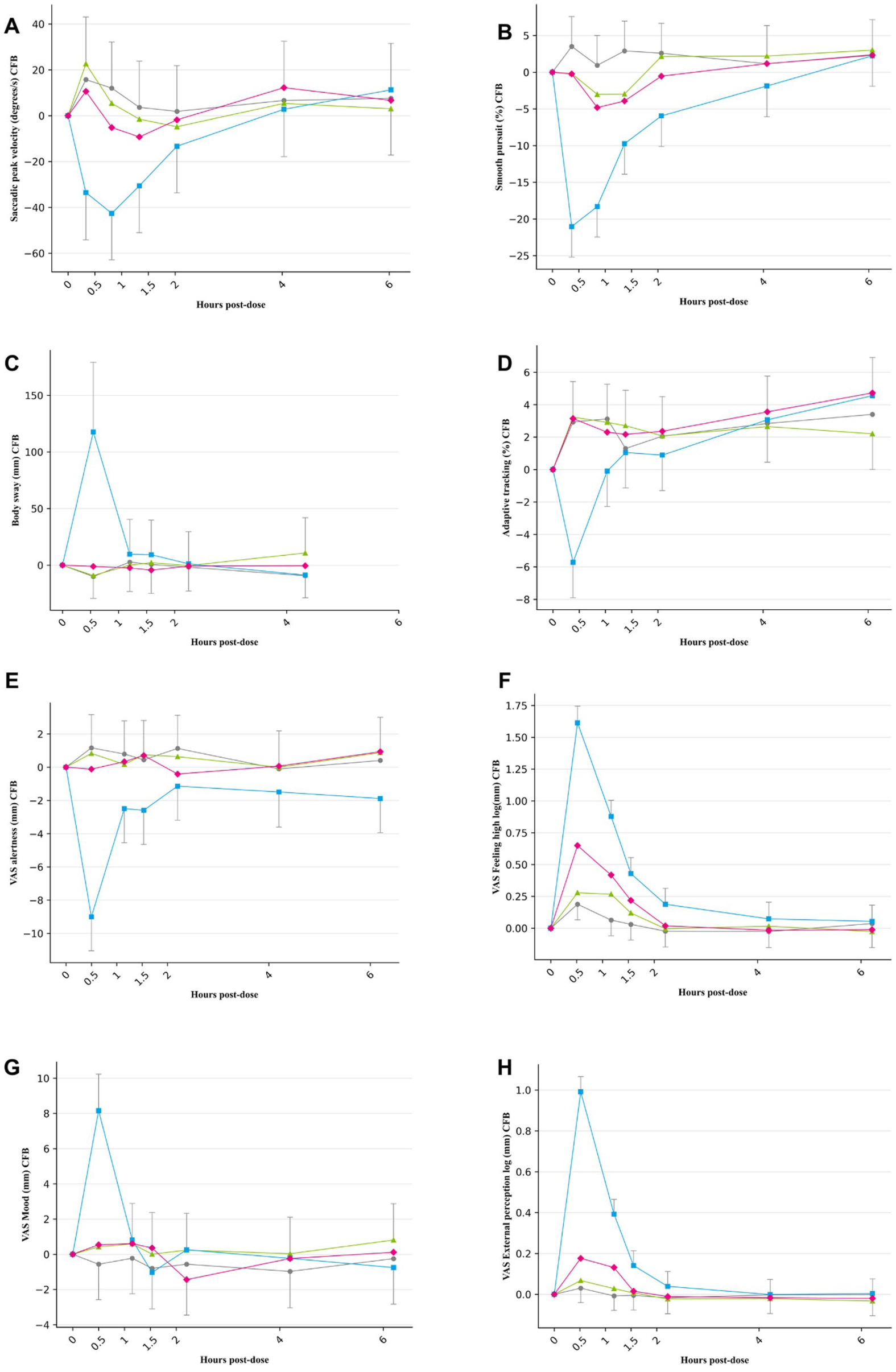
Average S-KET effects for Neurocart assessments up to 6 hours post-dose following administration of S-KETPO 0.20 mg/kg, S-KETPO 0.45 mg/kg, and S-KETIV 0.4 mg/kg. Average S-ketamine effects (LSM change from baseline (CFB)) up to 6 hours post-dose for (a) saccadic peak velocity; (b) smooth pursuit eye movements; (c) body sway; (d) adaptive tracking; (e) VAS alertness; (f) VAS feeling high; (g) VAS mood; and (h) VAS external perception, represented as LSM change from baseline, with 95% CI bars, and p-values representing statistically significant changes compared with placebo. Grey dot = placebo; blue dot = S-KETIV 0.40 mg/kg; pink diamond = S-KETPO 0.45 mg/kg; green triangle = S-KETPO 0.20 mg/kg.
