
Abstract
Regional blood flow within the brain is tightly coupled to regional neuronal activity, a process known as neurovascular coupling (NVC). In this study, we demonstrate the striking role of SUR2- and Kir6.1-dependent ATP-sensitive potassium (KATP) channels in control of NVC in the sensory cortex of conscious mice, in response to mechanical stimuli. We demonstrate that either globally increased (pinacidil-activated) or decreased (glibenclamide-inhibited) KATP activity markedly disrupts NVC; pinacidil-activation is capable of completely abolishing stimulus-evoked cortical hemodynamic responses, while glibenclamide slows and reduces the response. The response is similarly slowed and reduced in SUR2 KO animals, while animals expressing gain-of-function (GOF) mutations in Kir6.1, which underlie Cantú syndrome, exhibit baseline reduction of NVC as well as increased sensitivity to pinacidil. In revealing the dramatic effects of either increasing or decreasing SUR2/Kir6.1-dependent KATP activity on NVC, whether pharmacologically or genetically induced, the study has important implications both for monogenic KATP channel diseases and for more common brain pathologies.
Introduction
Neural activity is closely related to cerebral blood flow, and the close spatial and temporal relationship between the two, termed neurovascular coupling (NVC), is the basis for functional magnetic resonance imaging (fMRI). 1 Appropriate NVC is generally assumed to be necessary for temporal matching of blood supply to neuronal demand, and disruption of NVC in various brain pathologies may represent a causal basis for neuronal dysfunction. 1 Multiple cell types, including capillary endothelial cells, astrocytes, and pericytes, have been implicated in the coupling of neuronal activity to increased blood flow, but underlying molecular players remain unclear. Various studies have elucidated mechanisms by which metabolic by-products of neuronal activity may trigger electrical responses in nearby vascular smooth muscle (VSM) and pericytes that reduce their contractile state, leading to arteriolar and capillary dilation, and to increased blood flow.2,3 One potential metabolic by-product signal is elevated external [K+], a rapid and potent vasodilator. 4 Local release of K+ can activate Kir2 channels in vascular endothelium leading, via gap junctional coupling, to hyperpolarization and consequent relaxation of upstream contractile cells, resulting in locally increased blood flow. 5
ATP-sensitive potassium (KATP) channels are inhibited by intracellular ATP and activated by MgADP, thereby linking membrane potential to metabolic activity in cells that express them. 6 Throughout the vascular system, KATP channels are formed as heteroctameric complexes, with 4 pore-forming Kir6.1 subunits, responsible for ATP inhibition, and 4 regulatory SUR2B subunits responsible for Mg-ADP activation and sensitivity to KATP channel openers (e.g. diazoxide/pinacidil) and inhibitors (e.g. glibenclamide). 7 These KATP channel isoforms are encoded by a pairs of genes,8,9 ABCC9 and KCNJ8 which encode SUR2B and Kir6.1, respectively, and are widely expressed in vascular smooth muscle and in cerebral pericytes.10 –12 Recent studies have implicated ATP-sensitive potassium (KATP) channels as additional transducers of NVC, within the neurovascular unit,11,13 although their ubiquitous presence throughout the vascular system raises the possibility that they may also affect neurovascular coupling systemically.
In the present study we set out to test the role of Kir6.1/SUR2B-type KATP channels in NVC in vivo, in conscious mice. Intriguingly, we demonstrate that either global increase or decrease of KATP activity reduces NVC, with pharmacological KATP activation by pinacidil being capable of completely abolishing the response, while pharmacological inhibition of KATP with glibenclamide or genetic knockout of the SUR2 subunit both result in slowed and diminished NVC responses. Severe loss–of-function (LOF) mutations in ABCC9 underlie the very rare channelopathy ABCC9-dependent Intellectual disability Myopathy Syndrome (AIMS), 14 and the present results thus suggest a potential role for altered NVC in AIMS disease etiology. Conversely, gain-of-function (GOF) mutations in KCNJ8 and ABCC9 underlie the rare channelopathy, Cantú syndrome.9,15,16 Characterized by cardiomegaly, hypertrichosis and distinct facial features, Cantú patients also exhibit dilated and tortuous cerebral vasculature and neurological abnormalities, as well as high incidence of migraines with aura. Using a mouse model of KCNJ8-dependent Cantú syndrome, we further demonstrate a reduction in baseline NVC and increased sensitivity to pinacidil, providing support for disrupted NVC, as a result of KATP GOF, being a potential causal mechanism of migraines in Cantú syndrome, or in subjects exposed to potassium channel opener drugs.
Material and methods
Mouse lines used in this study
All studies were performed in compliance with the standards for the care and use of animal subjects defined in the NIH Guide for the Care and Use of Laboratory Animals and were reviewed and approved by the Washington University Institutional Animal Care and Use Committee. For optical imaging experiments including GCaMP, Thy1-GCaMP6f mice (JAX 024276) were crossed with the mouse genotype of interest. Littermate mice carrying only the GCaMP6 transgene were used for comparisons and mice were between 16 and 20 weeks old during experiments. Generation of Cantú mice using CRISPR/Cas9 genome editing was previously reported: briefly, KCNJ8 [c.193G>A/195A>G] mutations were introduced to generate Kir6.1[V65M] (Kir6.1wt/VM) mice, which model the autosomal-dominant Kir6.1[V65M] observed in human CS. 17 Mutations were originally introduced on various genetic backgrounds, but all animals used in the present study had been bred back to C57/Bl6 strain for >6 generations. SUR2 KO mice contain a premature stop mutation at position 475 in transmembrane domain 1 of SUR2 resulting in complete loss of KATP currents in cells expressing Kir6.1/SUR2. 18 Although we have seen no appreciable sex differences in any of the genetic lines used, and are unaware of any such reports from others, approximately even numbers of male and female mice were used for all experimental groups. Data reporting has followed the ARRIVE 2.0 guidelines. 19 Unless otherwise noted, all drugs (Sigma-Aldrich Inc.) were administered intravenously. Data are described as (n) number of samples from (N) number of animals. No a priori sample size calculation was done. No criteria were set for including or excluding animals. To minimize potential confounders, the order of treatments and measurements was randomized. Because anonymizing animals was not practical, investigators knew the group allocation during the experiment and while assessing outcome. Statistical methods used for each analysis, including software used are noted. Species, strain, sex and age are noted.
Intact skull windows for optical imaging
Mice were anesthetized with isoflurane (3.5 liters per minute for 4 minutes, 3–5% induction, 1.5% maintenance), and head-fixed in a stereotaxic frame (Figure 1). Temperature was maintained using a thermostatic heating pad. After fixation on a bite bar and lubrication of the eyes, the hair on the scalp was shaved and the scalp was cleaned with betadine and ethanol. A local injection of lidocaine was administered to the site of incision. The scalp was then incised and retracted along the midline. Metabond dental cement (C&B Metabond, Parkell Inc.) was used to secure a custom-designed Plexiglas window to the skull, completely covering the surgical opening (Figure 1). Mice were then placed in a temperature-controlled incubator and allowed to recover for at least 5 days. No effect of window implantation on behavior was noted.
