
Abstract
Background:
Psychosis, as a comorbidity, is seen in many neuropsychiatric and neurodegenerative disorders and is often treated with dopamine D2 receptor (D2R) antagonists, which could aggravate Alzheimer’s disease and Parkinson’s disease (PD) symptoms. Hence, there is a need for new treatments with a non-D2R antagonist-related mechanism of action. Recently approved drugs for psychosis in PD, including pimavanserin (PIM), and for schizophrenia, xanomeline-trospium, offer advances since they lack D2R antagonism but both carry associated risks. The highly-purified, plant-derived form of cannabidiol (hpCBD; Epidiolex®) has a different safety profile, does not inhibit D2R and has demonstrated antipsychotic effects in patients with psychosis.
Aims:
To systematically assess the potential antipsychotic-like effects of hpCBD, alongside newer non-D2 antagonist drugs approved for PD psychosis (PIM) and schizophrenia psychosis (xanomeline oxalate (XAN)) in the same set of standardised preclinical assays.
Methods:
MK-801-induced hyperlocomotion and pre-pulse inhibition (PPI) deficits in male C57BL/6J mice were assessed following PIM (0.1, 0.3, 1 mg/kg s.c.), XAN (1, 3, 10 mg/kg s.c.) or hpCBD (50, 100, 200 mg/kg i.p.) administration. Locomotor activity was measured by infrared photobeams and Laboratory Animal Behaviour Observation Registration and Analysis System, and PPI was measured in acoustic startle chambers using a variable pre-pulse intensity protocol.
Results:
PIM, XAN (all doses) and hpCBD (200 mg/kg) attenuated MK-801 hyperlocomotion (p < 0.01). PPI deficits, at various pre-pulse intensities, were attenuated by PIM (all doses), XAN 10 mg/kg and hpCBD 200 mg/kg (p < 0.05).
Conclusion:
These results suggest that hpCBD, like PIM and XAN, demonstrates putative antipsychotic-like activity in classic mouse assays relevant to psychosis, consistent with positive clinical data in patients with psychosis.
Introduction
Psychosis is characterised by delusions, hallucinations and disordered thoughts (Arciniegas, 2015; American Psychiatric Association, 2013) and is a symptom present in many neuropsychiatric disorders, such as schizophrenia spectrum disorders. Psychosis is also associated with neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease (PD; Factor and McDonald, 2016; Goldman and Holden, 2014). Psychosis is often treated with drugs that have a rich pharmacology and notably act at D2 receptors (D2R; dopamine antagonists) and at 5HT2A receptors (serotonin antagonists), for example risperidone (Risperdal®), which is a well-established drug for psychosis, and is also broadly used as a reference compound in preclinical assays of antipsychotic efficacy (Jones et al., 2011; Su et al., 2007; Varty and Higgins, 1995). However, antagonism of dopamine D2R may lead to blockade of the nigrostriatal pathway that can create parkinsonian symptoms such as akathisia (restlessness) and extra-pyramidal symptoms in patients with schizophrenia; thus, current drugs for psychosis can be treatment limiting. Of particular concern, treatment of psychosis associated with neurodegenerative disorders, for example, PD, with current drugs for psychosis can lead to exacerbation of the core motor symptoms resulting in worsening of the condition (Abdul-Rahman et al., 2024). Furthermore, a U.S. Food and Drug Administration (FDA) black box warning links the use of these drugs in the elderly with dementia to increased risk of death due to pneumonia (Knol et al., 2008; Steinberg and Lyketsos, 2012). Hence, there is a need for therapies with a mechanism of action without D2 antagonism and with an improved safety profile, for alleviating psychosis in neurodegenerative disorders without exacerbating other symptoms.
Currently, only two predominantly non-D2 antagonist drugs (hereafter referred to as non-D2) for psychosis are approved by the FDA: pimavanserin (PIM; Nuplazid®), a 5HT2A inverse agonist for PD psychosis, and xanomeline-trospium (Cobenfy®), a muscarinic M1/M4 agonist for psychosis in schizophrenia (Hawkins and Berman, 2017; Lobo et al., 2022). However, they both have significant known side effects and associated risks. The prescribing information for PIM displays a black box warning of mortality in elderly patients with psychosis associated with dementia similar to other drugs for psychosis, and a warning of cardiovascular adverse events based on clinical trial safety data (Cummings et al., 2014; Tariot et al., 2021). Whilst xanomeline-trospium does not have the warnings and precautions associated with standard drugs for psychosis, in particular the black box warning of mortality, it still has a number of safety warnings and gastrointestinal side effects reported in clinical trials related to its activity on the cholinergic system (Kaul et al., 2024a, 2024b). Although these newer drugs represent a step in the right direction, their safety profile is still concerning particularly for patients with psychosis associated with neurodegenerative diseases, and an effective treatment with improved safety is needed.
Different formulations of cannabidiol (CBD), one of the main constituents of the plant Cannabis sativa, have been investigated for their therapeutic potential in neurological and psychiatric disorders for over 30 years (Davies and Bhattacharyya, 2019; Noel, 2017; Zuardi et al., 2012). Although the actual mechanism of action in the brain is still to be elucidated, CBD does not show antagonism at D2R (internal binding data; also see De Almeida et al., 2020 (for review)), although partial agonist activity has been noted (Seeman, 2016). Evidence that CBD may have antipsychotic properties dates back to 1982 when healthy volunteers co-administered with Δ9-tetrahydrocannabinol (THC) (the main psychoactive component in C. sativa) and CBD showed less anxiety and psychomimetic symptoms than those administered with THC alone, as measured by self-rating scales (Zuardi et al., 1982). These data suggest that CBD can attenuate some of the psychomimetic effects of THC, indicating that CBD may have potential in treating psychosis. More recent studies in patients with schizophrenia have suggested that CBD as adjunctive therapy has beneficial effects when used in addition to current drug treatments for psychosis, with further reductions in aspects of the Positive and Negative Syndrome Scale (PANSS) for schizophrenia score (Leweke et al., 2012, McGuire et al., 2018). Additionally, a small study in patients with psychosis associated with PD demonstrated a significant reduction in both Brief Psychiatric Rating Scale (BPRS) and the Parkinson Psychosis Questionnaire (PPQ) following CBD administration (Zuardi et al., 2009). Moreover, compared with PIM and xanomeline-trospium, the only highly-purified plant-derived FDA-approved form of CBD, Epidiolex® (hpCBD), has a more favourable safety profile according to the comparison of the clinical trial safety data that supports the product label (Devinsky et al., 2018; Thiele et al., 2018, 2021).
Two commonly used early stage preclinical assays in psychosis research are stimulant-induced hyperlocomotion (locomotor activity; LMA) and deficits in pre-pulse inhibition (PPI; van den Buuse, 2010). Stimulant-induced assays of hyperlocomotion represent signalling dysfunction of the dopamine system with the use of stimulants such as amphetamine (Minassian et al., 2016), or of the glutamate system if N-methyl-D-aspaertate (NMDA) antagonists, such as phencyclidine (PCP), ketamine or MK-801, are used (Carter, 1995). In rodents, locomotion can be measured via beam-break infrared (LMA) or Laboratory Animal Behaviour Observation Registration and Analysis System (LABORAS) equipment (Quinn et al., 2003). PPI is used as a translationally relevant measure of sensorimotor gating (Braff et al., 2001), which is often dysfunctional in psychosis and can be mimicked preclinically with the use of the aforementioned stimulant/antagonists (Geyer et al., 2001). However, it should be noted that PPI deficits are not limited to diseases with psychosis symptoms but also have been demonstrated in other neuropsychiatric disorders such as anxiety- and stress-related disorders, obsessive-compulsive and tic disorders (Santos-Carrasco and Casa De la, 2023). Nevertheless, preclinically, both assays demonstrate face and predictive validity for assessing potential antipsychotic-like activity of novel compounds. We consider that hyperlocomotion represents the most suitable primary screening assay to be used for identifying antipsychotic-like activity followed by confirmation in the PPI assay. We chose to disrupt the behaviours in these assays with the non-competitive NMDA receptor antagonist, MK-801, because it is not a direct dopamine agonist and it is more selective and potent than PCP or ketamine (Tricklebank et al., 1989). Although MK-801-induced hyperlocomotor activity in rodents has been consistently attenuated by typical (predominantly dopamine receptor antagonists; Behrens and Gattaz, 1992; Corbett et al., 1995; O’Neill and Shaw, 1999; Tiedtke et al., 1990) and atypical (dopamine/serotonin receptor antagonists; Corbett et al., 1995; Gardell et al., 2007; Meltzer et al., 2012) drugs for psychosis, MK-801-induced PPI disruption in rodents has not generally been found to be ameliorated by drugs for psychosis thought to predominantly antagonise D2R (Bast et al., 2000; Varty and Higgins, 1995). Despite this apparent lack of effect by drugs that principally act as D2 antagonists, MK-801-induced PPI deficits in rodents appear to be more sensitive to drugs for psychosis with mixed dopamine/serotonin antagonist activity, such as risperidone and potentially novel antipsychotic-like drugs (Geyer et al., 2001), which do not primarily act via D2R antagonism, although it should also be noted that clozapine, often used as a gold standard to reverse PPI deficits, has mixed effects preclinically (Bast et al., 2000; Geyer et al., 2001; Varty and Higgins, 1995). Further, the use of both LMA and LABORAS in the present study enabled the comparison of sensitivity in measuring hyperlocomotion modulation in two distinct systems. Together, these assays should provide a complete early screening cascade to identify compounds with antipsychotic-like drug properties.
Evidence for potential antipsychotic-like properties of CBD in preclinical assays is conflicting (Zuardi et al., 2012). Some groups have shown that CBD can attenuate amphetamine and ketamine-induced hyperlocomotion and MK-801-induced PPI deficits, whilst others have shown no such effect of CBD upon amphetamine-induced hyperlocomotion in mice or MK-801-induced-hyperlocomotion and PPI deficits in rats (see Jaehne and van den Buuse, 2019; Zuardi et al., 2012). The sources of variation between these studies are numerous, for example, species, strain, dose studied, pretreatment time, stimulant type and source and purity of CBD.
To assess whether the potential antipsychotic-like effects of CBD noted in the clinic could also be demonstrated in classic preclinical assays relevant to psychosis, we embarked on a systematic series of studies using hpCBD (the same form of CBD used in the clinical studies with schizophrenia patients; Leweke et al., 2012, McGuire et al., 2018). Initial studies validated that the assays were fit for the purpose of assessing non-D2 related mechanism of action by testing PIM and xanomeline oxalate (XAN). Once the efficacy of PIM and XAN was characterised, hpCBD was tested in the same LMA, LABORAS and PPI assays.
