
Abstract
The therapeutic potential of psychedelics is currently being explored in a range of neuropsychiatric conditions. Nevertheless, their designation as Schedule 1 substances causes substantial regulatory and economic barriers to research. Rigorous human mechanistic studies are required to address the critical knowledge gaps surrounding the mechanisms underlying the putative treatment effects of psychedelics. This article presents practical guidance for navigating challenges relating to study design, legislation, and drug sourcing, to assist researchers in navigating the complex process of setting up human psychedelic studies, drawing on our experience of setting up non-clinical studies in the United Kingdom, while acknowledging that some of the content will be relevant for investigator-initiated studies more broadly.
Introduction
Classical psychedelics, such as psilocybin and lysergic acid diethylamide (LSD), are partial serotonin-2A (5-HT2A) receptor agonists. Psychedelics are emerging as potential treatments for a range of neuropsychiatric conditions (Andersen et al., 2021). The classification of psychedelics as Schedule 1 drugs in the United Kingdom (UK), meaning they have no recognised medical use and are subject to the highest level of regulatory control, has led to significant legislative and economic barriers to conducting research (Howard et al., 2021).
In the UK, Home Office 1 approval is required for any possession, storage, or administration of psychedelics in research settings. However, recent policy developments suggest the landscape may be shifting, with the Advisory Council on the Misuse of Drugs (ACMD) recommending that research using Schedule 1 drugs in universities and hospitals may be conducted in accordance with Schedule 2 drug requirements, with a pilot expected to follow soon (ACMD, 2025).
Despite these barriers, there are several ongoing and completed clinical and research studies involving psychedelics internationally, including in the UK (Bogenschutz et al., 2015; Butler et al., 2025; Carhart-Harris et al., 2018a; Davis et al., 2021; Gasser et al., 2014; Griffiths et al., 2016; Grob et al., 2011; Johnson et al., 2017; Moreno et al., 2006; Palhano-Fontes et al., 2019; Ross et al., 2016; Rucker et al., 2024a; Sanches et al., 2016). These studies are directly informing discussions on potential patient benefits. For example, preliminary results from a phase 3 trial investigating psilocybin versus placebo as a treatment for treatment-resistant depression suggest modest but significant efficacy (ClinicalTrials.gov, 2025).
There are, however, substantial knowledge gaps in our understanding of the mechanisms of psychedelics and their apparent efficacy across different diagnoses. There is a pressing need for new studies to ascertain the role of the psychedelic experience in therapeutic effects, characterise cognitive changes, determine the neural and psychological mechanisms of the experiences reported, and establish long-term effects, including safety.
To address these knowledge gaps, researchers are undertaking healthy volunteer and basic science studies in patients (which we refer to as ‘non-Clinical Trial of an Investigational Medicinal Product’ (non-CTIMP) studies; see Suggestion 4) with psychedelics, using broad, multidisciplinary methodologies. These are particularly important given the challenges of conducting psychedelic studies within the gold-standard randomised clinical trial format (Butler et al., 2022).
Likely in part due to the economic and legislative barriers to obtaining legal psychedelics for scientific studies, some researchers have developed protocols which capitalise on existing psychedelic use in real-world settings, known as naturalistic studies (Carvalho et al., 2025). Naturalistic approaches may be particularly useful for studies involving patterns of use that are difficult to implement within existing regulatory frameworks, for example, microdosing studies, and benefit from understanding psychedelic use in real-world contexts. The present article instead focusses on controlled studies conducted within research centres, where the drug product used is supplied and dosed by the research team.
While non-CTIMP studies have different legislative and economic constraints to clinical trials, the administrative burden can be challenging to navigate. In this article, we propose ten suggestions, which serve as a practical introduction to setting up a psychedelic study, with a focus on non-CTIMP human research in the UK. The authors are clinical and scientific researchers who have been involved in over 50 pharmacological studies, including non-CTIMP psychedelic studies in healthy populations and patients. This article is aimed at new or prospective researchers, and those who are considering applying for funding in this area, with the goal of demystifying this complex – and sometimes arcane – field, with the intention of encouraging researchers to conduct much-needed psychedelic mechanistic studies in humans.