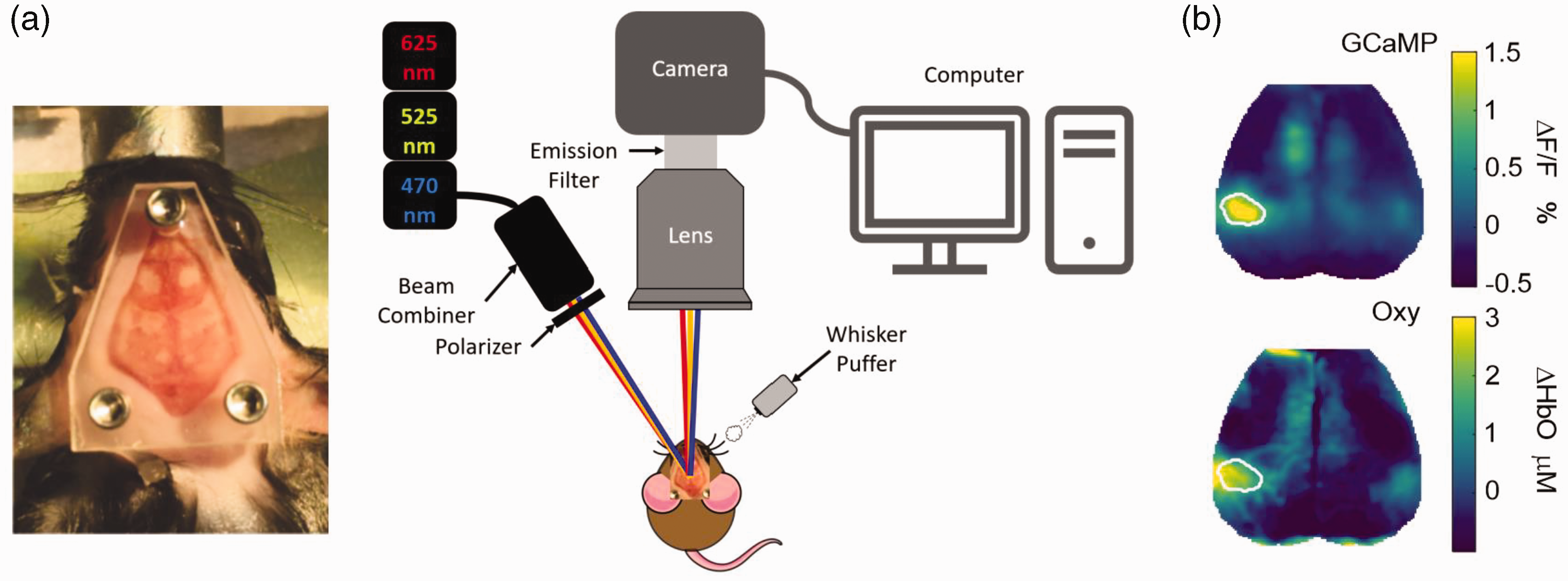
Experimental setup. (a) To place the imaging window under anesthesia, the scalp was incised and retracted along the midline and a custom-designed Plexiglas window was secured with dental cement to completely cover the surgical opening (left). For imaging, the conscious mouse was head-fixed on the stage using non-reflective screws. LEDs illuminated the cortex sequentially and reflected light levels were captured by the camera during optical imaging recordings. Whisker stimulation was provided by an air puffer fixed to blow air on either side of the whiskers. Changes in light levels were then processed and analyzed using MATLAB (right). (b) Representative image showing average GCaMP and oxygenated hemoglobin (Oxy) signals during right whisker stimulation in control mouse.
Wide-field optical imaging
Wide-field optical imaging was performed to measure cortical calcium and hemodynamic activity, as previously.20 –23 The dorsal neocortex was illuminated sequentially by three light emitting diodes (LEDs) of wavelengths 470 nm (Mightex Systems, LCS-0470-50-22), 525 nm (Mightex Systems, LCS-0525-60-22), and 625 nm (Mightex Systems, LCS-0625-07-22). The 470 nm LED was used for GCaMP excitation and a 500-nm long pass filter (Semrock, FF01-500/LP-25) in front of the camera blocked residual excitation light from reaching the camera sensor. An additional 460/60 nm bandpass filter (FF01-460/60-25) was placed in front of the excitation source to spectrally separate fluorescence excitation light from fluorescence emission. The remaining LEDs provided diffuse reflectance illumination for multispectral hemodynamic imaging. A custom light engine was built such that all LED beams were collimated using plano-convex lenses (Thorlabs, LA1433) and combined with dichroic beam combiners (Mightex Systems, LCS-BC25-0480, LCS-BC25-0605). Image detection was performed with a cooled sCMOS camera (Zyla 5.5-USB3, Andor) with an 85-mm f/1.4 camera lens and 75 Hz acquisition framerate (25 Hz acquisition rate for each channel). The field of view was approximately 1 cm × 1 cm, covering the dorsal neocortex, and contained a 512 × 512-pixel array. To ensure a high SNR and acquisition rate, images were binned 4 × 4 on camera, resulting in images containing 128 × 128 pixels, with each pixel being approximately 78 µm × 78 µm. A series of linear polarizers were used in front of both the LED sources and the camera lens to reduce specular reflection from the mouse skull.
Image acquisition
LED illumination sequences and camera triggering were controlled by a data acquisition board (PCI-6733, National Instruments) using MATLAB (MathWorks). Solis (Oxford Instruments) was used for image acquisition.
Prior to data collection, mice were acclimated to head fixation while wrapped in a felt blanket for 20 minutes before any imaging recordings were performed. Whisker stimulation was performed in awake mice for 5 minutes on the left and right whisker pads using computer controlled, 40 PSI air puffs in a block design (5 s rest; 5 s of 1 Hz, 0.1 s pulses; 10 s rest; 15 blocks/5 minutes recording per mouse).
Image processing
Brain masks were created for each imaging timepoint for each mouse, and all analyses that followed were performed only on pixels labeled as brain. Brain masks and image sequences of brain pixels were affine transformed to Paxinos atlas space using the cranial landmarks of the intersection of the frontonasal and interfrontal suture and the sagittal and lambdoid suture (lambda). Five seconds of dark frames were subtracted from all raw data prior to analysis. Images were temporally detrended as previously described. 24 Changes in 525 nm and 625 nm reflectance were used for hemodynamic spectroscopy, using the modified Beer-Lambert Law, 25 and oxygenated hemoglobin concentration changes were calculated from changes in light intensity as previously described. 26 Briefly, changes in the absorption coefficient due to hemodynamic activity were calculated from changes in light intensity using the equation: Φ(r,t) = Φ0 × exp(−Δμa(r,t) × L), where Φ(r,t) is measured light intensity, Φ0 is baseline light intensity, Δμa(r, t) is the change in absorption coefficient due to hemodynamic activity changes, and L is the optical path length of photons in the tissue being imaged. Changes in oxygenated and deoxygenated hemoglobin (which are assumed to obtain total hemoglobin) were calculated by the inversion of the system of equations Δμa,λ (r,t) = Eλ,i Δ[Hbi](r,t), where E is the matrix of extinction coefficients and i iterates through the two different hemoglobin species.