Materials and methods
Locomotor activity was measured independently in two different laboratories using different analysis forms: by LABORAS (Metris, The Netherlands) at Transpharmation Ltd., UK, and by infrared (IR) photobeam at Melior Discovery Inc., USA. PPI was also conducted at Melior Discovery Inc., USA.
Animals
A total of 782 animals were used in these studies. All studies involved separate cohorts of male C57BL/6J mice; 7–8 weeks old, at study start, for LMA (n = 14 per treatment group) and LABORAS (n = 10 per treatment group) studies and 6–7 weeks old for PPI studies (n = 16 per treatment group); see Supplemental Table S1 for details of individual cohorts. Younger mice were used for the PPI studies due to hearing loss/changes being evident from 8 weeks of age in C57BL/6J mice (Ison and Allen, 2003; Willott et al., 1993). LMA and PPI animals were supplied by Jackson Labs (USA) and LABORAS animals were from Charles River (UK). Mice were group housed (4–6 per cage) under standard laboratory conditions (including enrichment such as nesting material) with free access to rodent chow and water. Mice at Melior Discovery were housed in individually ventilated cages (approx. 30.48 cm × 18.64 cm × 15.24 cm); mice at Transpharmation UK were housed in GM500 cages (approx. 32 cm × 17 cm × 17 cm). Lights were on a 12-hour light/dark cycle with lights on at 07:00; all experiments were conducted during the light phase (approx. 8:30 a.m.–4:00 p.m. with treatment balanced across time). Lux levels for the LABORAS procedure and holding room were 150, whilst for the LMA and PPI procedure rooms, the lux levels were 200–600. Animals did not have access to food or water whilst in the LMA, LABORAS or PPI chambers.
Group sizes, animal sex and strain were based on previous studies conducted by each laboratory.
Facilities in the US were accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) (National Research Council, 2011), and studies complied with all US regulations and Guide requirements for mice and rats. Studies performed in the UK complied with the Home Office Guidance on the operation of the UK Animals (Scientific Procedures) Act 1986 and were awarded ethical approval by Agenda Life Sciences – Discovery Park Animal Welfare and Ethical Review Body.
Beam-break LMA
LMA was measured using an automated activity monitoring system (Med Associates; 27.9 cm × 27.9 cm) in which horizontal activity was monitored within open field chambers by an IR photobeam system. The open field chamber had clear acrylic walls and a solid bottom floor, with no bedding. Chambers were cleaned between animals with a soapy water solution (1:10 ratio of liquid antimicrobial soap (active ingredient para-chloro-meta-xylenol (PCMX)) to water).
All animals were acclimatised to the testing room for at least 30 minutes prior to study start on the same day as the test. All animals were placed individually into locomotor chambers for 60 minutes prior to MK-801 dosing, regardless of pre-treatment time of the test agent (pre-stimulant period) and recording started. Following this 60-minute habituation period, mice were dosed with vehicle (0.9% saline) or MK-801 and placed back in the LMA chambers (same chamber as previously), with LMA being recorded, uninterrupted, for a further 120 minutes. The pre-treatment time of each test agent prior to MK-801 administration (PIM and hpBCD was 60 minutes, XAN and risperidone was 30 minutes) was dependent on their individual pharmacokinetic and/or pharmacodynamic profile and aimed to target their known time of maximal plasma/brain concentration (Tmax) and/or peak pharmacodynamic effects (Deiana et al., 2012; McFarland et al., 2011; Montani et al., 2021; Rana et al., 2023; Schotte et al., 1996; Su et al., 2007; Varty and Higgins, 1995; Vanover at al., 2006). Animals did not have access to food or water during the habituation or test periods, n = 14 per treatment group.
Laboratory Animal Behaviour Observation Registration and Analysis System
LABORAS, a monitoring tool used to measure animal behaviours such as locomotion, immobility, rearing, eating and drinking, utilises a carbon fibre platform to detect unique vibrations produced by the animal as it moves around a clear polycarbonate cage. Advanced pattern recognition technologies and artificial intelligence (AI) convert these vibrations into behaviours and tracking information (Quinn et al., 2003). The LABORAS cages used were distinct from the animals’ home cage, that is, Plex type II cages with wire lid (floor area: 352 cm2, height: 14 cm/height to food hopper: 7.5 cm) and had a thin layer of sawdust on the floor of the cage.
Data produced from the LMA parameter only were collected for the studies presented here.
Testing conditions were the same as beam-break LMA studies, except that animals were allowed to acclimatise to the experimental room for at least 60 minutes prior to study start (due to differing laboratory protocols). LABORAS equipment was calibrated before study start according to the manufacturer’s instructions, n = 10 mice per treatment group.
Pre-pulse inhibition
PPI used acoustic startle monitoring chambers (35.56 cm W × 27.62 cm D × 49.53 cm H) from Kinder-Scientific. Each chamber was capable of delivering 35–120 dB white noise with the animal restrainer placed evenly over a sensing transducer capable of measuring the force response of each animal (N). The restrainers allow free movement without ambulation.
All animals were acclimatised to an ante-room with 65-dB white noise 1 hour before dosing and moved to the testing room, also with 65-dB white noise, 20 minutes prior to the start of testing.
Each PPI chamber was calibrated (to 0.25 N) before each study using 25 g of weight.
Treatment groups (n = 16 mice per group) were balanced further based on body weight and baseline acoustic startle performance. Baseline acoustic startle sessions utilised additional mice (see Supplemental Table S1) to enable selection based on exclusion criteria of high or low responders (upper limit was ⩾0.1 N; lower limit was ⩽0.037 N, for the 120 dB level only) and were performed at least 3 days prior to PPI testing.
Baseline acoustic startle: Mice received a total of 40 trials of white noise at 80, 100 and 120 dB for 40 ms with randomly varied intertrial intervals (ITIs; 13–30 seconds) in a quasi-random order; that is, 2 blocks of 5 120-dB trails at the beginning and end of each session with 10 trials of each dB level randomised between these blocks.
PPI testing: The following parameters were used with background white noise set at 65 dB; 5 minutes habituation in the chambers; house light: off; 20-ms interstimulus interval (ISI; time from end of pre-pulse (PP) to start of pulse); randomised 13–30 second ITI; PP duration = 20 ms; pulse (P) duration = 40 ms; P intensity = 120 dB; PP intensity = 4, 8, 16 and 20 above background (AB), that is, 69, 73, 81 and 85 dB.
In total, there were 90 trials in a quasi-random order: 10 no stimulus trials, 20 P alone trials of 120 dB (block of 5 trials at the beginning and end of each session that were omitted from the analysis), 20 PP alone trials (5 of each intensity: 4, 8, 16 and 20 dB above background) and 40 trials of PP + P at each PP intensity (10 trials of each).
PPI parameters and study design were based on the findings of Paylor and Crawley (1997), Ralph et al. (1999), Long et al. (2006) and Young et al. (2010) and previously demonstrated by Melior Discovery Inc., to produce robust PPI deficits.
Compounds
All compounds were administered in a 10-ml/kg dose volume. All doses were expressed as base equivalent. Group sizes were as follows: LMA n = 14, LABORAS n = 10 and PPI n = 16. Treatments were randomised within home cages and balanced across equipment chambers, test session, test day and time of day. All animals received two treatments, that is, test compound or its vehicle plus MK-801 or its vehicle (see Tables S2a and S2b).
MK-801
(+)-MK-801 hydrogen maleate (Sigma-Aldrich) was formulated in 0.9% saline, vortexed and sonicated until dissolved. MK-801 was administered subcutaneously (s.c.) at 0.1 mg/kg for LMA and LABORAS studies. For PPI studies, a dose of 0.6 mg/kg was administered interperitoneally (i.p.) 30 minutes prior to the first startle trial. Doses and route of administration for MK-801 were based on the experimental literature (Geter-Douglass and Witkin, 1999; O’Neill and Shaw, 1999; Wu et al., 2005) and consistent with those doses and routes previously demonstrated by each laboratory to induce hyperlocomotion and PPI deficits.
Cannabidiol
Highly-purified plant-derived CBD (hpCBD ⩾ 99%, Epidiolex®; GW Research Ltd., part of Jazz Pharmaceuticals, Kent, UK) was formulated in 1:1:18 Ethanol: Kolliphor EL: 0.9% Physiological Saline. Excipients were added to hpCBD slowly whilst stirring and in the order stated. Kolliphor EL was warmed to 60°C. 0.9% saline was only added once the solution was clear. CBD was administered i.p. All studies evaluated hpCBD at 50, 100 and 200 mg/kg (i.p.) 60 minutes prior to MK-801 administration for the LMA and LABORAS studies or prior to the first startle trial for the PPI study. Doses were selected based on known anti-seizure efficacy data in male mice (Rana et al., 2023).
Pimavanserin
Pimavanserin (Tocris) was formulated in 1% acetic acid (0.17 M diluted in water); vortexed and sonicated until dissolved, buffered to pH 5–6 with 10 M NaOH and administered s.c. 60 minutes prior to MK-801 administration for the LMA and LABORAS studies or prior to the first startle trial for the PPI study. Doses of 0.1, 0.3 and 1 mg/kg (s.c.) were selected according to published studies for evaluation in the LMA and LABORAS studies whilst doses of 0.3, 1 and 3 mg/kg (s.c.) were assessed in the PPI study (McFarland et al., 2011; Vanover at al., 2006).
Xanomeline
Xanomeline oxalate (Tocris) was formulated in 0.9% saline; vortexed and sonicated until dissolved and administered s.c. 30 minutes prior to MK-801 administration for the LMA and LABORAS studies or prior to the first startle trial for the PPI study. LMA and LABORAS studies utilised doses of 1, 3 and 10 mg/kg (s.c.), whilst the PPI study evaluated 3, 10 and 30 mg/kg (s.c.). Doses were selected according to published studies (Montani et al., 2021).
Risperidone (RIS)
Risperidone (Sigma-Aldrich) was formulated in 1% acetic acid (0.17 M diluted in water); vortexed and sonicated until dissolved and buffered to pH 5–6 with 10 M NaOH. Risperidone was administered s.c. at 0.032 mg/kg 30 minutes prior to MK-801 administration for the LMA and LABORAS studies and at 0.32 mg/kg 60 minutes before the first startle trial for PPI. Doses were selected according to published studies (Schotte et al., 1996; Su et al., 2007; Varty and Higgins, 1995) and prior studies performed at each laboratory.