This article does not provide regulatory advice or an in-depth review of regulatory processes; recently, experts in the field have summarised in detail the processes required to set up and run clinical trials with psychedelics (Barnett et al., 2024; Glennon et al., 2026). We acknowledge that regulatory landscapes continue to evolve – with recommendations for change being actively considered (Drug Science, 2025) – and that institutional processes may vary.
Suggestions
Suggestion 1: Formulate a clear and testable research question that addresses methodological pitfalls
The first step in setting up a psychedelic study is to identify a clear research question, followed by a feasible study design that addresses some of the methodological challenges faced in the field. While many questions concerning psychedelic effects may require recognised techniques in psychological, cognitive, computational, or neuroimaging sciences, research questions have and can also encompass philosophical, political, and developmental areas. Cross-disciplinary studies combining techniques are to be encouraged (for review, see (Bartlett et al., 2023).
There are several methodological challenges (and potential solutions) to be considered when designing psychedelic studies. High levels of public interest in psychedelic research may contribute to selection bias, as individuals who volunteer for these studies may have pre-existing interest in or perceptions of psychedelics, which may not be representative of the wider population. Generic recruitment for research studies with specific studies or study details highlighted only after a volunteer shows interest is a possible solution. Adequate blinding is difficult to implement due to the profound shifts in consciousness experienced by participants and observed by researchers; previous suggestions to mitigate such effects have been the use of independent and blinded outcome measurers, although this does little to mitigate functional unblinding at the participant level. This functional unblinding may lead to inflation of effects in the active group, particularly in ‘high expectancy’ conditions (Butler et al., 2022; Hudson and Pope, 2026). Similarly, selecting an appropriate placebo condition is challenging. While previous studies have used ‘active placebos’, such as benzodiazepines, nicotinamide (niacin), and methylphenidate (Aday et al., 2025), these drugs induce distinct subjective effects. Low-dose comparisons (such as microdoses as the placebo condition) may be a partial solution to expectancy and unblinding effects; however, it is unlikely to ever fully mitigate unblinding (particularly at lower relative doses) and may not be a valid placebo (at higher relative doses).
Another important consideration for study design is the timing of outcome measurements. As the field shifts towards examining post-acute and long-term psychedelic effects, a clearer definition of these temporal windows, and justification of measurements within them, is recommended. For example, the immediate post-acute period should not be conflated with long-term effects, and the relationship among acute, post-acute, and long-term changes requires further examination. For reviews, see Aday et al., 2020, Evens et al., 2023, and Goldberg et al., 2020.
It is advisable to seek input from colleagues with experience in psychedelic studies to ensure your aims are feasible within your institutional setup, and paid consultancy services are also available. With your study design in place, begin drafting the study protocol. This is a detailed document outlining your methods and procedures. The protocol will form the foundation of your ethics and governance applications, so it is advisable to begin at an early stage. Templates are often available within institutions, and an increasing number of protocols are uploaded to public domains (Butler et al., 2025; Husain et al., 2023; Rucker et al., 2021; Spriggs et al., 2021).
Suggestion 2: Account for additional time and costs in research planning
Psychedelic research places financial and logistical demands on researchers and institutions that extend beyond those of conventional studies. For example, additional costs include the study drug itself, Home Office licences due to the Schedule 1 classification, staff time to cover the intensive set-up process, and, if required, expenses for third-party regulatory support such as repackaging, importation, and storage. 2 It is advisable to obtain a range of quotes and negotiate where possible. If your protocol involves therapeutic support, further costs must be considered for hiring or training specialist therapists and securing dedicated therapy spaces.
The legislative constraints placed on psychedelic studies can substantially lengthen the setup time, and the process may be fraught with unexpected hurdles. For example, obtaining a Home Office licence alone can take several months, with unpredictable delays and restricted operating hours making efficient communication challenging. Even with efficient progress, a 1-year setup period is a realistic minimum.
Careful planning of the data-collection period can help mitigate the heavy time burden of setup. Realistic expectations are essential, and consideration should be given to factors such as ease of recruitment (are you recruiting from a broad ‘healthy volunteer’ sample pool or a restricted patient pool?), the number of participant visits, and the size and capacity of your research team. That said, the psychedelic field needs larger studies (Drummond et al., 2024), so if you are able to assemble a team, be ambitious.