The raw GCaMP fluorescence signal was corrected for artifacts due to hemoglobin absorption using 525-nm reflectance, as previously described.24,27 For each mouse, maps of cortical responses to peripheral whisker stimulation were created by averaging responses to each of the 5 air puffs per block. Regions of interest (ROIs) were defined using the GCaMP signal within right and left somatosensory whisker barrel cortex, by thresholding individual and group-averaged evoked response maps, averaged across blocks and peaks at 50% of the maximum pixel intensity at each time point. The ROIs defined using GCaMP were used for analysis of hemoglobin signaling.
Inter-contrast analysis
Cross-covariance between calcium and oxygenated hemoglobin (OxyHb) signals was used to estimate coupling between the mean calcium and oxygenated hemoglobin signals within the individual ROIs described above. To calculate the cross-covariance a lag was applied to the OxyHb signal, relative to the GCaMP signal, from −15 s (GCaMP signal is delayed 15 seconds) to 15 s (Oxy Hb signal is delayed 15 s), in 0.5 s intervals. At each lag, the covariance of was calculated between the adjusted signals using the COVARIANCE.S function in Microsoft Excel©. This was done for each recording and then averaged within and across mice. From the cross-covariance, peak amplitude, time-of-peak amplitude (Peak lag), full width at half maximum, and energy of cross-covariance (area under the squared covariance curves) were calculated.
Measurement of blood pressure and cerebral blood flow rates
Long-term implanted catheter measurements of blood pressure were not available for our study, and intra-arterial blood pressure measurements were performed in anesthetized mice to ensure accuracy of blood pressure readings, as correlations between awake tail cuff blood pressure measurements (indirect) and anesthetized intra-arterial blood pressure measurements (direct) have not been reproducible, 28 likely due to confounds of stress, increased sympathetic tone, and the inherent properties of peripheral arteries. Mice were anesthetized as indicated above in Material and Methods. After attainment of appropriate level of anesthesia, 70% isopropyl rubbing alcohol and Betadine scrub were performed three times to sterilize the surgical area. The surgical area was shaved and then a skin incision was made and the femoral artery exposed. A silicone catheter was inserted into the vessel, advanced 2 mm, and secured with silk which remained subcutaneous. The catheter was filled with saline and tunneled, and attached to a Digi-Med Blood Pressure Analyzer 400. Mice were then transferred to a Kopf frame and head fixed. Blood pressure was allowed to stabilize such that fluctuations in blood pressure did not exceed 1 mm Hg over 5 minutes. Blood pressure and cerebral blood cell flow rate were then measured every 5 minutes beginning at the time of injection of each drug (t = 0). Cerebral blood flow rates were measured in primary somatosensory cortex by laser doppler flowmetry while mice were head-fixed with ear bars. The flow probe was stabilized using a micromanipulator attached to the Kopf frame on which the mouse was mounted.
After blood pressure readings ceased, mice were euthanized using pentobarbital.
Statistics
Data are presented as mean ± 95% CI, unless otherwise noted. Normality was tested for each data set used in statistical comparisons using the D’Agostino & Pearson test. Differences between two groups were tested using 2-tailed t-tests and among several groups were tested using analysis of variance (ANOVA) and post-hoc Turkey’s test, when data were normally distributed. For data that were not found to be normally distributed the difference between two groups was tested using a Wilcoxon test and the difference between multiple groups where one or more was not normally distributed was tested using the Kruskal-Wallis test. Significant differences among groups are indicated with p-values, and non-significant (p > 0.05) differences are not shown. Effect sizes for statistically significant comparisons are listed in figure legends.
Results
Systemic pharmacological modulation of KATP markedly alters neurovascular coupling