Data analysis
All statistical analyses were performed in GraphPad Prism, version 9.5.1. (GraphPad Software, San Diego, California, USA).
All data were assessed for normality (D’Agostino and Pearson normality test or Shapiro–Wilks test for n < 8 or unequal groups) and outliers (Robust regression and Outlier removal method (ROUT (Q = 1%)). Where required, and if possible, data were transformed to achieve normality (log10 or square root).
All data are represented as mean ± SEM.
Following a significant two-way repeated measures analysis of variance (ANOVA) or one-way ANOVA, all post-hoc comparisons were conducted across groups in a between-subjects design using the Vehicle + MK-801 group as the reference. Statistical significance is defined as p < 0.05.
LMA and LABORAS
The data from the 60–65 (LMA) or 60–70 (LABORAS) minute time epochs, which corresponded to the dosing of MK-801 or its vehicle, were excluded from the analysis.
Average total distance travelled, measured in centimetres (cm) for LMA and metres (m) for LABORAS per 5 minute epoch were subjected to two-way repeated measures ANOVA (time and treatment) followed by pairwise comparisons of the treatment groups at each time point using Dunnett’s multiple comparison test.
The accumulated total distance travelled data were also grouped into pre- and post-MK-801 phases with each phase being analysed by one-way ANOVA followed by Dunnett’s multiple comparison test or Kruskal–Wallis followed by Dunn’s multiple comparison test depending on the distribution of the data.
No outliers were removed from the analysis.
In addition, an ANCOVA analysis of covariance (ANCOVA) of log-transformed change in distance travelled from 30 to 60 minutes for PIM and XAN and 0–60 minutes for hpCBD to 70–180 minutes was performed on each dataset. The ANCOVA model included log-transformed distance travelled prior to MK-801 dosing as a covariate and treatment as a fixed factor. For all ANCOVA analyses, the least squares means, treatment differences and 95% confidence intervals were back transformed (data available as Supplemental information).
Pre-pulse inhibition
Outliers were identified using ROUT (Q = 1%) on the startle amplitude data only, and the performance of that animal(s) across all parameters was assessed. If these responses fell below or above the group mean ± 2 SD for more than 50% of all trials, that animal(s) was removed from both the startle and percent PPI parameters.
In the PIM study, two outliers were removed from the Vehicle + MK-801 group; no outliers were removed from the XAN study and one animal was removed from the hpCBD study (RIS group) due to in-cage fighting.
Startle amplitude did not follow normal distribution even after log10 or square root transformation and so was assessed by Kruskal–Wallis followed by Dunn’s multiple comparison test.
Percent PPI was calculated as: 100 − [(startle magnitude for PP + pulse/startle magnitude for pulse alone) × 100]; (Ralph et al., 1999, Young et al., 2010) and subjected to a two-way repeated measures ANOVA (PP intensity and treatment) followed by Dunnett’s multiple comparison test.
Results
PIM and XAN effects on MK-801-induced hyperlocomotion and PPI disruption
PIM attenuates MK-801-induced hyperlocomotion
To test the ability of PIM to reduce MK-801-induced hyperlocomotion, PIM was dosed at 0.1, 0.3 and 1 mg/kg s.c. in the LMA and LABORAS assays, using RIS (0.032 mg/kg s.c.) as a positive control. RIS has previously shown efficacy in these assays (Su et al., 2007; Varty and Higgins, 1995).
Two-way ANOVA of average distance travelled (cm) per 5 minute epoch revealed a significant effect of treatment (LMA: F(5, 78) = 14.49, p < 0.0001; LABORAS: F(5, 54) = 14.26, p < 0.0001), time (LMA: F(7.874, 614.2) = 89.63, p < 0.0001); LABORAS: (F(4.143, 223.7) = 65.25, p < 0.0001 and of the interaction, treatment × time (LMA: F(39.37, 614.2) = 9.51, p < 0.0001; LABORAS: F(20.72, 223.7) = 8.59, p < 0.0001). Post hoc analysis showed a significant increase in distance travelled for the Vehicle + MK-801 group at 85–145 and 155–160 minutes epochs (p < 0.05–0.0001 compared with Vehicle + Vehicle, Dunnett’s multiple comparison test; Figure 1(a), Supplement Table S3). Post hoc analysis also revealed a significant effect of RIS upon distance travelled, that is, an increase at 5 and 20–25 minute epochs (prior to administration) and a decrease at the 40–45, 55–140 and 155 minute epochs (p < 0.05–0.0001 compared with Vehicle + MK-801 group; Dunnett’s multiple comparison test; Figure 1(a), Supplemental Table 3). For PIM 0.1 mg/kg s.c, post hoc analysis revealed a significant decrease in distance travelled at 70, 115–120 minute epochs and also for PIM 0.3 mg/kg s.c. at 20–25, 35–45, 55–60, 75–145, 155 and 170 min and at 20–145 minute for PIM 1 mg/kg (p < 0.05–0.0001 compared with Vehicle + MK-801 group, Dunnett’s multiple comparison test; Figure 1(a)). Similarly, for the LABORAS study, post-hoc analysis revealed a significant increase in distance travelled in the vehicle group following MK-801 administration at the 90–155 minute epochs (p < 0.05–0.001 compared with Vehicle + Vehicle group; Dunnett’s multiple comparison test; Figure S1, Supplemental Table S9). RIS decreased the distance travelled at 40, 50–60 and 80–150 minute epochs, whilst PIM at 0.1 mg/kg p.o. decreased distance travelled at 70, 95, 140–145 minute epochs (p < 0.05–0.001 compared with Vehicle + MK-801 group; Dunnett’s multiple comparison test; Figure S1, Supplemental Table S9). PIM at 0.3 mg/kg showed significant decreases in distance travelled at 35, 55, 65–70, 80, 90–150 minute epochs and PIM at 1 mg/kg showed effects at 30–40, 55–70, 80–155 minute epochs (p < 0.05–0.001 compared with Vehicle + MK-801, Dunnett’s multiple comparison test; Figure S1, Supplemental Table S9).
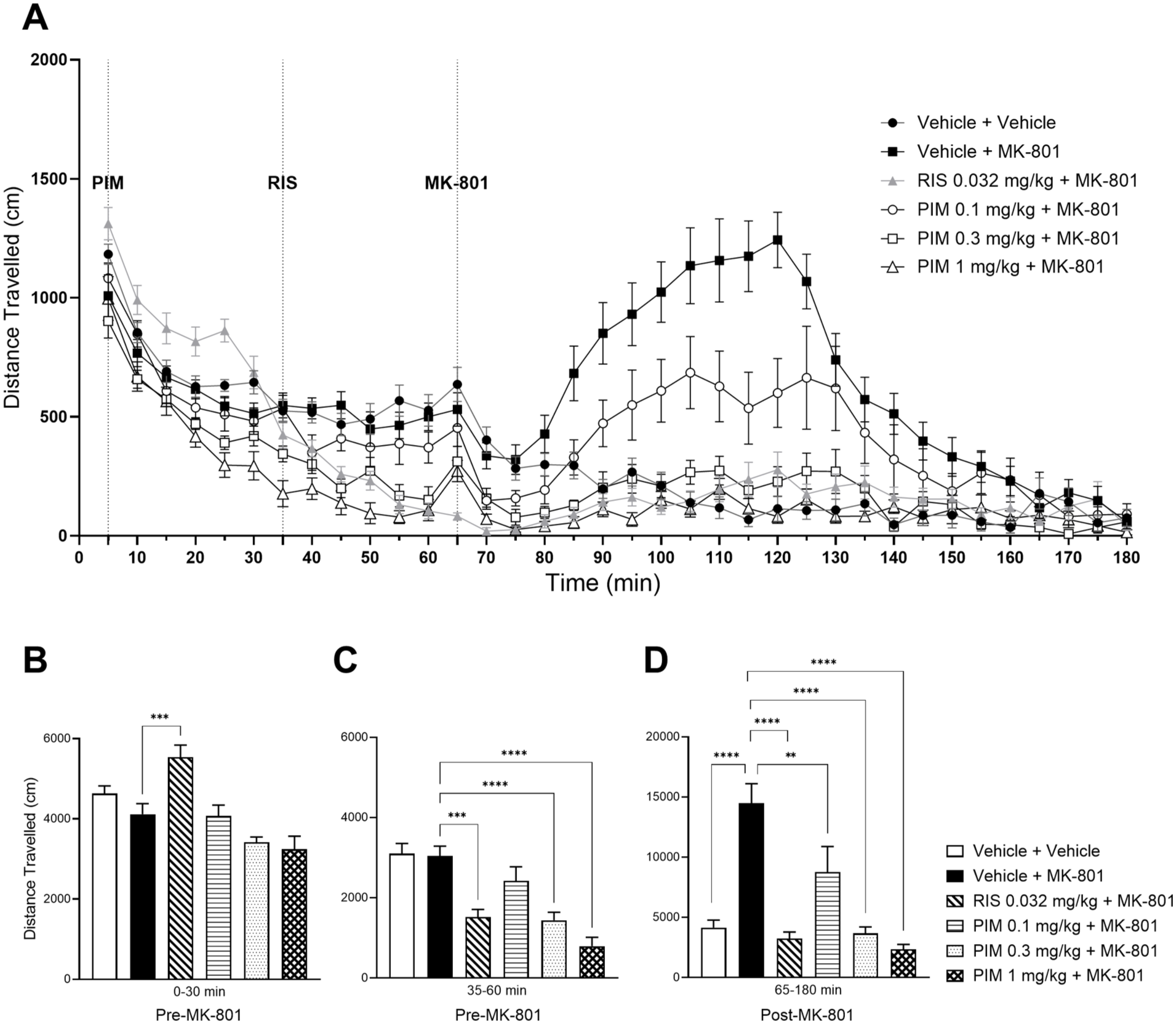
MK-801-induced hyperlocomotion is reduced by pimavanserin (PIM) administration. (a) Effect of PIM (0.1, 0.3, 1 mg/kg s.c.; 60 minute before MK-801 administration) and risperidone (0.032 mg/kg s.c.; 30 minute before MK-801 administration) upon MK-801 (0.1 mg/kg s.c.)-induced hyperlocomotion as measured in the photobeam LMA apparatus. (b) Mean LMA data for the pre-stimulant period (0–30 minute), (c) the pre-stimulant period (30–60 minute) corresponding to the start of dosing with Risperidone and (d) the post-MK-801 period (65–180 minute). Data shown are mean ± SEM total distance travelled (cm); n = 14; **p < 0.01, ***p<0.001,****p < 0.0001 compared with Vehicle + MK-801.