Suggestion 3: Give thought to your choice of psychedelic
Psychedelics differ widely in their receptor profiles, pharmacokinetics, routes of administration, and subjective effects (Caulfield et al., 2025; Holze et al., 2024), and these pharmacological differences have important implications for study design and feasibility. For a recently developed comprehensive pharmacological classification system describing the various psychedelics and doses, see Nutt et al., 2025.
Psilocybin is the most commonly used compound in modern-era research (Siegel et al., 2021), largely due to its oral bioavailability and moderate duration of action – subjective effects typically last around 5 hours, making it well-suited for day-case administration (Ley et al., 2023). In contrast, short-acting psychedelics (such as N,N-dimethyltryptamine (DMT) and 5-methoxy-N,N-dimethyltryptamine (5-MeO-DMT)) produce a shorter (approximately 30 minutes), but more intense subjective experience (at doses used in clinical trials; Good et al., 2023; Rucker et al., 2024b), and generally require intravenous or intranasal administration. When administered orally, the subjective effects of LSD last up to 14 hours, which presents additional logistical challenges for day-case studies (Holze et al., 2021).
It may seem tempting to choose a short-acting psychedelic for its potential time-saving benefits; however, your choice of drug should be driven by your specific research hypotheses. For example, recent preclinical evidence suggests that the duration of the subjective effects may correlate with the persistence of subsequent behavioural change (Nardou et al., 2023). Activities conducted during the acute phase is an additional consideration: completion of behavioural tasks would be highly challenging during a DMT or 5-MeO-DMT session.
If a therapeutic intervention during the psychedelic experience is relevant for your study, the gradual onset and offset of psilocybin or LSD may be more conducive to intention-setting and building a therapeutic narrative than a short-acting psychedelic. It is also important to weigh the risks and benefits of exposing participants to a more intense, albeit shorter, experience, although comparative risk profiles for the various psychedelics have not been well established.
Suggestion 4: Determine early whether your study is a clinical trial
An important early step in planning your study is to establish whether your study qualifies as a CTIMP or a non-CTIMP. According to the World Health Organization, a clinical trial is ‘any research study that prospectively assigns human participants or groups of humans to one or more health-related interventions to evaluate the effects on health outcomes’, with health outcomes including both safety and efficacy (World Health Organization, 2020). Studies aiming to evaluate safety or efficacy are always CTIMPs; however, studies evaluating other outcomes such as mechanisms of action or biomarker studies may also be classified as CTIMPs. The Medicines and Healthcare products Regulatory Agency (MHRA) provides an algorithm to help determine CTIMP status (Medicines and Healthcare products Regulatory Agency, 2014), and your local Clinical Trials Office or sponsor should confirm this in writing to support your ethics application.
CTIMP classification will substantially influence your study setup, as CTIMPs are subject to several additional processes, including MHRA approval, clinical trials registration, specifying a database management process, and incorporating monitoring visits and organising trial management and trial oversight groups. These can substantially alter timelines, costs, and volume of approvals required. The study sponsor (the institution taking full responsibility for the study, often a university) would need to have clinical trials insurance and assume full Good Clinical Practice responsibilities. The study drug would be classified as an Investigational Medicinal Product (IMP), requiring Good Manufacturing Practice (GMP) labelling and Qualified Person release – a formal certification by a legally designated expert that each batch meets all regulatory and quality standards before use in the trial. Early clarification of your study’s CTIMP status will streamline regulatory planning and help avoid costly delays later in the process.
Suggestion 5: Select your study population with safety as a priority
As with any psychoactive substance, there are risks associated with psychedelic use. These should be considered when defining your study’s eligibility criteria. While a detailed review of all potential risks is beyond the scope of this article, emerging evidence from recent studies is reassuring. While a recent meta-analysis of 3504 participants from 114 studies reported no serious adverse events in healthy volunteers and 4% in participants with pre-existing neuropsychiatric conditions (Hinkle et al., 2024), the meta-analysis highlighted concern for incomplete adverse event reporting. A more recent clinical trial has reported hallucinogen persisting perception disorder (HPPD) as a serious adverse event (Mertens et al., 2026), and accurate prevalence rates are difficult to ascertain. Furthermore, the evidence base remains limited for certain populations due to the strict eligibility criteria commonly applied in psychedelic studies.