Safety
No SAEs occurred, and the majority of reported TEAEs was mild in intensity and self-limiting in nature. The most frequently reported TEAEs for S-KETIV were dissociation, nausea/vomiting and euphoria in 70%, 47% and 47% of participants, respectively. Dissociation and nausea/vomiting were absent for S-KETPO (0.20 and 0.45 mg/kg), while euphoria was reported in 12.5% of participants receiving S-KETPO 0.20 mg/kg and in 29.4% of participants receiving S-KETPO 0.45 mg/kg, respectively. S-KETIV modestly increased the average systolic and diastolic blood pressure (BP) with maximal increases of approximately 16 and 14 mmHg, respectively. Both systolic and diastolic BP returned to baseline within 2 hours after start of the infusion. S-KETIV modestly increased the resting heart rate (HR) with maximally 16 beats/min, which also resolved within 2 hours, while neither S-KETPO 0.20 nor 0.45 mg/kg affected BP or HR. No clinically significant changes were observed in laboratory tests or on C-SSRS.
Discussion
The current study in healthy participants characterized the PD and safety profiles of S-KETPO in relation to its PK profile, compared head-to-head with an S-KETIV dose that had previously demonstrated antidepressant effects in TRD. Following PO administration, the first-pass effect was anticipated to significantly reduce S-KET’s bioavailabilty and alter systemic exposure of its pharmacolologically active metabolites, necessitating the current comprehensive pharmacological characterization for adequate design, dose selection and interpretation of safety and therapeutic outcomes in future studies with S-KETPO in TRD.
PO administration of S-KET resulted in a distinct PK profile of S-KET and its pharmacologically active metabolites compared with IV administration. Most importantly, S-KET was absorbed poorly, demonstrating an absolute oral bioavailability of 9–12% compared with S-KETIV. In contrast, maximal S-NOR concentrations for S-KETPO 0.20 mg/kg and 0.45 mg/kg were respectively factor 1.1 (62.1 ng/mL vs 55.2 ng/mL) and 1.3 (127 ng/mL vs 55.2 ng/mL) higher than that of S-KETIV. Similarly, maximal S-HNK plasma concentrations for S-KETPO 0.45 mg/kg were factor 1.9 higher than that of S-KETIV (62.0 ng/mL vs 32.2 ng/mL), while maximal plasma concentrations for both S-NOR (62.1 ng/mL vs 55.2 ng/mL) and S-HNK (29.5 vs 32.2), were relatively comparable between S-KETPO 0.20 mg/kg and S-KETIV. As a result, S-NOR and S-HNK to S-KET metabolite-to-parent compound ratios (MPRs) were markedly higher for both PO doses (MPR = 3.70–7.50) compared with IV administration (MPR = 0.43). These MPRs are line with S-KET MPR ratios (approximately 0.41) reported in studies that characterized the PK of KETIV in patients with therapy resistant bipolar depression (Zhao et al., 2012), and in patients with severe therapy resistant depression receiving S-KETPO in a dose range of 0.75–4 mg/kg twice weekly for 6 weeks (MPR = 7.96; Veraart et al., 2025). Our findings therefore not only confirm S-KETPO yielding poor systemic S-KET availability, but also demonstrate an exposure profile dominated by S-NOR and S-HNK, as opposed to S-KETIV’s S-KET-dominated exposure profile. Considering that both S-NOR and S-HNK demonstrated pharmacological activity preclinically (Kamp et al., 2020; Zanos et al., 2016), alterations in MPRs following PO administration could potentially impact its safety profile and/or potential antidepressant efficacy relative to S-KETIV in humans. Although alterations in S-NOR and S-HNK to S-KET MPR ratios, combined with its poor bioavailability and significant PK variability, complicates S-KETPO dose selection for therapeutic studies, literature suggests that higher S-KETPO doses that the ones administered in this study were safe and well tolerated in TRD (Smith-Apeldoorn et al., 2022b). Together, our findings advocate routine determination and reporting plasma S-KET, S-NOR, and S-HNK concentrations in relation to PD and safety findings in future clinical studies with S-KETPO, as this will facilitate reliable interpretation of efficacy and safety outcomes relative to S-KETIV, and to improve between-study comparison.