One-way ANOVA of the cumulative distance travelled (cm) during the pre-stimulant period (0–30 minute prior to MK-801 administration) for the LMA study showed a significant effect of treatment (F(5, 78) = 10.75, p < 0.0001) with post hoc comparisons revealing a significant increase in distance travelled of the RIS group, compared with Vehicle + MK-801 group (both arbitrary as RIS and MK-801 have yet to be administered; p < 0.001; Figure 1(b)), indicating inherent variability between treatment groups in baseline LMA. For the 35–60 min pre-stimulant phase, one-way ANOVA revealed a significant effect of treatment (F(5, 78) = 14.55, p < 0.0001) with post hoc Dunnett’s multiple comparisons test showing a significant decrease in cumulative distance travelled for the RIS and PIM 0.3 and 1 mg/kg groups (p = 0.0002, p < 0.0001 and p < 0.0001, respectively, compared with the Vehicle + MK-801 group (arbitrary as MK-801 has yet to be dosed), indicating possible sedative-like effects of RIS and PIM (Figure 1(c)). For the post-stimulant phase (65–180 minutre), one-way ANOVA showed a significant effect of treatment (square root transformed data; F(5, 78) = 17.29, p = 0.0001) with Dunnett’s multiple comparison test revealing a significant increase in cumulative distance travelled following MK-801 administration (p < 0.0001, compared with Vehicle + Vehicle), whilst RIS + MK-801 and PIM + MK-801 at all doses showed a significant decrease (p < 0.0001, p = 0.0030, p < 0.0001, p < 0.0001, respectively, Figure 1(d)). These results indicate that pre-treatment with RIS and PIM can attenuate MK-801-induced hyperlocomotion.
For the LABORAS study, there was no significant effect of treatment during the 0–30 minute pre-stimulant period on cumulative distance travelled (one-way ANOVA; F(5, 54) = 0.6300, p = 0.6776; Figure S1B). One-way ANOVA of the 35–60 minute pre-stimulant period showed a significant effect of treatment (F(5, 54) = 7.195, p < 0.0001) with Dunnett’s multiple comparisons test revealing a significant decrease in cumulated distance travelled for the RIS, PIM 0.3 mg/kg and 1 mg/kg groups (p = 0.0016, p = 0.0057, p = 0.0001, respectively; Figure S1C). Like the LMA study, this effect is prior to MK-801 administration and may possibly indicate sedative-like effects of these compounds. For the post-stimulant period (70–180 minute), one-way ANOVA showed a significant effect of treatment (log10 transformed data; F(5, 54) = 13.62, p < 0.0001), with post hoc analysis revealing a significant increase in cumulative distance travelled following MK-801 administration (p < 0.0001, compared with Vehicle + Vehicle; Dunnett’s multiple comparison test; Figure S1D) and a significant decrease in the RIS + MK-801, PIM 0.3 mg/kg + MK-801 and PIM 1 mg/kg + MK-801 groups (p < 0.0001, p = 0.0042, p < 0.0001, respectively, compared with Vehicle + MK-801; Dunnett’s multiple comparison test; Figure S1D), indicating attenuation of MK-801-induced hyperlocomotion by the test compounds.
Further, ANCOVA analysis of both the LMA and LABORAS data confirmed a significant reduction in distance travelled in the RIS + MK-801 and PIM (all doses, with the exception of the 0.1 mg/kg dose in the LABORAS study) + MK-801 groups (p < 0.05; Supplemental Tables S4 and S10). By accounting for pre-MK-801 LMA, this analysis indicates that possible sedative-like effects during the pre-stimulant phase do not confound the attenuation of MK-801-induced hyperlocomotion.
Since PIM significantly reduced MK-801-induced hyperlocomotion in both LMA and LABORAS, the submaximal dose of PIM, 0.3 mg/kg (s.c.), was selected as the positive control in the subsequent LMA and LABORAS studies.
XAN attenuates MK-801-induced hyperlocomotion
XAN was then assessed in the same assays of MK-801-induced hyperlocomotion (LMA and LABORAS) dosed at 1, 3 and 10 mg/kg (s.c.).
Two-way ANOVA of average distance travelled per 5 minute epoch revealed a significant effect of treatment (LMA: F(5, 78) = 14.28, p < 0.0001; LABORAS: F(5, 54) = 13.43, p < 0.0001), time (LMA: F(7.818, 609.8) = 190.8, p < 0.0001; LABORAS: F(3.965, 214.1) = 95.88, p < 0.0001) and of the interaction, treatment × time (LMA: F(39.09, 609.8) = 12.46, p < 0.0001; LABORAS: F(19.82, 214.1) = 7.63, p < 0.0001). Post hoc analysis of the LMA data showed a significant increase in distance travelled at 80–155 minute epochs in the vehicle group following MK-801 administration (p < 0.05–0.0001, compared with Vehicle + Vehicle; Dunnett’s multiple comparison test; Figure 2(a), Supplemental Table S5). The positive control PIM significantly decreased distance travelled at the 20 and 80–145 minute epochs (p < 0.05–0.0001, compared with Vehicle + MK-801; Dunnett’s multiple comparison test; Figure 2(a), Supplemental Table S5). Significant decreases in distance travelled were seen at 40–60, 70, 90–100 and 140 minutes epochs for XAN at 1 mg/kg (p < 0.05–0.0001, compared with Vehicle + MK-801; Dunnett’s multiple comparison test; Figure 2(a), Supplemental Table S5). For XAN at 3 mg/kg, significant decreases in distance travelled occurred at the 35–145 minute epochs and for the 10 mg/kg group, at 35–135 minute epochs (p < 0.05–0.0001 compared with Vehicle + MK-801, Dunnett’s multiple comparisons test; Figure 2(a), Table S5). For the LABORAS study, post hoc analysis revealed a significant increase in distance travelled for the vehicle group following MK-801 administration at the 85–150 minute epochs (p < 0.05–0.0.001 compared with Vehicle + Vehicle; Dunnett’s multiple comparison test; Figure S2A, Supplemental Table S11). PIM showed significantly decreased distance travelled at the 10, 25–30, 40–60, 85–150 minute epochs (p < 0.05–0.0.001 compared with Vehicle + MK-801; Dunnett’s multiple comparison test; Figure S2A, Supplemental Table S11). For the XAN 1 mg/kg group, significant decreases were seen at the 40–60 and 85–105 minute epochs; at 40–65, 85–100, 115 and 125 minute epochs for the XAN 3 mg/kg group and at 35–65, 80–130 minute epochs for the XAN 10 mg/kg group (p < 0.05–0.0001 compared with Vehicle + MK-801, Dunnett’s multiple comparisons test; Figure S2A, Table S11).
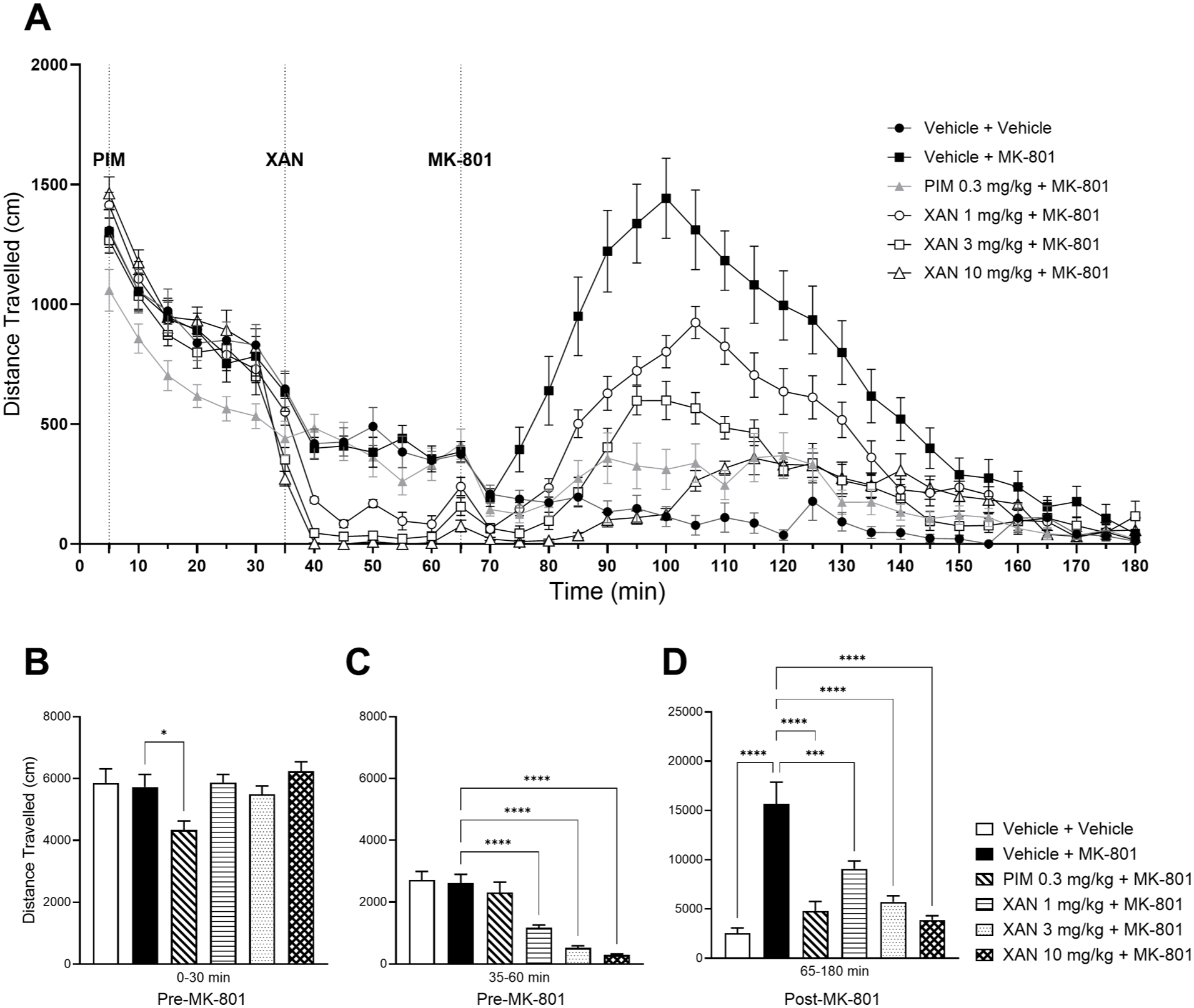
MK-801-induced hyperlocomotion is reduced by xanomeline (XAN) administration. (a) Effect of XAN (1, 3, 10 mg/kg s.c.; 30 min before MK-801 administration) and pimavanserin (0.3 mg/kg s.c.; 60 minute before MK-801 administration) upon MK-801 (0.1 mg/kg s.c.)-induced hyperlocomotion as measured in the photobeam LMA apparatus. (b) Mean LMA data for the pre-stimulant period (0–30 min), (c) the pre-stimulant period (30–60 minute) corresponding to the start of dosing with XAN and (d) the post-MK801 period (65–180 minute). Data shown are mean ± SEM total distance travelled (cm); n = 14; *p < 0.05, ***p<0.001,****p < 0.0001 compared with Vehicle + MK-801.