For example, while it appears that psychedelics do not increase seizure risk in people without epilepsy, the effects on seizure threshold in people with epilepsy require further research (Soto-Angona et al., 2024). People with a personal or family history of psychotic conditions (including bipolar disorder) are typically excluded from studies. The risk of precipitating psychosis or mania in these populations remains unclear; however, it is possible that those with genetic vulnerabilities have a greater risk (Simonsson et al., 2024). Similarly, participants with a history of suicidal ideation are often excluded due to concerns about psychological destabilisation. The interaction between psychedelics and concomitant medications is also uncertain, so participants are usually required to discontinue other medications or are excluded altogether.
One risk which is more difficult to anticipate is HPPD, characterised by persistent and distressing perceptual disturbances. HPPD has been reported once in modern clinical studies (Mertens et al., 2026). It has been reported in approximately 4% of recreational users (Baggott et al., 2011), with non-distressing visual changes occurring much more frequently – up to 61% in one survey (Baggott et al., 2011). While one interpretation of this may be that the controlled research environment is protective, another is that by excluding people at risk or using dosing regimens which pose low risks of HPPD, this is falsely reassuring. Indeed, many studies enrol only participants with prior psychedelic experience who have previously tolerated these substances well. The aetiology of HPPD is complex (Butler et al., 2026; Halpern and Pope, 2003; Halpern et al., 2018; Vis et al., 2021; Zhou et al., 2025), but is best conceptualised as a functional or somatic disorder with symptoms modulated by anxiety (Butler et al., 2026). Further research is required to delineate risk factors for developing the disorder.
Psychedelics have acute sympathomimetic effects, and there is some experimental evidence suggesting that chronic use may lead to cardiac effects such as arrhythmias, cardiotoxicity, and platelet aggregation (for review, see Wsół, 2023). Although the low frequency of doses in clinical trials is very likely to be safe, further research is needed to explore the potential link with chronic use, again particularly in people with underlying cardiac conditions or cardiac risk factors (for review, see Nahlawi et al., 2025).
Given the incomplete reporting to date (Hinkle et al., 2024), collecting data on adverse events is of critical importance to the field. When obtaining informed consent from participants, be transparent about the limits in predicting the precise experience people will have. It remains unclear whether different psychedelics have distinct risk profiles, and, therefore, careful consideration should be given to the specific psychedelic used and the characteristics of your target population.
Finally, ensure your participants can realistically complete the study protocol. For example, if the study requires extended periods without nicotine or caffeine, consider whether this is feasible for your population. If participants are undergoing neuroimaging during the acute psychedelic experience, consider whether they can tolerate, for example, the sensory experience of being in an MRI scanner. Individuals’ familiarity with psychedelics would be an important consideration in this case; while in our experience, experienced users manage this without significant issue, it may be challenging for those without prior experience, who may become overwhelmed.
Suggestion 6: Develop a feasible and ethical recruitment strategy
There are many important factors to feasible and ethical recruitment strategies in human drug studies. While not exclusive to psychedelic studies, the time commitment required from participants is often substantial, particularly in studies involving longer-acting drugs. Considering methods to minimise inconvenience is important. For example, implementing efficient pre-screening procedures, such as remote interviews, reduces the number of participants invited for in-person screening, and enriches eligibility. Electronic pre-screening tools, such as pre-screening surveys, can further streamline this process and reduce administrative burden.
Pre-screening interest in psychedelic studies can be high, which presents both opportunities and challenges. When advertising your study, it is best to avoid listing detailed inclusion and exclusion criteria. This reduces the risk of potential participants tailoring their responses to qualify and is especially important to maintain both scientific and participant safety-related integrity.
Psychedelic studies have historically had a lack of participant diversity (Hughes and Garcia-Romeu, 2024), and under-reporting of certain protected characteristics. Given the significant public interest in this field, there is both an opportunity and a responsibility to improve representation through proactive and inclusive recruitment strategies.
Finally, be mindful of potential disappointment among those who are not selected, as well as those who may receive placebo. Clear communication about study procedures, expectations, and the likelihood of placebo assignment is essential to managing expectations and maintaining trust.