We first assessed the impact of systemic pharmacological KATP channel activation and inhibition on NVC in awake conscious animals (Figure 1) by IP injection of the KATP channel activator pinacidil (400 mg/kg), or of the inhibitor glibenclamide (25 mg/kg). The neuronal (GCaMP) and vascular (oxygenated hemoglobin, OxyHb) evoked response signals (stimulus: 1 Hz, 0.1-second, 40 PSI air puffs) within the whisker barrel cortex were measured before and 25 minutes after drug injection, to determine baseline and drug-modulated responses (Figure 2(a)). In GCaMP-positive mice with wild type KATP (WT), discrete whisker barrel GCaMP responses to each stimulus were reproducibly associated with a rapid local increase of OxyHb that was maintained during the stimulation period and then declined over 5–10 seconds following the end of stimulation. Glibenclamide treatment resulted in a delayed rise of the OxyHb signal and ∼50% reduction in the maximum change in oxygenated hemoglobin (ΔHb AUC, Figure 2(a) and (b)). Pinacidil had a more dramatic effect, essentially abolishing the OxyHb response (Figure 2(a) and (b)). Interestingly, both treatments slightly reduced the GCaMP signal by ∼20% (Figure 2(a) and (b)). As an estimate of NVC, the relationship between GCaMP and OxyHb was quantified using cross-covariance. A significant reduction in coupling is demonstrated by reduced peak amplitude, as well as a delay to peak, in animals treated with glibenclamide and more dramatically in animals treated with pinacidil (Figure 2(c) and (d)).
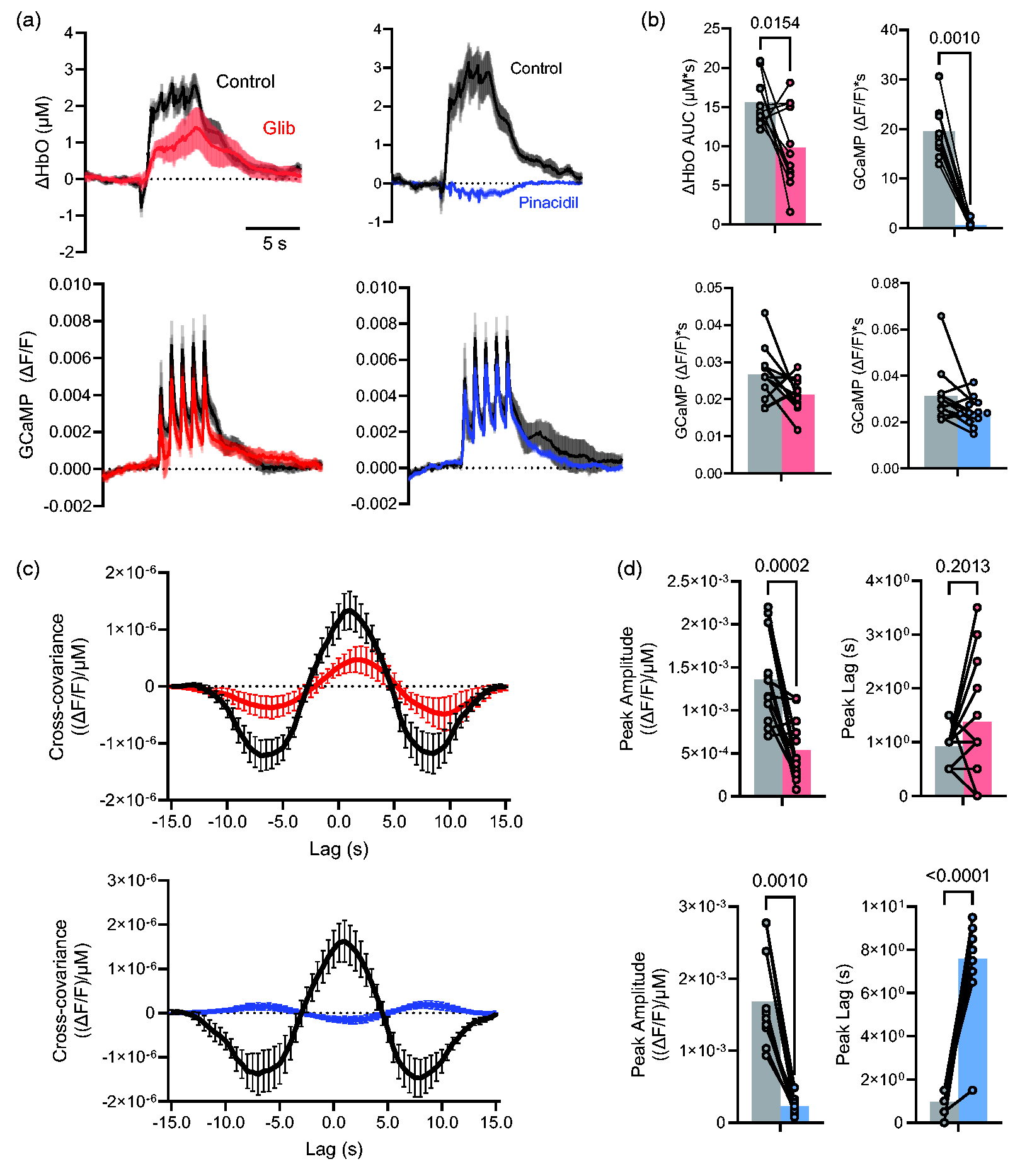
Effect of pharmacological KATP modulation on neurovascular coupling and neuronal activity. (a) Change in oxygenated hemoglobin (OxyHb, above) and GCaMP (below) signals during whisker stimulation (arrowheads) in control and in the presence of 25 mg/kg glibenclamide (left) or 400 mg/kg pinacidil treatment (n = 12 hemispheres from 7 mice in each case, mean ± 95% CI. for each time point). (b) Mean area under the curve (±95% CI.) of oxygenated hemoglobin (ΔHbO AUC, above) and GCaMP (below) signals, measured from ROIs as in Figure 1(b). (c) Average cross-covariance of GCaMP and Oxy signals from mice before (black) and after (color) treatment with glibenclamide (above) and pinacidil (below) and (d) Average peak amplitude (±95% CI.) of cross covariance in the positive direction as well as average peak lag (±95% CI.) are shown for glibenclamide (above) and pinacidil (below).
KATP knockout or Cantú syndrome KATP GOF also reduce neurovascular coupling
Vascular KATP channels are generated by Kir6.1 and SUR2B subunits, encoded by KCNJ8 and ABCC9, respectively.29,30 Loss of ABCC9-dependent (and potentially KCNJ8-dependent) KATP channels results in a distinct complex disorder which we have termed ABCC9-dependent intellectual disability myopathy syndrome (AIMS). 14 It has previously been demonstrated that loss of KCNJ8-encoded Kir6.1 disrupts NVC, with a delayed and reduced blood flow response to whisker stimulation in anesthetized animals. 12 Loss of SUR2 has similarly been shown to attenuate vasodilation induced by oxygen and glucose deprivation, although NVC has not previously been tested in these animals. We observe a delayed rise and ∼30% reduction in the maximum amplitude of OxyHb in ABCC9 (SUR2) KO mice (n = 10, purple in Figure 3(a) and (b)), very similar to that seen in the presence of glibenclamide in WT animals (Figure 2). In contrast to the abolition of NVC by pinacidil in WT mice, pinacidil (400 mg/kg) caused only a relatively small reduction of NVC in these KO mice (Figure 3(a) and (b)), confirming that the striking abolition of NVC in WT mice (Figure 2(b)) is through activation of SUR2-containing KATP channels. Cross-covariance between OxyHb and GCaMP (Figure 3(c) and (d)) shows that, as with glibenclamide-treated WT, stimulus-evoked NVC is significantly delayed in SUR2 KO animals relative to WT control animals (Figure 3(c), Time delays of ∼1 s in WT animals and ∼2.5 s in SUR2 KO animals), but cross-covariance plots were similar in SUR2 KO before and after pinacidil treatment (Figure 3(d)).
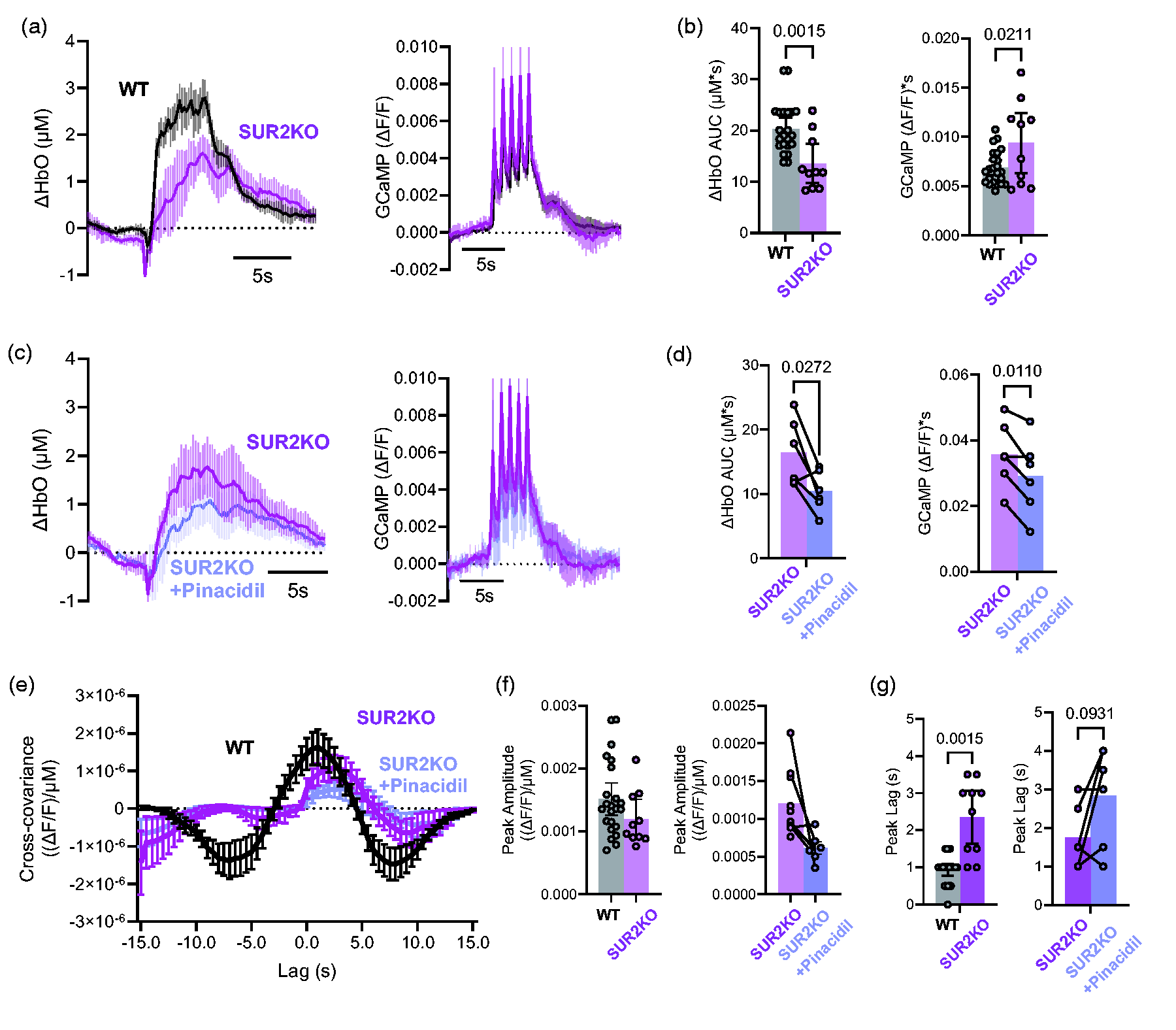
Effect of KATP KO on neurovascular coupling. (a) Mean change (±95% CI.) of oxygenated hemoglobin (ΔHbO AUC, left) and GCaMP (right) signals, measured from ROIs as in Figure 1(b), in WT (n = 23 hemispheres from 13 mice, and SUR2 KO mice (n = 10 hemispheres from 5 mice). (b) Mean area under the curve (±95% CI.) of oxygenated hemoglobin (ΔHbO AUC, above) and GCaMP (below) signals, measured from ROIs as in A. (c) Mean change (±95% CI.) of oxygenated hemoglobin (ΔHbO AUC, left) and GCaMP (right) signals, measured from ROIs as in Figure 1(b), in SUR2 KO mice (n = 6 hemispheres from 3 mice), before and after treatment with 400 mg/kg pinacidil. (d) Mean area under the curve (±95% CI.) of oxygenated hemoglobin (ΔHbO AUC, above) and GCaMP (below) signals, measured from ROIs as in C. (e) Cross-covariance calculated from the experiments above. (f) Average (±95% CI.) peak amplitude and (g) peak lag from cross-covariance in E.