One-way ANOVA of the LMA data showed a significant effect of treatment for the pre-stimulant period, 0–30 min (prior to XAN and MK-801 but post PIM administration; F(5, 78) = 3.738, p = 0.0044). Post hoc analysis of this period revealed a significant decrease in cumulative distance travelled for the PIM group (p = 0.0213 compared with Vehicle + MK-801 (arbitrary as MK-0801 has yet to be dosed); Dunnett’s multiple comparison test; Figure 2(b)), once again suggesting possible sedative-like effects of PIM during this period. During the pre-stimulant period, 35–60 minute, one-way ANOVA showed a significant effect of treatment (F(5, 78) = 24.81, p < 0.0001) with post hoc analysis revealing a significant decrease in cumulative distance travelled following XAN at all doses tested (all doses p < 0.0001, compared with Vehicle + MK-801 group (arbitrary as MK-801 is yet to be dosed); Dunnett’s multiple comparisons test; Figure 2(c)). This could be attributed to possible sedative-like effects or known cholinergic actions of M1/M4 receptor agonists. Following MK-801 administration (65–180 minute), one-way ANOVA revealed a significant effect of treatment (F(5, 78) = 19.46, p < 0.0001). Post hoc analysis showed a significant increase in cumulative distance travelled during this period by the Vehicle + MK-801 group (p < 0.0001 compared with Vehicle + Vehicle group; Dunnett’s multiple comparison test; Figure 2(d)) and a significant decrease for the PIM and all doses of XAN (p < 0.0001, p = 0.0003, p < 0.0001, p < 0.0001, respectively, compared with Vehicle + MK-801; Dunnett’s multiple comparison test; Figure 2(d)). These data suggest that PIM and XAN can attenuate the hyperlocomotion induced by MK-801.
Similar to the LMA data, LABORAS cumulative distance travelled data showed a significant effect of treatment for both pre- and post-stimulant phases (0–30 minute: F(5, 54) = 5.516, p = 0.0004; 35–60 minute: log10 transformed; F(5, 54) = 27.05, p < 0.0001; 70–180 minute: log10 transformed; F(5, 54) = 15.30, p < 0.0001). Post hoc analysis of the 0–30 minute pre-stimulant period revealed a significant decrease in cumulative distance travelled following PIM pre-treatment (p = 0.0094; compared with Vehicle + MK-801 group (arbitrary as MK-801 is yet to be dosed); Dunnett’s multiple comparison test; Figure S2B), which was still evident in the 35–60 minute pre-stimulant period (log10 transformed data; p < 0.0001; compared with Vehicle + MK-801 group (arbitrary as MK-801 is yet to be dosed); Dunnett’s multiple comparison test; Figure S2C). Also during this period, a significant decrease in cumulated distance travelled was seen following administration of XAN at all doses (log10 transformed data; p = 0.0002, p < 0.0001, p < 0.0001, respectively; compared with Vehicle + MK-801 group (arbitrary as MK-801 is yet to be dosed); Dunnett’s multiple comparison test; Figure S2C). For the 70–180 minute post-stimulant phase, Dunnett’s multiple comparison test revealed a significant increase in cumulative distance travelled following MK-801 treatment (log10 transformed data; p < 0.0001, compared with Vehicle + Vehicle; Dunnett’s multiple comparison test; Figure S2D) and a significant decrease in the PIM + MK-801, XAN 3 mg/kg + MK-801 and XAN 10 mg/kg + MK-801 groups (log10 transformed data; p < 0.0001, p = 0.0243, p = 0.0062, respectively; compared with Vehicle + MK-801; Dunnett’s multiple comparison test Figure S2D).
The ability of XAN to attenuate MK-801-induced hyperlocomotion was further confirmed by ANCOVA analysis of the LMA data showing a significant effect at 3 mg/kg despite the pre-stimulant effects (p < 0.05; Supplemental Table S6); however, this was not demonstrated with the LABORAS data (Supplemental Table S12).
These data collectively confirm that the non-D2 drugs for psychosis, PIM and XAN can attenuate MK-801-induced hyperlocomotion, although both demonstrated effects in the pre-stimulant periods.
PIM and XAN both attenuate MK-801-induced PPI deficits
To confirm the antipsychotic-like effects of PIM and XAN observed in the assays of stimulant-induced hyperlocomotion, their efficacy was further assessed in the PPI assay. Like the LMA and LABORAS studies, RIS was primarily used as the positive control, albeit at a higher dose following in-house observations that the hyperlocomotion assay is more sensitive than the PPI assay.
In the study assessing the effects of PIM upon PPI, two-way repeated measures ANOVA showed a significant effect of treatment (F(5, 88) = 4.943, p = 0.0005), PP intensity (F(2.678, 235.6) = 434, p < 0.0001) and the interaction, treatment × PP intensity (F(15, 264) = 5.175, p < 0.0001). Post hoc comparisons revealed a significant decrease in % PPI of the Vehicle + MK-801 group at 4 dB (p < 0.01), 8 dB (p < 0.0001), 16 dB (p = 0.001) and 20 dB (p < 0.01; compared with Vehicle + Vehicle; Dunnett’s multiple comparison test; Figure 3(a)). RIS (0.32 mg/kg s.c.) significantly attenuated the MK-801-induced PPI deficit at PP intensities 8 dB (p < 0.05) and 16 dB (p < 0.05), whilst PIM at all doses tested significantly attenuated the MK-801-induced deficit in % PPI (0.3 mg/kg s.c. at PP intensity 20 dB (p = 0.0242; Dunnett’s multiple comparison); 1 mg/kg s.c. at PP intensities 16 and 20 dB (p = 0.0134; p = 0.0095, respectively; Dunnett’s multiple comparison; Figure 3(a)); 3 mg/kg s.c. at PP intensity 20 dB (p = 0.0068; Dunnett’s multiple comparison; Figure 3(a)). Kruskal–Wallis analysis showed that startle amplitude significantly differed between treatment groups (H(5) = 19.13, p = 0.0018). Post hoc comparisons revealed a significant decrease in startle amplitude by RIS (p < 0.01 compared with Vehicle + MK-801, Dunn’s multiple comparison test; Figure 3(a)). PIM at the doses tested had no effect upon startle amplitude (Figure 3(a)).
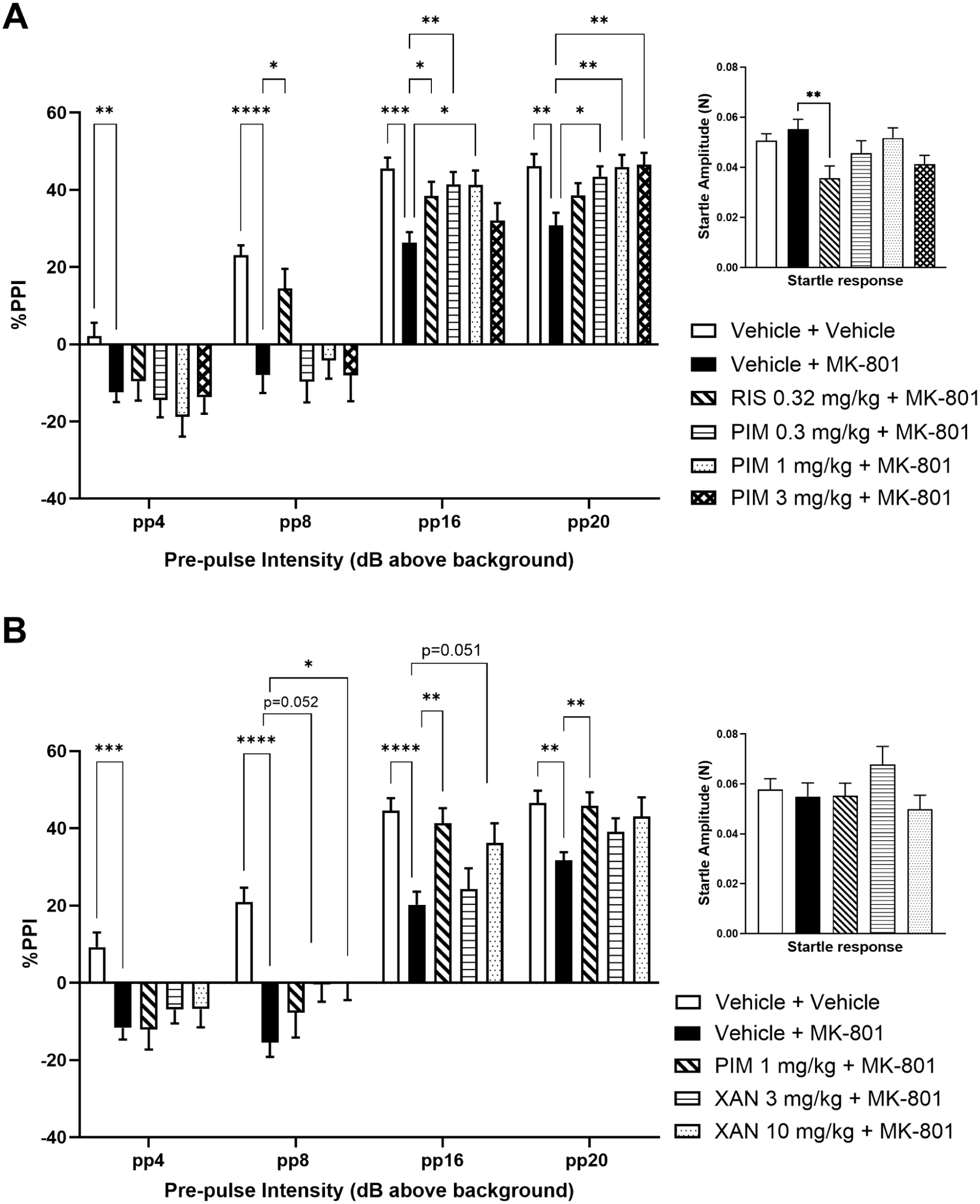
Pimavanserin (PIM) and xanomeline (XAN) attenuate MK-801-induced PPI deficits. (a) Effect of PIM (0.3, 1, 3 mg/kg s.c.; 60 min before first startle trial) and risperidone (0.32 mg/kg s.c.; 60 minute before first startle trial) and (b) XAN (3, 10 mg/kg s.c.; 30 min before first startle trial) and PIM (1 mg/kg s.c.; 60 minute before first startle trial), upon MK-801 (0.6 mg/kg i.p.; 30 min before first startle trial)-induced deficits in PPI using variable pre-pulse (pp) intensities (above background; 65 dB). Inserted graph depicts the startle amplitude response. Data shown are mean ± S.E.M % PPI; n = 14–15; *p < 0.05, **p<0.01, ***p<0.001, ****p < 0.0001 compared with Vehicle + MK-801.