Given the context-sensitive state of mind induced by psychedelics, and the possibility of psychological distress (known as a ‘bad trip’), attention must be paid to mitigating these risks. While an adequate discussion of this is beyond the scope of this article, relevant safeguards include excluding participants with a personal or family history of psychotic conditions; excluding those who have had previous adverse psychoactive drug experiences; developing a trusting rapport between the participant and researchers before the psychedelic session; and having a full discussion regarding participants’ previous experiences with mind-altering substances. For a comprehensive discussion of risks and mitigation strategies, see (Johnson et al., 2008).
Suggestion 7: Ask pharmaceutical companies the right questions
Once your study design and drug have been selected, the next step is to find a pharmaceutical supplier. Despite growing global interest, sourcing psychedelics for research remains challenging. Knowing the right questions to ask in advance can help focus your search:
Does the company manufacture to GMP standard? This is the recommended standard of drug manufacturing for human studies (Department of Health and Social Care, 2014), and the supplier should be able to provide documentation confirming their GMP status.
Do they manufacture your required doses, including matched placebos where required? This is not always the case; many suppliers have pipelines with specific dosages, and some do not produce matched placebos. If you are unable to find a company which meets your requirements, it may be possible to purchase the drug (active pharmaceutical ingredient) in bulk powder form and have it weighed and encapsulated by a licensed manufacturing facility, such as a pharmacy. At the time of writing, this facility must hold a Schedule 1 manufacturing licence and be able to weigh and prepare your full dose range – do check early that they have protocols to weigh your required dose.
What is the shelf-life of the product? Once you know your projected data collection period, ensure the product’s shelf-life will cover that duration and will not expire prior to study completion. Ask specifically about shelf-life from the point of purchase, not just the date of manufacture, and determine if you need multiple batches. Ask what the lead-in time is for a new batch.
If the company is outside the UK, do they have a process in place for import, and a preferred import pharmacy in the UK? This route may be necessary if your institution is unable to import the drug directly.
Once you have secured the drug product, packaging and labelling may present further challenges. Packaging and labelling requirements 3 vary by institution and depend on whether your study is a CTIMP. If your study involves the use of pharmacy services, early engagement with your pharmacy team to understand their policies is advisable. If your pharmacy requires each dose to be individually packaged, and your candidate pharmaceutical company does not provide this, you may require the manufacturing services of a third party, and this can come at a significant cost. For blinded studies, labels will need to be designed to support blinding and dispensing procedures, so be sure to review the label templates with your collaborating pharmacist. Many companies are unable to alter their label templates, or can do, but at a significant cost. 4
This is all in addition to the cost of the drug which may be supplied in bulk or ready weighed and prepared as a tablet or capsule (if for oral administration). The number of suppliers is growing so it is worth checking with your pharmacy and other researchers on current sources.
Your institution will need to enter into a series of agreements with the pharmaceutical company, which is not unique to psychedelic research. At first contact, most pharmaceutical companies will request a non-disclosure agreement (NDA), or confidential disclosure agreement (CDA), which should be reviewed and signed by your institution’s legal department. If you proceed, a licence agreement will follow, covering intellectual property, data use, and commercialisation terms. This will be reviewed by the institution’s legal team and, where necessary, amended in negotiation with the company. A separate quality agreement may also be required, defining responsibilities for manufacturing standards and regulatory compliance. This will be reviewed by both your institution’s legal team and likely institutional pharmacy staff, if applicable.