Both treatment with KATP channel openers such as pinacidil and Cantú syndrome, which results from GOF mutations in ABCC9 or KCNJ8, are associated with various neurological features, including prominently increased risk of migraines. Such effects may be linked to inappropriate blood flow control. In Kir6.1[V65M] mice, which recapitulate multiple key Cantú syndrome features, 9 the maximum OxyHb signal was significantly reduced compared to littermate controls (Figure 4(a) and (b)). As with WT mice, glibenclamide treatment reduced the maximum OxyHb response for V65M mice, and cross covariance analysis revealed a significant reduction in coupling, with a peak amplitude of 1.5 × 10−6 in WT and 9.7 × 10−7 in V65M (Figure 4(c) and (d)). We measured the dose response of pinacidil on maximal oxygenated hemoglobin in Control and V65M mice. There was a significant increase in pinacidil sensitivity in the latter (Figure 4(e)).
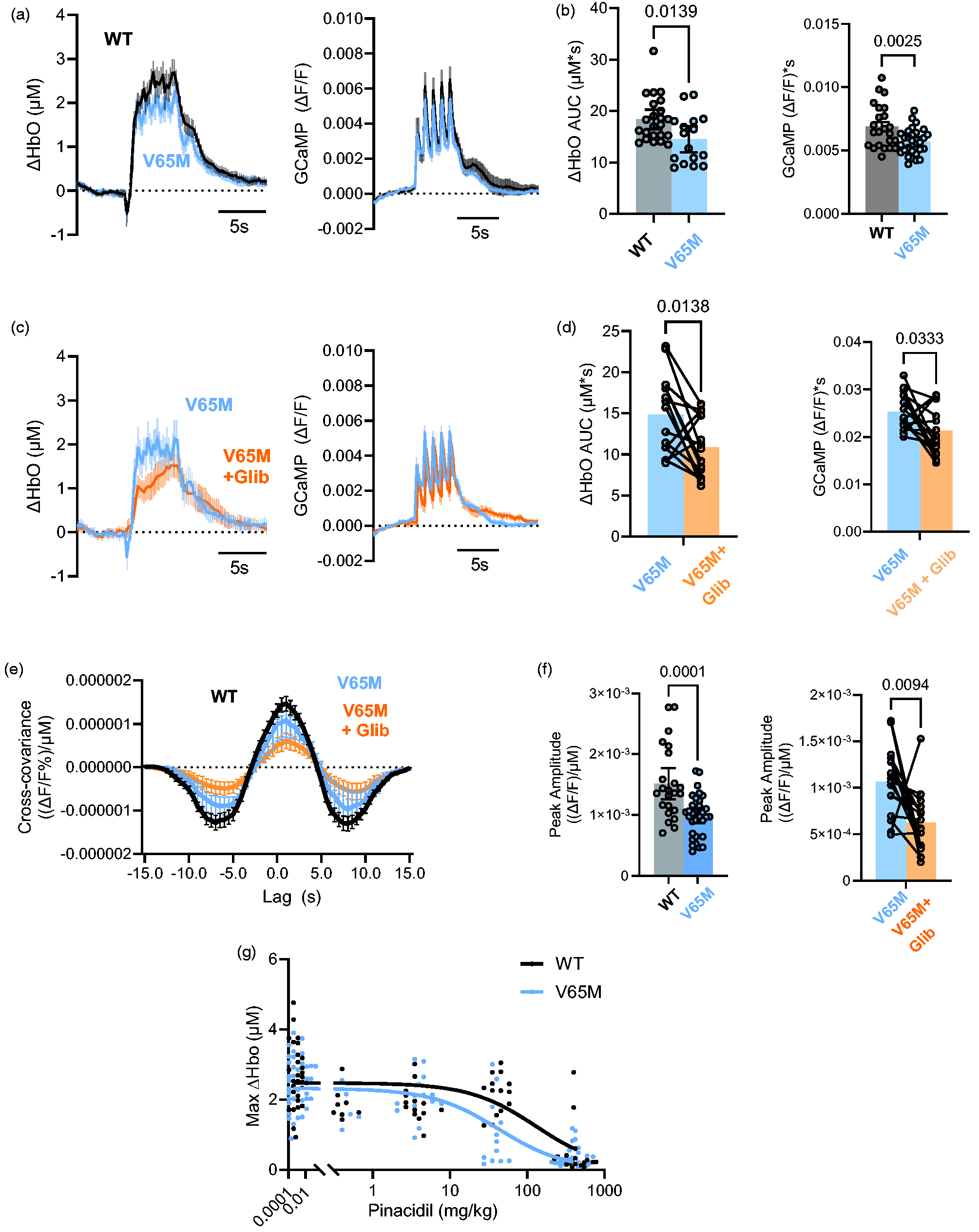
Effect of KATP GOF mutation on neurovascular coupling. (a) Mean change (±95% CI.) of oxygenated hemoglobin (ΔHbO AUC, left) and GCaMP, right) signals in WT (n = 25 hemispheres from 13 mice) and Kir6.1[V65M] mice (n = 15 hemispheres from 8 mice) (b) Average (±95% CI.) AUC of oxygenated hemoglobin (left) and GCaMP (right) signals from experiments in A. (c) Mean change (±95% CI.) of oxygenated hemoglobin (ΔHbO AUC, left) and GCaMP, right) signals in V65M (n = 15 hemispheres from Continued.8 mice) before and after treatment with glibenclamnide (25 mg/kg). (d) Average (±95% CI.) AUC of oxygenated hemoglobin (left) and GCaMP (right) signals from experiments in C. (e) Cross-covariance calculated from V65M experiments in A and C. (f) Comparison of average (±95% CI.) peak amplitude of the cross-covariance between WT and V65M (left) as well as a paired comparison between V65M mice before and after glibenclamide treatment (right) and (g) Average (±95% CI.) maximum change in oxygenated hemoglobin (hemoglobin (Max ΔHbO) signals as a function of injected [pinacidil], in WT and Kir6.1[V65M] mice. Mean data at each concentration fit with sigmoidal function, K1/2 = 130.0 mg/kg and 42.7 mg/kg, for WT and V65M, respectively (p = 0.0122, F-test), H fixed at 1 in each case.