As PIM at 1 mg/kg showed a consistent effect size at the two PP intensities at which the positive control, RIS, also demonstrated attenuation of the MK-801-induced deficit in PPI, this dose of PIM was selected as the positive control in the subsequent PPI studies.
In the study assessing XAN efficacy in the PPI assay, two-way repeated measures ANOVA showed a significant effect of treatment (F(4, 68) = 6.833, p = 0.0001), PP intensity (F(2.359, 160.4) = 352, p < 0.0001) and of the interaction, treatment × PP intensity (F(12, 204) = 4.379, p < 0.0001). Post hoc comparisons revealed a significant decrease in % PPI for the Vehicle + MK-801 group at PP intensities of 4 dB (p < 0.001), 8 dB (p < 0.0001), 16 dB (p < 0.0001) and 20 dB (p < 0.01; compared with Vehicle + Vehicle, Dunnett’s multiple comparison test; Figure 3(b)). PIM significantly attenuated the MK-801-induced PPI deficit at 16 and 20 dB PP intensities (p < 0.01 compared with Vehicle + MK-801, Dunnett’s multiple comparison test; Figure 3(b)). XAN 10 mg/kg s.c. significantly attenuated the MK-801-induced deficit in % PPI at the 8 dB PP intensity (p < 0.05 compared with Vehicle + MK-801; Dunnett’s multiple comparison test; Figure 3(b)). Post hoc analyses also showed that % PPI at the 8 and 16 dB PP intensities following XAN at 3 and 10 mg/kg s.c. were −0.44 ± 4.47 compared with −15.47 ± 3.70 (Vehicle + MK-801; p = 0.052) and 36.19 ± 5.11 compared with 20.23 ± 3.36 (Vehicle + MK-801; p = 0.051), respectively (Figure 3(b)). Startle amplitude was not altered by XAN, PIM (positive control) or MK-801 in this study (H(4) = 6.003, p = 0.199); Figure 3(b)).
In this study, XAN was also dosed at 30 mg/kg s.c. but the data were not analysed due to a pronounced reduction in LMA that could potentially confound the PPI measure. Overall, the findings of this study indicate an antipsychotic-like effect of XAN in this assay.
The effect of hpCBD on MK-801-induced hyperlocomotion and PPI disruption
hpCBD attenuates MK-801-induced hyperlocomotion
Combining the results from LMA, LABORAS and PPI assays, we confirmed that both PIM and XAN elicit antipsychotic-like properties in these assays in line with clinical efficacy (Isaacson et al., 2021; Kaul et al., 2024a, 2024b; Figures 1–3 and Supplemental Figures 1 and 2). Therefore, the testing conditions established in these assays were fit for the purpose of assessing molecules lacking D2 antagonism actions for antipsychotic-like potential. Hence, hpCBD was evaluated in these preclinical assays.
Two-way ANOVA of average distance travelled per 5 min epoch revealed significant effects of treatment (LMA: F(5, 78) = 11.73, p < 0.0001; LABORAS: F(5, 54) = 6.465, p < 0.0001), of time (F(4.698, 366.5) = 89.99, p < 0.0001; LABORAS; F(3.228, 174.3) = 36.77, p < 0.0001) and of the interaction, treatment × time (LMA: F(23.49, 366.5) = 7.008, p < 0.0001; LABORAS: F(16.4, 174.3) = 4.466, p < 0.0001). Post hoc analysis revealed a significant increase in distance travelled by the Vehicle + MK-801 group at the 85–140 minute epochs (p < 0.01–0.0001, compared with Vehicle + Vehicle group; Dunnett’s multiple comparison test; Figure 4(a)), whilst a significant decrease in distance travelled was seen by PIM at the 5, 20, 30, 40, 60–65, 75–130 minute epochs and by hpCBD 200 mg/kg at 30, 40–115 minute epochs (p < 0.05–0.0001, compared with Vehicle + MK-801 group; Dunnett’s multiple comparison test; Figure 4(a) and Table S7). For the LABORAS study, post-hoc comparisons showed a significant increase in distance travelled by the Vehicle + MK-801 group at 85–135 minute epochs (p < 0.05–0.01, compared with Vehicle + Vehicle group; Dunnett’s multiple comparison test; Figure S3A and Table S13) and a significant decrease in distance travelled following PIM (0.3 mg/kg) administration at the 45–65, 90–125 minute epochs and by hpCBD 200 mg/kg at the 30–65, 85–130 minute epochs (p < 0.05–0.001, compared with the Vehicle + MK-801 group; Dunnett’s multiple comparison test; Figure S3A and Table S13).
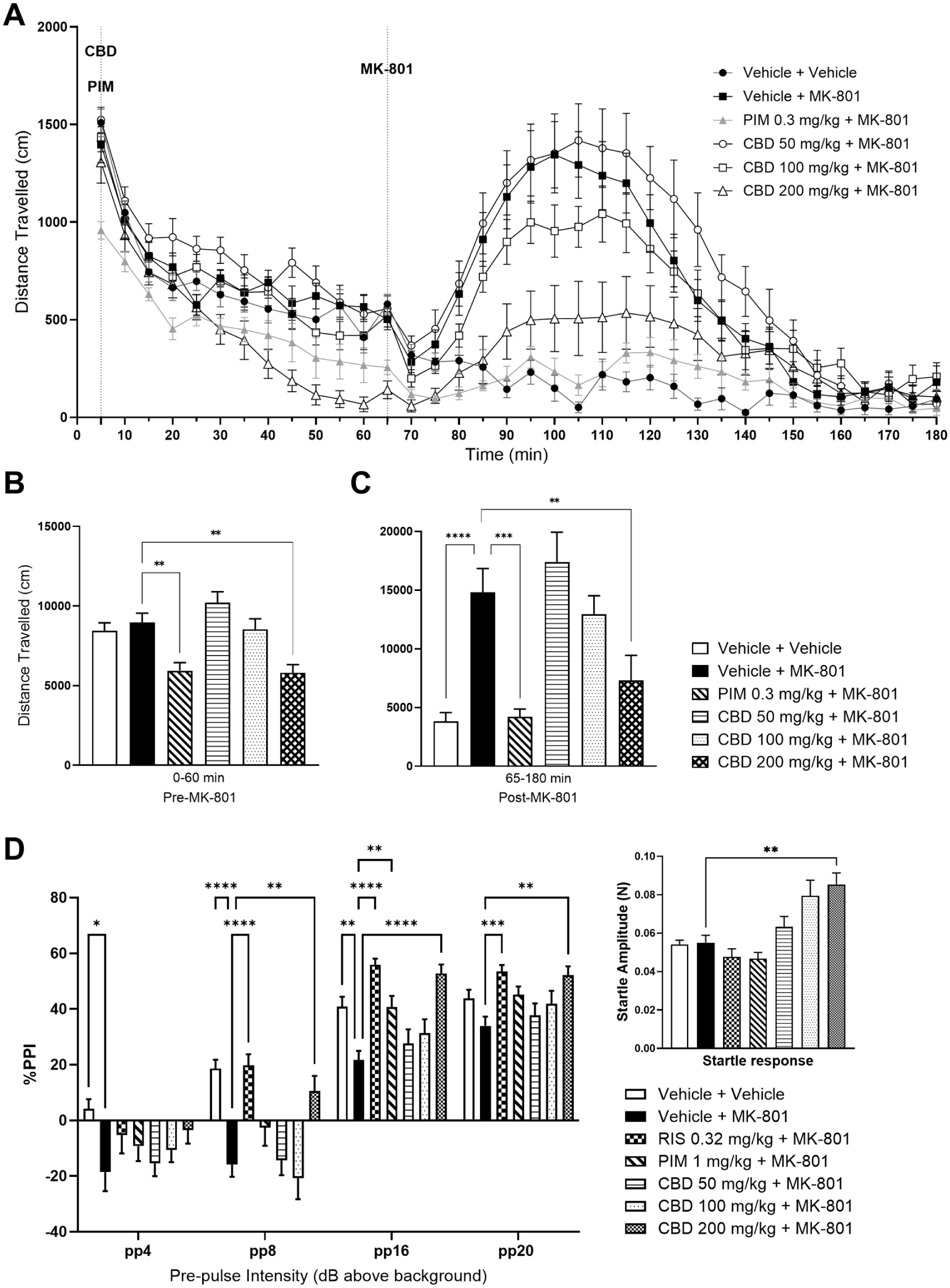
hpCBD (CBD) attenuates MK-801-induced hyperlocomotion and PPI deficits. (a) Effect of CBD (50, 100, 200 mg/kg i.p.; 60 min before MK-801 administration) and pimavanserin (0.3 mg/kg s.c.; 60 minute before MK-801 administration) upon MK-801 (0.1 mg/kg s.c.)-induced hyperlocomotion as measured in the photobeam LMA apparatus. (b) Mean LMA data for the pre-stimulant period (0–60 minute) and (c) the post-MK801 period (65–180 minute). Data shown are mean ± SEM total distance travelled (cm); n = 14. (D) Effect of CBD (50, 100, 200 mg/kg i.p.; 60 minute before first startle trial), risperidone (0.32 mg/kg s.c.; 60 minutes before first startle) and pimavanserin (1 mg/kg s.c.; 60 minutes before first startle trial) upon MK-801 (0.6 mg/kg i.p.; 30 minutes before first startle trial)-induced deficits in PPI using variable pre-pulse (PP) intensities (above background; 65 dB). Inserted graph depicts the startle amplitude response. Data shown are mean ± SEM% PPI; n = 15–16; *p < 0.05, **p<0.01, ***p<0.001,****p < 0.0001 compared with Vehicle + MK-801.