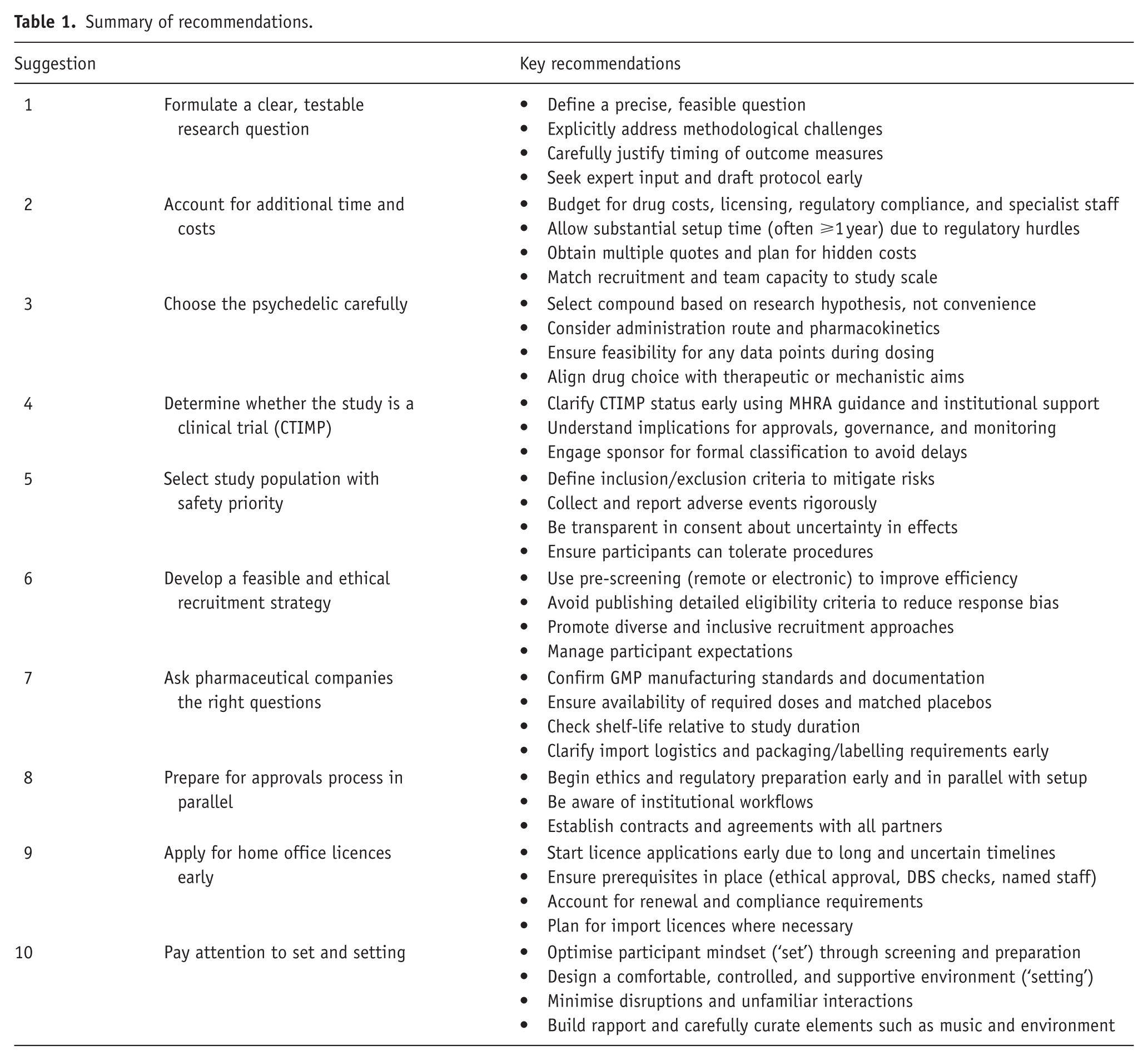
Summary of recommendations.
Suggestion 8: Prepare for the approvals process in parallel to the above
As with all scientific studies in people, the regulatory and ethical approvals process must be navigated. Ideally, much of the preparatory work can proceed in parallel with other study setup steps; for example, finalising drug supply is not a prerequisite for submitting your ethics application. The time taken for each stage of the approvals process can vary widely, so it is wise to begin preparing necessary documents in advance. These typically include the study protocol, participant information sheet, consent form, advertising materials, and screening questionnaires.
The approvals process varies between institutions. The likely first step is the risk assessment, where the study protocol is reviewed by the institution’s Risk Assessment Committee to evaluate whether the proposed study poses an acceptable level of risk. Once this has been approved, the study moves to sponsorship review, usually conducted by the university’s Research and Development office. In the case of non-CTIMPs, the sponsor is often the substantive employer of the principal investigator and sponsorship provides institutional indemnity.
Once sponsorship has been granted, you may proceed to apply for ethical approval. The specific route depends on factors such as CTIMP status, whether National Health Service (NHS) patients or infrastructure are being used, and local institutional policies. The Health Research Authority Health Research Authority (HRA) decision tool can help determine the appropriate route (Health Research Authority, 2022). For example, if your study uses NHS infrastructure (such as a pharmacy for storage or dispensing the drug) but does not involve NHS patients, you may require HRA governance approval without a full NHS Research Ethics Committee (REC) review. This must be specified on the Integrated Research Application System platform, the platform on which you would apply for MHRA/HRA ethical approval, whereas university internal ethics submissions typically use separate platforms.