As might be expected considering the poor bioavailability of S-KET following PO administration, S-KETPO 0.20 mg/kg lacked PD effects altogether, while S-KETPO 0.45 mg/kg demonstrated inconsistent effects on vigilance and arousal and induced subjective high. In fact, VAS ‘feeling high’ and smooth pursuit eye movement effects for S-KETPO 0.45 mg/kg were approximately 30% that of KETIV. In contrast, S-KETIV robustly affected virtually all functional CNS domains, including vigilance, arousal, subjective high and euphoria, psychotomimetic effects, mystical experiences, processing speed, executive function, and memory. Although both S-KETPO 0.45 mg/kg and S-KETIV reduced qEEG alpha, beta, and delta power in parieto-occipital electrodes, reductions for S-KETPO 0.45 mg/kg amounted to 70–80% of those associated with KETIV. Considering that maximal S-KET exposures for S-KETIV were roughly 15-fold that of S-KETPO 0.20 mg/kg (146.00 ng/mL vs 9.81 ng/mL), while maximal S-NOR exposures were comparable (55.20 ng/mL vs 62.00 ng/mL), S-KET appears to be the primary mediator of CNS effects up to 6 hours post-dose. However relative to S-KETIV, S-KETPO 0.45 mg/kg was associated with significantly higher S-NOR and S-HNK but lower S-KET concentrations, nonetheless demonstrating limited, but not absent PD effects as was the case for S-KETPO 0.2 mg/kg. It therefore seems fair to state that active metabolites may contribute to CNS effects in humans, despite their relatively lower NMDAR potency compared with S-KET. However, PK-PD modelling is considered necessary to reliably elucidate the relative contributions of S-NOR and/or S-HNK to the observed PD effects, which is currently precluded by unavailability of S-NOR and S-HNK for human administration. At any rate, our data presented opportunities to tentatively capture the individual contributions of S-KET and its pharmacologically active metabolites, as the study design allowed for relatively large contrasts in plasma concentrations between S-KET on the one hand and S-NOR and S-HNK on the other, sampled within the same individual.
S-KETIV and S-KETPO were safe, with dissociation and nausea or vomiting being absent or significantly less for S-KETPO compared with S-KETIV. However, S-KETIV showed significant undesired sedative, psychomotor and psychotomimetic effects, whereas S-KETPO 0.45 mg/kg demonstrated inconsistent undesired PD effects, and S-KETPO 0.20 mg/kg lacked PD effects altogether. Although such findings might suggest a superior safety and tolerability profile for S-KETPO relative to S-KETIV as potential maintenance treatment for TRD, comparison of adverse effect profiles at therapeutically equivalent doses of S-KETIV and S-KETPO is indicated prior to making such assumptions, especially when considering S-KETPO’s limited bioavailability and substantial interindividual PK variability.
Altered dose-response relationships and/or PK characteristics for S-KET, S-NOR, and/or S-HNK in patients with TRD could raise questions on the relevance of the present healthy participant findings. To address that, comparing current findings to TRD patients at equivalent doses or exposures could have been helpful. However, comparison of PD effects is precluded as the Neurocart CNS test battery has not been applied across research groups. Alternatively, as the Clinician-Administered Dissociative States Scale (CADSS) is widely applied in both healthy participant and patient studies with S-KET, it would have potentially allowed comparison of dissociative effects between the present healthy participants and TRD patients at equipotent doses. However, its absence in the present study precludes this approach. Also, reliable comparison of the present PK data to S-KETIV in TRD is not feasible as no previous study has quantified PK in TRD patients. Published data on intranasally administered esketamine (S-KETIN) can however serve as a tentative underpinning (Perez-Ruixo et al., 2021). For example, in comparison, Cmax MPR’s for S-KETPO and S-KETIV in the present study were 7.0–7.5 and 0.4, respectively, while S-KETIN 28–84 mg administered to healthy volunteers and patients, demonstrated relatively consistent MPRs of 1.3–1.8, thereby lining up between the observed MPR’s for S-KETPO and S-KETIV. Hence, considering the concordant MPRs for S-KETIN and S-KETPO mentioned earlier, it seems reasonable to assume that large MPR differences between healthy individuals and TRD patients receiving S-KETPO are unlikely, confirming the relevance of our healthy participant data to support dose selection in depression studies and adequate interpretation of outcomes. Moreover, integration of relevant PD assessments and adequate PK sampling in future clinical studies with S-KETPO should be prioritized, as this will facilitate comparison of pharmacological profiles between different clinical populations.