One-way ANOVA of the cumulative distance travelled (cm) during the pre-stimulant period (0–60 minute prior to MK-801 administration) showed a significant effect of treatment (F(5, 75) = 9.218, p < 0.0001) with post hoc comparisons revealing a significant decrease in distance travelled of the PIM and hpCBD 200 mg/kg group (p < 0.01, compared with Vehicle + MK-801 group (arbitrary as MK-801 has yet to be administered); Dunnett’s multiple comparison test; Figure 4(b)). For the post-stimulant phase (65–180 minute), one-way ANOVA of cumulative distance travelled (cm) showed a significant effect of treatment (F(5, 78) = 11.93, p < 0.0001; square root transformed data), with post hoc analysis revealing a significant increase in LMA for the Vehicle + MK-801 group (p < 0.0001, compared with Vehicle + Vehicle group; Dunnett’s multiple comparison test; Figure 4(c)) and a significant decrease in LMA for the PIM + MK-801 and hpCBD 200 mg/kg + MK-801 group (p < 0.001 and p < 0.01, respectively; compared with Vehicle + MK-801; Dunnett’s multiple comparison test; Figure 4(c)). Similar to the LMA study, the LABORAS study showed a significant effect of treatment (F(5, 54) = 5.293, p = 0.0005; one-way ANOVA) during the pre-stimulant (0–60 minute) phase with post hoc comparisons revealing a significant decrease in distance travelled in the PIM and hpCBD 100 and 200 mg/kg groups (p < 0.01, p < 0.05, p < 0.001, respectively, compared with the Vehicle + MK-801 group (arbitrary as MK-801 is yet to be dosed; Figure S3B). Following a significant effect of treatment during the post-stimulant phase (70–180 minute; one-way ANOVA: F(5, 54) = 10.73, p < 0.0001; square root transformed data), post hoc analysis revealed a significant increase in LMA of the Vehicle + MK-801 group (p < 0.001) and a significant decrease in the PIM + MK-801 and hpCBD 200 mg/kg + MK-801 groups (p < 0.001, compared with Vehicle + Mk-801; Dunnett’s multiple comparison test; Figure S3C).
These data suggest that PIM (positive control) and hpCBD at 200 mg/kg can attenuate the hyperlocomotion induced by MK-801 administration. The effect of hpCBD 200 mg/kg is further confirmed by ANCOVA analysis of both the LMA (p = 0.0286) and LABORAS (p = 0.0007) data (Supplemental Tables S8 and S14).
hpCBD attenuates MK-801-induced PPI deficits
Similar to PIM and XAN, following assessment of hyperlocomotion, hpCBD was assessed in the PPI assay. In this study, both RIS (0.32 mg/kg s.c.) and PIM (1 mg/kg s.c.) were used as positive controls.
Two-way repeated measures ANOVA of % PPI showed significant effects of treatment (F(6, 104) = 9.627, p < 0.0001), PP intensity (F(2.308, 240.1) = 381.6, p < 0.0001) and the interaction, treatment × PP intensity (F(13.85, 240.1) = 3.261, p < 0.0001). Post hoc analysis revealed a decrease in % PPI in the Vehicle + MK-801 group at 4, 8 and 16 dB (p < 0.05, p < 0.0001, p < 0.01; respectively, compared with Vehicle + Vehicle group; Dunnett’s multiple comparison test; Figure 4(d)). RIS significantly attenuated the deficit in % PPI induced by MK-801 (0.6 mg/kg s.c.) at three of the four PP intensities tested (8 dB: p < 0.0001; 16 dB: p < 0001 and 20 dB: p < 0.001; Dunnett’s multiple comparison test; Figure 4(d)) A significant effect of PIM was demonstrated at the 16 dB above background PP intensity (p < 0.01, compared with Vehicle + MK-801; Dunnett’s multiple comparison; Figure 4(d)). hpCBD at 200 mg/kg (i.p.) also significantly attenuated the deficit in % PPI induced by MK-801 at three of the four PP intensities tested (8 dB: p = 0.0031; 16 dB: p < 0.0001 and 20 dB: p = 0.0026; compared with Vehicle + MK-801; Dunnett’s multiple comparison; Figure 4(d)).
Kruskal–Wallis revealed that startle amplitude differed significantly between treatment groups (H(6) = 41.49, p < 0.0001), with post-hoc comparisons showing increased startle amplitude following hpCBD at 200 mg/kg (i.p.; p = 0.0047, compared with Vehicle + MK-801 group; Dunn’s multiple comparison test; Figure 4(d)).
Discussion
The present findings show that hpCBD demonstrates antipsychotic-like activity in classic mouse assays relevant to psychosis, namely NMDAR antagonist-induced hyperlocomotion and PPI deficits, consistent with its clinical efficacy and other non-D2 agents. The highest dose of hpCBD tested (200 mg/kg i.p.) consistently reversed MK-801-induced hyperlocomotion in the LMA and LABORAS assays and deficits in the PPI assay to a similar level as the new generation of non-D2 drugs for psychosis PIM, a 5HT2A inverse agonist approved for the treatment of PD psychosis, and XAN, an active component (a muscarinic M1/M4 agonist) of the recent combination therapy (xanomeline-tropsium) approved for schizophrenia.
CBD’s mechanism of action has yet to be confirmed, but putative molecular targets include G protein-coupled receptor 55, transient receptor potential vanilloid 1 and the adenosine transporter, ENT1 (Devinsky et al., 2024; Gray and Whalley, 2020). Whilst further work is required to understand precisely how these targets and CBD affect the pathways involved in psychosis, it is known that hpCBD does not have any dopamine D2 antagonist activity and thus is distinct from the conventional antipsychotic mode of action. Our internal data (unpublished) suggest that hpCBD does not significantly bind to dopamine D2R when tested in in vitro screening panels (broad binding screening panels). However, one published report has suggested CBD (source not cited) has some dopamine D2 partial agonist activity (Seeman, 2016), which is yet to be replicated. Moreover, if CBD is primarily acting on dopaminergic pathways, it is surprising that it does not cause akathisia in patients, an effect which is observed with partial agonists (Frankel and Schwartz, 2017).
Several studies have previously tested the effect of CBD in similar rodent assays commonly used to screen for antipsychotic activity with mixed results; with some showing no effect of CBD, whilst others did show a positive effect (see Zuardi et al., 2012). For example, Moreira and Guimarães (2005) demonstrated attenuation of amphetamine-induced hyperlocomotion in Swiss mice by CBD (Hebrew University Israel; 2% Tween 80 in saline) at 30 and 60 mg/kg (i.p.) and of ketamine-induced hyperlocomotion at a dose of 30 mg/kg (i.p.). With regard to PPI studies, Long et al. (2006) showed attenuation of MK-801-induced PPI deficits in Swiss mice by CBD (5 mg/kg, i.p.; Tocris; 1:1:18 Cremophor EL: ethanol: saline) as did Pedrazzi et al. (2021 and 2024) with CBD at doses of 30 and 60 mg/kg (i.p.; BSPG Laboratories; 2% Tween 80 in saline), which also attenuated amphetamine-induced PPI deficits in the same study, whilst Gururajan et al. (2012) showed no effect of CBD (3–30 mg/kg, i.p.; THC Pharm; 1:1:18, Cremophor EL: ethanol: saline) upon MK-801-induced hyperlocomotion or PPI deficits in Sprague–Dawley rats. Although the most notable difference in the aforementioned studies is the species used (mice vs rat), another important explanation for the variability in published data could be the dosing regimen used, pretreatment time and the type, source and formulation of the CBD tested. It is also unclear whether those studies used synthetic or botanical sources of CBD, whereas the current study tested highly-purified plant-derived CBD that contains additional cannabinoids that may contribute to the overall efficacy. Indeed, there have been studies that have shown an entourage effect of botanical mixtures where the different constituents may act synergistically that is, CBD exerts its therapeutic effects via synergy with other cannabinoids and metabolites also found in C. sativa (André et al., 2024; Russo, 2011). Thus, the purity and composition of the CBD used may influence the results obtained. The rationale for using plant-derived hpCBD in the current studies was based on it being the only FDA-approved form of CBD licenced to treat seizures associated with specific epilepsies, and is also the same form used in the previous clinical studies of schizophrenia psychosis (McGuire et al., 2018) as well as the ongoing follow-up clinical trials. There is currently no other form of CBD, synthetic or otherwise, approved for clinical use in any indication. In addition, there were also differences in the NMDA antagonist used to induce the ‘psychotic’ state (MK-801, ketamine or PCP) and the specific dose level, as well as species, strain and age, all of which could contribute to variability in the CBD response.
Known anti-seizure efficacy and pharmacokinetic properties of hpCBD in preclinical species drove the dose selection used in the present studies (50, 100 and 200 mg/kg i.p.) where a dose of 100–200 mg/kg i.p. consistently produces a clear anti-seizure effect in male mice (Deiana et al., 2012; Rana et al., 2023). We note that regardless of the source and type of CBD used in other published studies, antipsychotic-like efficacy was reported at lower doses than observed in the present study, where we saw significant attenuation of the MK-801 response at 200 mg/kg i.p. However, since the anti-seizure and antipsychotic-like activity of hpCBD in our hands were in a similar dose range in mice, we are confident that the specific formulation and resulting pharmacokinetic properties of this form of CBD are consistent with the anticonvulsant preclinical data package for the approved clinical drug. There is a known disconnect between the clinical dose (oral administration of 5–25 mg/kg/day in Dravet syndrome (DS), Lennox–Gastaut syndrome (LGS) and tuberous sclerosis complex (TSC)) and preclinical effective dose (100–200 mg/kg i.p.) of hpCBD, possibly due to species differences in its pharmacokinetic and pharmacodynamic profiles. Nonetheless, the dose effective in clinical studies of schizophrenia psychosis (1000 mg/day), albeit as adjunctive therapy, is in a similar range as the anticonvulsant clinical dose when taking body weight into consideration (~12 mg/kg/day; McGuire et al., 2018; Millar et al., 2019).