Systemic consequences of altered vascular resistance
Conceivably, the striking abolition of NVC in WT mice by pinacidil might result from a systemic action of pinacidil to maximally increase KATP channel activity such that vascular tone is reduced to such an extent that further local dilation is not possible. Conversely, as a small uncharged molecule, pinacidil would be expected to diffuse across the blood-brain barrier, although the extent of accumulation within the brain is unknown. Both pinacidil (400 mg/kg) and the vasodilator adenosine (10 mg/kg) significantly lowered blood pressure and decreased cerebral red blood cell velocity in anesthetized WT mice (Figure 5(a) to (d)) but, in distinct contrast to pinacidil, adenosine had no effect on NVC in the conscious WT mouse (Figure 5(e)), consistent with a specific effect of pinacidil on NVC.
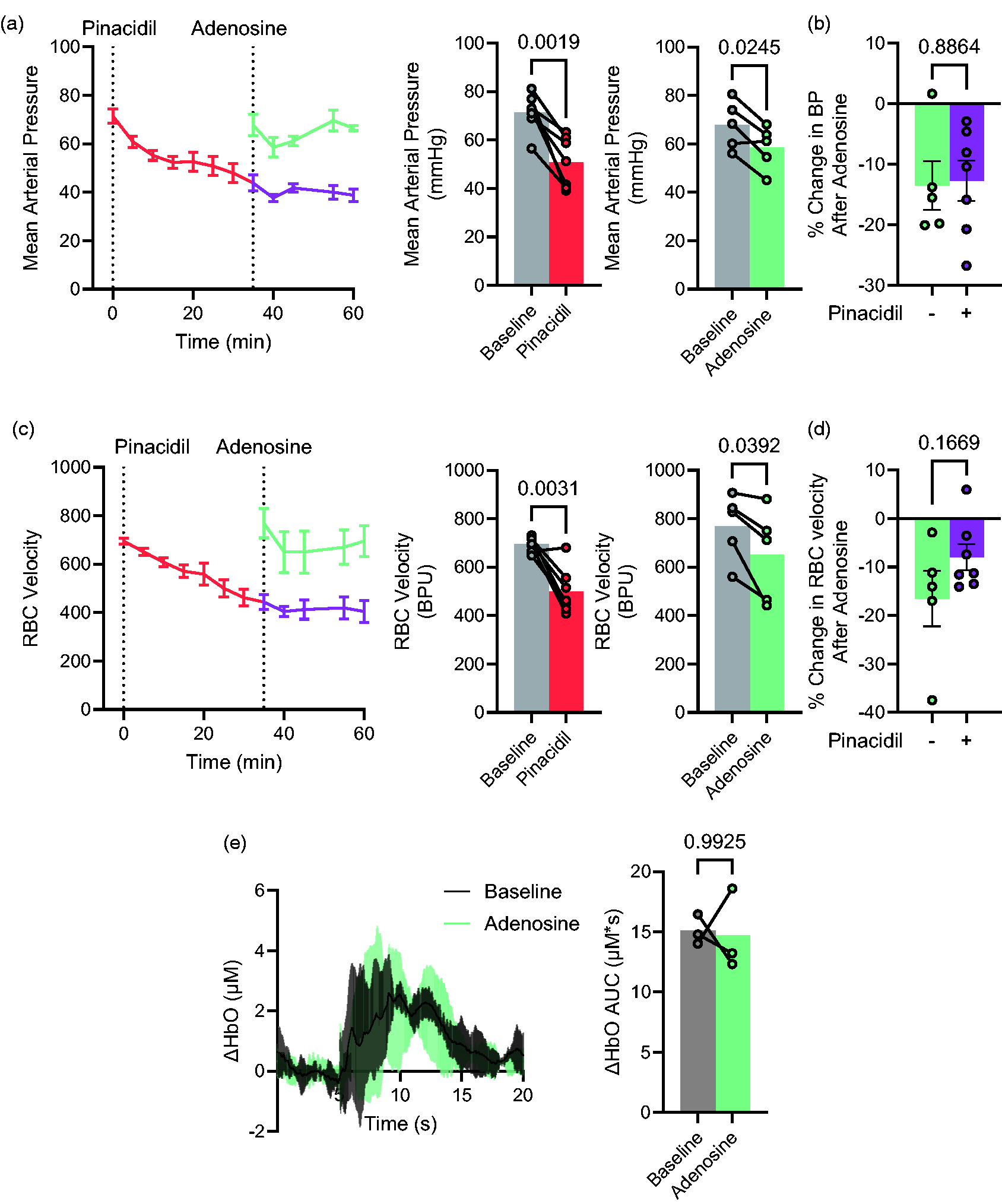
Systemic consequences of pinacidil and adenosine. (a) Mean arterial pressure under baseline conditions and following addition of 400 mg/kg pinacidil (red, n = 7 animals) or 10 mg/kg adenosine (green, n = 7 animals), or adenosine at t = 35 min after pinacidil (purple, n = 7) in anesthetized mice (left). Mean arterial pressure at baseline or versus 25 min after pinacidil or 5 min after adenosine. (b) Percent change in BP after adenosine in absence or presence of pinacidil from A. (c) Red blood cell velocity, measured in blood perfusion units (BPU), under baseline conditions and following addition of pinacidil (red) or adenosine (green) alone, or adenosine at t = 35 min after pinacidil (purple) in anesthetized mice (left). Mean RBC velocity at baseline or versus 25 min after pinacidil or 5 min after adenosine. (d) Percent change in RBC velocity after adenosine in absence or presence of pinacidil from A and (e) Mean change (±95% CI.) of oxygenated hemoglobin (ΔHbO AUC, left) and GCaMP (right) signals, measured from ROIs as in Figure 1(b), at baseline and 10 minutes after adenosine (10 mg/kg) in WT mice. Data show mean (±95% CI.) from n = 3 hemispheres from 3 mice.