Several strengths and weaknesses of the current study deserve mentioning. The study design enabled a head-to-head comparison of S-KETPO and S-KETIV, by guaranteeing similar S-NOR exposures for S-KETPO 0.20 mg/kg and S-KETIV 0.40 mg/kg but significantly higher S-KET exposures for S-KETIV 0.40 mg/kg compared to S-KETPO 0.20 mg/kg, and in addition, S-KET and S-NOR exposures for S-KETPO 0.45 roughly double that of S-KETPO 0.20 mg/kg. In addition, data-rich PK sampling and repeated PD measures using standardized assessments that have previously been shown to be sensitive to the PD effects of S-KET were applied, permitting the identification of differences in PD effects over time for S-KETIV versus S-KETPO. In fact, the PD profile of S-KETIV was comparable to a previous healthy volunteer study demonstrating robust effects at similar S-KET exposures (120 ng/mL) on SPV, smooth pursuit eye movements, adaptive tracking, body sway and VAS, which underscores the reliability of Neurocart to characterize S-KET’s effects in humans (Fanta et al., 2015; Kleinloog et al., 2015; Singh et al., 2016). Finally, dose selection for S-KETPO was informed by a S-KET population PK model which predicted expected exposures of S-KET and S-NOR with a high level of accuracy (Otto et al., 2021). Nevertheless, S-KET exposures tended to be underpredicted for both S-KETIV and S-KETPO, possibly indicating a higher absolute bioavailability of the parent compound than predicted, while the amount of S-NOR directly formed through first-pass metabolism may have been less than anticipated in advance. The most important and obvious weakness is omission of the CADSS, which was a conscious decision to reduce participant burden due to the number and frequency of planned assessments. As a consequence, dissociation was only assessed and reported as an AE. In retrospect however, the CADSS should have deserved more priority since it could have served as potential conduit for inter-comparison of S-KETIV, S-KETPO and S-KETIN in (patient) studies where CADSS had been administered regularly.
To the best of our knowledge, this is the first study in healthy humans to comprehensively characterize the PD and safety profiles of S-KETPO in relation to its PK , compared head-to-head to an S-KETIV dose that had previously demonstrated antidepressant effects in TRD. Relative to IV administration, PO administration results in poor bioavailability of S-KET, inverted S-NOR and S-HNK to S-KET metabolite-to-parent compound ratios (being higher for S-KETPO and lower for S-KETIV), and limited to complete absence of PD effects relative to intravenous administration. Based in these findings, significantly higher oral doses are required to achieve systemic S-KET exposures that would approximate those reached with 0.4 mg/kg S-KETIV. However, as S-NOR and/or S-HNK seem to contribute to CNS effects based on the S-KETPO 0.45 mg/kg dose, additional studies will have to reveal whether the dramatically increased exposures of pharmacologically active metabolites impact S-KETPO’s efficacy and/or safety profile in treating TRD. For the time being, the observed differential PK profile for S-KETPO relative to that of S-KETIV should not only inform dose selection for future studies, but also support the interpretation of emerging safety and therapeutic outcomes with S-KETPO in TRD.
Supplemental Material
sj-docx-1-jop-10.1177_02698811261456187 – Supplemental material for Pharmacokinetics and pharmacodynamics of orally administered S-ketamine in healthy participants
Supplemental material, sj-docx-1-jop-10.1177_02698811261456187 for Pharmacokinetics and pharmacodynamics of orally administered S-ketamine in healthy participants by Joost. C. van Mechelen, Tobias A. Wieles, Laura G. J. M. Borghans, Marije E. Otto, Maria J. Juachon, Amy L. Gillespie, Martijn S. van Noorden, Catherine J. Harmer, Albert M. van Hemert, Joop M. A. van Gerven and Gabriël E. Jacobs in Journal of Psychopharmacology
Footnotes
Acknowledgements
We thank Karen Broekhuizen who provided medical writing services on behalf of the Centre for Human Drug Research.
Ethical considerations
The study was approved by the BEBO Foundation for the Assessment of Ethics of Biomedical Research (Assen, The Netherlands), was conducted according to the Dutch Act on Medical Research Involving Human Subjects (WMO) and in compliance with all International Conference on Harmonisation-Good Clinical Practice (ICH-GCP) guidelines and the Declaration of Helsinki. This study was registered in the ‘Overview of Medical Research in the Netherlands’ (OMON) under NL-OMON55261. Participants provided written informed consent before any screening procedures were performed. The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors contributions
All authors (i.e., JCM, TAW, LGJMB, MEO, MJJ, ALG, MSN, CJH, AMH, JMAG, and GEJ) participated in research design. JCM, LGJMB, MEO, ALG, JMAG, and GEJ performed data analysis. JCM drafted the initial version of the article. LGJMB, MEO, JMAG, and GEJ were involved in critically revising the draft of the initial version of the article and gave input on different versions of the manuscript. All authors have given final approval of the version to be published. All authors have agreed to be accountable for the article and to ensure that all questions regarding the accuracy or integrity of the article are investigated and resolved.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by: Centre for Human Drug Research, Leiden, The Netherlands.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.