In the present study, two distinct systems for measuring LMA in mice (beam-break LMA and LABORAS) were employed to evaluate their relative sensitivity, specificity and precision in detecting pharmacodynamic effects with MK-801 and the test compounds. The results demonstrated comparable outcomes across both systems, with neither showing clear superiority, thereby providing confidence of the reproducibility of the drug effects obtained across assay systems. Further, it should be noted that the LMA and LABORAS experiments in the current study were carried out in entirely different laboratories, in different countries, with mice from different suppliers/breeders but still produced consistent effects, strengthening the validity of the outcomes reported. These assays, which used LMA as the primary readout and MK-801-induced hyperlocomotion as a proxy for a ‘psychotic state’, also enabled assessment of the test compounds’ locomotor effects during the pre-stimulant phase, prior to MK-801 administration. Here, a consistent reduction in mouse LMA when receiving active doses of PIM, XAN and CBD to varying extents were observed and could be suggestive of possible sedative effects, which may confound the desired MK-801 hyperlocomotion attenuation. However, these assays were not designed to specifically assess sedation, and additional assays would be needed to confirm. Additionally, ANCOVA analysis that accounted for variations in pre-stimulant activity (30–60 minutes) confirmed a significant attenuating effect of the test compounds on MK-801-induced hyperlocomotion in the LMA and, with the exception of XAN, in the LABORAS studies, regardless of the pre-stimulant effects. The pre-stimulant effect of XAN was more pronounced compared to PIM and hpCBD, as revealed by the ANCOVA, suggesting that the overall reduction in hyperlocomotion could be confounded by potential side effects of XAN. The fact that XAN and PIM displayed a similar reduction in pre-stimulant activity and are known to have clinical antipsychotic activity suggests this phenomenon is not a critical confounding issue and will need to be confirmed in appropriately designed studies of sedation.
There are several limitations noted in the present study, the first being the potential sedative effect of the test compounds as previously discussed and how this may impact on the overall attenuation of the MK-801 hyperlocomotion response. The ANCOVA analysis confirmed that there was predominantly a significant effect despite baseline effects, but it should still be noted as a potential confound in studies of this design. Additionally, the test compounds were administered at varying timepoints in the habituation phase prior to MK-801 dosing based on their individual pharmacokinetic and/or pharmacodynamic profiles, to align with time of maximal plasma and/or brain concentrations or peak pharmacodynamic effects (Deiana et al., 2012; McFarland et al., 2011; Montani et al., 2021; Rana et al., 2023; Schotte et al., 1996; Su et al., 2007; Vanover at al., 2006; Varty and Higgins, 1995) at stimulant dosing. This resulted in some compound groups having a longer habituation period than others, which may have impacted on the level of LMA prior to MK-801 administration. Of note, the RIS + MK-801 group in the PIM study appeared to have higher activity prior to the dosing of any of the pharmacological agents, but this can only be attributed to normal experimental variation. Following habituation (i.e. 0–30 min), this appeared to be no longer the case, and levels were in line with the range of starting points across groups. Moreover, ANCOVA using the pre-stimulant (30–60 minute) data as a covariate identified a significant reduction in distance travelled following MK-801 administration in the RIS + MK-801 and PIM (all doses) + MK-801 groups (p < 0.05; Supplemental Tables S3 and S9). Together, these findings demonstrate robust pharmacological effects of the test agents relative to MK-801-induced increases in LMA, whilst also highlighting the analytical rigor applied to the evaluation of these data. Nevertheless, future studies could aim to design consistent habituation times for all animals across different studies, whilst accounting for the differing pharmacokinetic and pharmacodynamic profiles of the test drugs.
It is possible that changes in startle magnitude may be influencing the resulting PPI (see Bakshi et al., 1998; Cilia et al., 2001, Shoji and Miyakawa, 2018). However, it is interesting to note that risperidone at 0.32 mg/kg had differing effects upon startle magnitude in two of our studies, that is, decreased startle amplitude in the PIM study and no effect in the hpCBD study, at the same dose, route and pre-treatment time. It is also possible that sedative effects may be influencing the startle response at this dose of risperidone. However, this dose of risperidone was not assessed in the LMA studies presented here, nor was risperidone alone assessed, making it difficult to assess the effect of risperidone upon startle amplitude in the present studies. Similarly, whilst hpCBD at 200 mg/kg showed a decrease in LMA in the pre-stimulant phase, it must be noted that this group started at a lower activity level relative to the other treatment groups and that possible sedation here does not explain an increase in startle amplitude. Again, these studies did not include an hpCBD alone group, nor were the LMA and PPI studies assessed in the same animals, making interpretation of the influence of such effects difficult. Also of note is the seemingly short ISI used in the PPI studies presented here, 20 ms compared with 100 ms which is typically used, especially in pharmacological studies in rats. However, Varty et al. (2001) conducted an extensive PPI study with differing mouse strains under varying PP intensities and ISI intervals and found that some mouse strains, including C57BL/6, exhibited higher levels of PPI at 30 ms than at 100 ms ISI. Varty et al. (2001) also highlighted the need for optimising the parameters used according to the strain of mouse being studied and prior to pharmacological intervention. Indeed, the authors and collaborators found that a 20-ms ISI was optimal for C57BL/6J mice. Another key observation is that MK801 reliably disrupted PPI across all PP intensities in the present studies, although this effect was not observed at the highest PP intensity (20 dB) in the hpCBD study. Importantly, the ability of the test compounds to attenuate the MK801-induced PPI deficit was dependent on PP intensity. PIM was effective primarily at higher PP intensities (12 and 16 dB), whereas RISP, XAN and hpCBD restored PPI across a broader range of intensities, including 8, 12 and 16 dB. This intensity-dependent profile is consistent with the findings of Long et al. (2006), who showed in mice that attenuation of MK801-induced PPI disruption by CBD and clozapine was evident only at the highest PP intensity tested (12 dB). Despite methodological differences between studies, these findings collectively suggest that antipsychotic-like effects on MK801-induced PPI deficits are not uniformly expressed across all PP intensities. Rather, the sensitivity of PPI to pharmacological modulation may depend on both the PP intensity employed and the underlying mechanism(s) of action of the compounds studied.
An additional limitation of the current study is the fact that only male mice were used in all experiments presented. Relying solely on male mice may limit translational relevance because it overlooks sex specific biological differences that can affect therapeutic responses. However, many studies still use only males to avoid potential variability introduced by the oestrous cycle and to align with historical research practices that have prioritised male animals for perceived simplicity and consistency. To enhance translatability, future confirmatory studies should use mixed sex groups to determine whether there is a consistent effect in female mice.
Whilst there have been exciting recent advancements for patients with psychosis following the approval of newer non-D2 antagonist drugs for psychosis such as PIM for PD psychosis and xanomeline-trospium for schizophrenia, which offer improvements over the conventional treatments in terms of extrapyramidal and metabolic side effects, some safety concerns remain. As previously described, PIM has a black-box safety warning of mortality in elderly patients with psychosis associated with dementia and an additional warning of cardiovascular adverse events such as QT prolongation (Cummings et al., 2014; Tariot et al., 2021). Whilst xanomeline-trospium does not have a black-box safety warning for elderly mortality like the other drugs for psychosis, it does have safety warnings of urinary retention, increased heart rate, decreased gastric movement, angioedema of the face and lips, and risk of liver damage (Kaul et al., 2024a, 2024b). Within the combination drug xanomeline-trospium, trospium chloride is a peripherally-acting non-selective muscarinic antagonist that eliminates peripheral cholinergic side effects and is meant to improve the safety profile of the CNS active ingredient xanomeline. The current study used xanomeline oxylate since this is the active component of the drug and the readouts were of CNS efficacy rather than peripheral side effect observations. hpCBD, approved for the treatment of seizures associated with DS, LGS and TSC, has some safety warnings of hepatic injury, sedation/somnolence and suicidal behaviour (Devinsky et al., 2018; Thiele et al., 2018, 2021), but avoids any black box safety warnings and cholinergic side effects. No substantial new safety or tolerability issues were found with hpCBD in two (relatively short-term) clinical trials in schizophrenia patients with psychosis (Boggs et al., 2018; McGuire et al., 2018). The same is also observed with other formulations of CBD (Leweke et al., 2012). Therefore, with a more favourable safety profile, particularly in elderly patients with neurodegenerative disorders associated with psychosis, hpCBD may offer an improvement on currently available treatments for psychosis.
The clinical evidence for the antipsychotic potential of CBD is compelling with several clinical trials showing clear improvements in psychosis scores in different patient populations. A randomised controlled trial using hpCBD as adjunctive therapy showed a clear improvement in the PANSS score in schizophrenia patients (McGuire et al., 2018). Further, larger scale clinical studies (1000 participants, 38 sites, 11 countries, 3 trials) are now being conducted with hpCBD in patients with schizophrenia by academic groups (Oxford University & King’s College London, supported by the Wellcome Trust), to confirm and explore the utility of CBD in schizophrenia psychosis (STEP programme; STEP-ENHANCE: NCT06778564, STEP, PROMOTE and STEP-ASSIST yet to be registered). Additionally, the broader antipsychotic efficacy of CBD was supported by a smaller clinical study in PD patients with psychosis, where a significant reduction in both BPRS and the PPQ was demonstrated following CBD administration (Zuardi et al., 2009).
The present study confirms preclinical antipsychotic activity of hpCBD in classic mouse assays relevant to psychosis, consistent with the reported clinical efficacy in schizophrenia and PD psychosis. In addition, the effect of hpCBD in these assays was on par with that of the recently approved non-D2 antagonist drugs for PD psychosis (PIM) and schizophrenia (XAN), suggesting that these assays are useful for the screening of potential new psychosis treatments with differentiated mechanisms of action.
Supplemental Material
sj-docx-1-jop-10.1177_02698811261456216 – Supplemental material for Effects of pimavanserin, xanomeline and highly-purified plant-derived cannabidiol on MK-801-induced behaviours in mice relevant to psychosis
Supplemental material, sj-docx-1-jop-10.1177_02698811261456216 for Effects of pimavanserin, xanomeline and highly-purified plant-derived cannabidiol on MK-801-induced behaviours in mice relevant to psychosis by Jackie Cilia, Oriane Guillermin-Escobar, Andrew C. McCreary, David Virley and Jennifer Li in Journal of Psychopharmacology
Footnotes
Acknowledgements
The authors wish to thank Transpharmation Ltd, UK for performing the LABORAS studies, Melior Discovery Inc., USA for performing the LMA and PPI studies, Mr Richard Newman for his management of the studies and Dr Kerry Hyland for her help with the statistical analyses of the locomotor activity data.
Ethical considerations
All animal housing and experiments were conducted in strict accordance with the institutional Guidelines for Care and Use of Laboratory Animals at Melior Discovery Inc., and Animal Welfare and Ethical Review Body at Transpharmation Limited.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: all studies were funded by Jazz Pharmaceuticals.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: all authors were employees and shareholders of Jazz Pharmaceuticals at the time the studies were conducted.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.