Discussion
Ion channel players in neurovascular coupling
It has long been recognized that neurovascular coupling involves transduction of chemical signals released from active neurons to electrical signals in cells associated with the capillaries to upstream smooth muscle or other contractile cells. These processes lead to relaxation and dilation of the local vasculature and subsequent increases in blood flow and oxygenation. While underlying mechanisms remain unclear, there appears to be both early contributions from cell-specific vasoactive messaging, and then later responses to signals from by-products of neuronal activity.23,31 –34 A well-championed mechanism implicates increased external [K+], resulting from increased neuronal activity, causing increased inward rectifier Kir2 channel activity in capillary endothelial cells, which spreads retrograde via gap junctions to hyperpolarize upstream arteriolar smooth muscle or pericytes. 4
Recent work from several groups has also begun to implicate KATP channels in the local blood flow response to neuronal activity in isolated preparations,10 –12 although there have been few studies of the role of KATP channels in vivo and the existing studies are somewhat contradictory. In one study of anesthetized Kir6.1 KO mice, 12 a delayed and reduced neurovascular coupling (NVC) response to whisker stimulation was reported, while another study reported that resting cerebral blood flow and brain parenchymal partial pressure of oxygen were lower than in wildtype but blood oxygen level dependent responses triggered by visual stimuli were unaffected. 35 While not directly comparable, our own data in conscious animals are consistent with those of Ando et al, 12 with both pharmacologic inhibition of KATP with glibenclamide, or genetic knockout of SUR2 slowing and reducing the NVC response. Reduction of NVC by activation or inhibition of KATP channels, despite the opposing cellular consequences, indicates a reliance of NVC on a certain level of KATP activity and suggests that dynamic changes of KATP channel activity may normally be involved in the vascular response. We did not measure absolute levels of excitability or oxygenation in these experiments, and baseline levels could conceivably be different in the distinct genetic models, but the complete loss of NVC in pinacidil-treated mice demonstrates the power of KATP to obviate any vascular response to neuronal stimulation. In mice that lack SUR2 this dramatic abolition of NVC by pinacidil is absent, confirming that the drug effect in WT is specifically on KATP channels that contain the SUR2 subunit.
Mechanism of KATP involvement in neurovascular coupling
Both the genetic and pharmacologic interventions demonstrate that either global suppression of KATP channel activity (glibenclamide or SUR2 KO) or global activation of KATP channels (pinacidil or Kir6.1[V65M]) both significantly impair the NVC response in conscious mice; with glibenclamide reducing the local rise in oxygenated hemodynamic response to whisker stimulation, and pinacidil completely abolishing it. Conceivably, pinacidil may lower vascular tone throughout the vascular system, thereby obviating any possible local changes of flow. Adenosine is another vasodilator that can act on NVC when intracerebrally applied. 10 However, peripheral administration of adenosine also caused lowering of blood pressure, even on top if pinacidil, but with no effect on NVC (Figure 5). Glibenclamide caused a marked reduction of NVC but with no obvious effect on blood pressure (Supplementary Fig. 1). Previously, direct measurements have shown that glibenclamide was undetectable in cerebrospinal fluid (CSF) despite high plasma concentrations in glibenclamide pellet-implanted animals and that glibenclamide directly administered into the lateral ventricle of the brain was rapidly exported from the CSF. 36 However, both pinacidil and glibenclamide are hydrophobic and smaller than the recognized molecular weight cutoff for diffusion across the blood-brain barrier. 37 It is thus reasonable to expect that systemic application of these drugs will cause local CSF rises of these drugs within the vicinity of blood vessels, and that the effects we see are due to local effects on pericytes or other cells within the neurovascular unit.
Figure 6 illustrates our proposed role of KATP in NVC. The model assumes the VSM or pericytes that control blood flow will hyperpolarize and relax in response to increasing K conductance. WT cells under normal conditions exhibit low, but non-zero KATP conductance and are relatively depolarized. Since multiple K channels are present in VSM, polarization and hence contractility are dependent on overall K conductance. Because of polyamine-dependent rectification, Kir2 channels, located in capillary endothelial and smooth muscle cells, exhibit a unique, instantaneous, extracellular [K]-dependence of conductance, such that the membrane potential hyperpolarizes when [K] increases in the range of 3–8 mM, as may happen locally during high neuronal activity. 4 Recent studies have convincingly demonstrated that conduction of this hyperpolarizing signal to upstream arteriolar smooth muscle via gap junctions, is a primary driver of arteriolar relaxation, leading to increased blood flow in neurovascular coupling. 4 Neuronal activity results in K-dependent Kir2 conductance in capillary endothelium that is conducted upstream will add to the VSM KATP conductance, shifting rightwards along the overall K conductance-response curve, causing optimal hyperpolarization and relaxation in WT (Figure 6). When KATP is absent, VSM cells or pericytes will already be relatively depolarized and contracted under basal conditions, and the hyperpolarizing/relaxing effect of the same additional Kir2 conductance will then be reduced. Conversely, when KATP is increased above normal (i.e. in Cantú syndrome) the hyperpolarizing/relaxing effect of additional Kir2 conductance will be reduced, and when KATP is maximal (pinacidil), VSM will already be maximally hyperpolarized/relaxed and there may be no effect of additional Kir2 conductance (Figure 6).
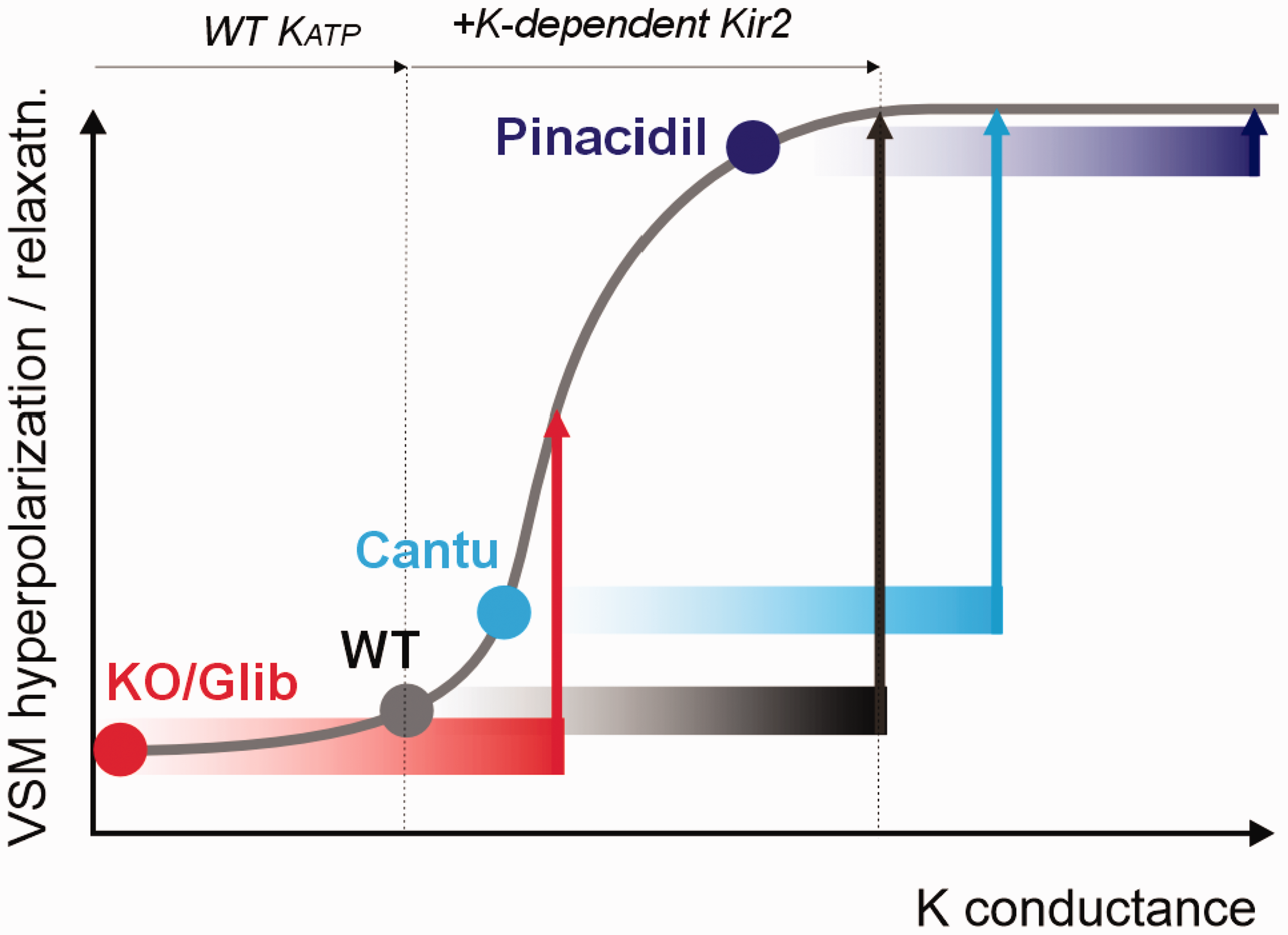