In parallel with ethical and regulatory submissions, you must set up agreements with the various parties involved in your research (where relevant). This includes licensing for assessment tools and questionnaires, contracts for services (e.g. data collection platforms, laboratories for blood tests), and finalising legal agreements with the pharmaceutical company or companies supplying the psychedelic drug and placebo.
Suggestion 9: Apply for home office licences early
Conducting psychedelic research currently requires obtaining the appropriate Home Office licences, a process that is both time-consuming and expensive. The Home Office website lists step-by-step instructions on how to apply (Home Office, 2025). For non-CTIMPs, a domestic research licence is required which covers Schedule 1 research activities under a named researcher. If your institution runs psychedelic studies regularly, there may already be a licence in place; however, the Home Office should be updated regarding any new studies which would fall under this licence. For completely new licences, a compliance officer visit may be required, which further adds to the timeline.
Before applying for a licence, you should have ethical approval in place, and all relevant staff named on the licence (including the Chief Investigator and those responsible for premises security) must have completed enhanced Disclosure and Barring Service (DBS) check. These DBS checks are separate to your institution’s standard DBS checks, and information is available on the Home Office website (Home Office, 2025). Any prescribers to be added to the licence should be communicated to the Home Office via email once the application has been submitted; however, this is only required for clinical trial licences. Once granted, such licences typically require annual renewal, with associated fees and potential inspections.
If your drug is manufactured abroad, an import licence must be obtained from the Home Office. The import licence may be applied for once there is a domestic licence in place (which covers possession or storage). 5 The Home Office website contains step-by-step instructions (Home Office, 2022). Generally, the import licence must be in place before the pharmaceutical company can apply for an export licence from their respective country.
Suggestion 10: Pay attention to set and setting
A unique consideration in psychedelic studies is the set and setting (Carhart-Harris et al., 2018b). ‘Set’ refers to the individual’s mindset going into the experience and can have profound implications for their subjective experience. Elements of the set may be selected for through eligibility criteria, for example, a lack of negative past psychedelic experiences, recent traumatic events, and mental health history. Additional factors less explicitly screened for include components of the participants’ personality, such as their relationship with control and ability to surrender to the experience.
The ‘setting’ refers to the physical and social environment in which the dosing takes place. Purposefully designing the setting can significantly impact participant comfort and safety. A comfortable and private dosing room is important, equipped with, for example, a bed, blanket, eye mask, and headphones. Avoiding unexpected noises such as slamming doors and interruptions is advised. Music can have a profound impact on experience, and curating a playlist which aligns with the level of therapeutic support given to the participant is important. Familiarity and rapport with the researcher supervising the participant is important, as well as minimising interactions with unfamiliar people. There are numerous factors to consider, and speaking with experienced psychedelic researchers can help guide you. For a comprehensive review on how aspects of set and setting may be optimised, see Johnson et al., 2008, and Pronovost-Morgan et al., 2025, which provides a comprehensive account of set and setting, as well as guidance on how to report these factors in psychedelic trials.
Conclusion
Psychedelic research continues to develop, with a clear need for well-powered studies employing diverse methodologies to investigate underlying mechanisms. Their setup involves navigating complex and, sometimes, unfamiliar regulatory and logistical landscapes. While the process can be challenging and resource-intensive, early planning and institutional support can make it achievable. We hope this guide serves as a practical starting point for researchers considering exploring this dynamic and rapidly evolving field.
Footnotes
Acknowledgements
The authors would like to acknowledge Catherine Bird for her advice on some topics included in this article.
Author contributions
Conceptualisation: AC. Writing – original draft: AC and MB. Writing – reviewing and editing: AC, MB, and MAM. Supervision: MAM.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: AC is a Wellcome Trust Doctoral Clinical Research Fellow (223486/Z/21/Z). MB is a Wellcome Trust Doctoral Clinical Research Fellow (227515/Z/23/Z). The funders played no role in the decision to publish or preparation of this article.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MAM has research funding from Nxera and Lundbeck and received in-kind contributions from Compass Pathways. He has consulted for Boehringer Ingelheim and Nxera and received speaker fees from Takeda. MB works as a clinical trial doctor on commercially and publicly funded psychedelic trials.