Proposed role of Kir6.1/SUR2-dependent KATP channels in neurovascular coupling KATP activity in resistance vessel smooth muscle or pericytes (VSM) determines the set-point for the hyperpolarizing and relaxing effects conducted to the arteriole by gap junctional coupling of K-driven Kir2 channel activation in capillary endothelial cells. Under normal conditions (WT), the added K-dependent Kir2 conductance will shift rightwards along the response curve causing optimal hyperpolarization and relaxation. Below a certain overall K conductance, VSM cells will be maximally depolarized and contracted; above a certain level, cells will be maximally hyperpolarized and relaxed. The model thus implies that the same level of [K]-induced Kir2 conductance will have a lesser hyperpolarizing effect when KATP is very low (KO, glibenclamide) or increased (Cantú syndrome), and essentially no effect when KATP is maximal (pinacidil).
Feedback vasculo-neural coupling?
The initial dip in oxygenated hemoglobin that we observe in these experiments is likely due to initial deoxygenation of hemoglobin at the start of neural activity upon stimulation, and has been previously been documented in rodents. 38 We consistently observed a small reduction in GCaMP signals, in the presence of glibenclamide or pinacidil, although we see no effect of repeated control measurements on HbO or GCaMP signals (Supplementary Fig. 2). The underlying causes are unclear, but could relate to an inhibitory consequence of impaired NVC on neuronal activity, or potentially to unwonted effects on KATP channels in neurons. We would note that there is little evidence for ABCC9 expression in cortical neurons 39 arguing against any direct neuronal action of pinacidil, although we cannot discount an effect of glibenclamide on ABCC8 (SUR1)-dependent neuronal KATP channels, potentially increasing baseline neuronal activity and consequently reducing the relative increase of neuronal activity following whisker stimulation. Although it is recognized that baseline oxygenation in the normal brain is probably sufficient to meet local metabolic demand, it is also conceivable that impaired vascular response may cause failure to restore full metabolic support, leading to loss of excitatory currents, or perhaps even metabolic activation of neuronally expressed Kir6.2/SUR1 KATP channels.
Pathological implications
Complementary to recent studies in isolated tissues,10,11 and in anesthetized mice, 12 our results provide direct evidence for the role of KATP channels in control of NVC in awake animals. A potential caveat to interpreting such experiments is that animals could be stressed due to the experimental conditions, which could lead to hyperventilation or altered blood pressure that could affect the interpretation, although we observed no obvious stress signs, such as attempts to run or vocalize.
Cantú Syndrome (CS), characterized by hypertrichosis, cardiomegaly, and multiple other organ issues, is caused by GOF mutations in the genes encoding SUR2 and Kir6.1. Cardiovascular issues include dilated, tortuous blood vessels, physiologically low blood pressure and low systemic vascular resistance, and greatly enlarged hearts with high cardiac output. 40 CS patients also have tortuous vessels in the cerebral vasculature and a relatively high incidence of migraines, as well as several other neurological abnormalities, along with persistent fetal brain blood vessels and enhanced diffuse white matter lesions.9,15,16 The dramatic loss of NVC with the KATP channel opener pinacidil in WT mice, as well as the reduced basal NVC and increased sensitivity to this pinacidil effect in Kir6.1[V65M] mice, suggests that the NVC response will be reduced in patients with CS. There is now mounting evidence that pinacidil and other K channel openers induce or exacerbate migraines in humans, supporting the hypothesis that disrupted NVC could underlie the enhanced migraine incidence in CS.
In contrast to CS, AIMS patients, lacking any SUR2 expression, exhibit significant intellectual disability. 14 Potential Putative LOF ABCC9 variants are also associated with hippocampal sclerosis of ageing (HSA).41,42 HSA indicates pathologic neuronal cell loss and gliosis in the hippocampus and is a common Alzheimer’s dementia-related disorder, associated with vascular cognitive impairment and dementia. 41 Given that we observed a pronounced slowing and reduction of the NVC response after global knockout of ABCC9 (SUR2) in mice, and scant evidence for SUR2 expression in anywhere other than vascular cells in the brain, we therefore suggest that intellectual disability in AIMS, and dementia in HSA may both be linked to a loss of NVC.
Conclusions
Our study demonstrates the significant role of Kir6.1/SUR2-dependent KATP in regulating functional hyperemia in response to neuronal activity in the conscious animal, with either pharmacologically- or genetically-driven increase or decrease of KATP function markedly impairing the coupling response. Disrupted NVC due to KATP GOF or LOF, in Cantú syndrome or AIMS respectively, may lead to altered neuronal activity underlying migraine development or intellectual disability in these conditions.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X251313906 - Supplemental material for Control of neurovascular coupling by ATP-sensitive potassium channels
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X251313906 for Control of neurovascular coupling by ATP-sensitive potassium channels by Ryan M Bowen, Nathaniel W York, Jonah Padawer-Curry, Adam Q Bauer, Jin-Moo Lee and Colin G Nichols in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These studies were supported by NIH grants R35 HL171542 (to CGN), R01NS126326 (to AQB) R01NS102870 (to AQB), RF1AG07950301 (to AQB and JML), 1F31NS122499 (to RMB), and T3EB014855 (salary support to RMB).
Acknowledgements
We are grateful to Ernest Gonzalez and Carmen M. Halabi for assistance with blood pressure and cerebral blood flow assays.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
The project was conceived by CGN and JML. Experimental work and analyses were carried out by RMB, NWY, CMN, JPC, AQB. The paper was written by RMB, NWY and CGN, and edited by all authors. All authors gave final approval for publication.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.