
Abstract
Children who experience cardiac arrest often suffer lasting neurological deficits, including impairments to learning and memory, due to global cerebral ischemia (GCI). Using a juvenile mouse model of cardiac arrest and resuscitation, we investigated the long-term effects of GCI and potential therapeutic interventions. Following juvenile GCI, long-term potentiation (LTP) and memory were impaired for several weeks followed by endogenous recovery coinciding with changes in brain-derived neurotrophic factor (BDNF) levels, an essential regulator of synaptic plasticity specifically in juveniles but not adults. Given that BDNF is unstable in plasma and cannot cross the blood–brain barrier, we explored the use of type II ampakines, positive allosteric modulators of AMPA receptors, to increase BDNF protein levels in the brain. In vivo administration of type II ampakines 14 days after GCI increased hippocampal BDNF levels, restored LTP, and improved hippocampal-dependent memory and learning behavior. These findings highlight the potential of type II ampakines as an innovative therapeutic intervention to restore synaptic and cognitive function at delayed time points after juvenile GCI.
Keywords
Introduction
Global cerebral ischemia (GCI), most often caused by cardiac arrest, affects individuals of all ages. In the United States alone, more than 20,000 children experience GCI each year. 1 Advances in emergency medicine and pediatric critical care have improved survival to discharge from historically low rates of 2%–6% to as high as 38%.2,3 However, most survivors sustain lasting neurological injury, with cognitive impairments—particularly in learning and memory—that can range from mild academic difficulties to complete dependence on caregivers.4,5
Most mechanistic insights into GCI have come from adult animal models, where research has focused on acute neuroprotection and improved resuscitation. Currently, therapeutic hypothermia is the only treatment proven to improve survival and neurological outcomes in adults and neonates treated within 6 h of injury.6,7 In contrast, large pediatric trials have shown no benefit for survival or neurologic function following therapeutic hypothermia after cardiac arrest,8,9 underscoring the urgent need for alternative, age-specific strategies. 10
Recent work in juvenile rodent models has revealed a unique therapeutic window not present in adults.11,12 After GCI, juveniles exhibit impaired hippocampal long-term potentiation (LTP), a cellular correlate of memory, 13 and memory deficits at 7 and 14 days post-injury, but show partial endogenous recovery by 30 days. 11 This recovery coincides with dynamic changes in brain-derived neurotrophic factor (BDNF), 11 a critical regulator of synaptic plasticity. 14 In the juvenile hippocampus, BDNF levels in CA1 fall at 7 days post-GCI, corresponding with LTP and behavioral deficits, but return to baseline by 30 days, 11 though this is a sex-specific response that only occurs in males. 15 It is well described that developmental timelines differ substantially between rodents and humans. Several weeks of postnatal development in mice can correspond to several years of maturation in children. 16 For example, postnatal days (P) 21–25 mice are estimated to approximate the neurodevelopmental stage of a 3–4-year-old child, whereas P35–40 mice are considered equivalent to a 12–14-year-old adolescent. 16 This time course suggests a transient period when synaptic plasticity is impaired but potentially reversible. Importantly, interventions targeting this phase, such as the TrkB agonist 7,8-DHF given at 7 days post-injury, can restore LTP and improve memory performance. 11 Evidence from neonatal hypoxic–ischemic models suggests BDNF supports both neuroprotection17,18 and regeneration. 19 However, direct BDNF delivery is clinically challenging due to poor plasma stability and inability to cross the blood–brain barrier,20,21 prompting interest in strategies that enhance endogenous BDNF production.
An alternative approach to enhance plasticity is to target AMPA receptor-mediated signaling, which is known to promote synaptic plasticity. 22 AMPA receptor activation increases dendritic depolarization and calcium influx, thereby triggering activity-dependent BDNF transcription. Ampakines, positive allosteric modulators of AMPA receptors, enhance excitatory postsynaptic potentials by slowing receptor deactivation (type I) or both slowing deactivation and reducing desensitization (type II). 22 Notably, certain type II ampakines increase BDNF expression in hippocampal and cortical neurons.23,24 In adult models of focal stroke, delayed type II ampakine administration has been shown to improve motor recovery.25,26 However, the effects of ampakines on hippocampal physiology following ischemia—whether focal or global—have not yet been studied. Importantly, their impact on the juvenile brain remains entirely unexplored, despite clear evidence that injury mechanisms and recovery trajectories differ substantially between juveniles and adults.27–29
Here, we test the hypothesis that delayed administration of type II ampakines can restore hippocampal synaptic and cognitive function after juvenile GCI via a BDNF/TrkB-dependent mechanism. Specifically, we aimed to: (1) assess the potential of neurorestorative intervention at 14 days post-injury, a time point well beyond acute neuronal death and (2) evaluate LY404187 and CX546 as candidate therapeutics to reverse hippocampal dysfunction and improve memory performance in a juvenile mouse model of GCI.
Methods
Experimental animals
All experimental protocols were approved by the University of Colorado Anschutz Institutional Animal Care and Use Committee (IACUC) and conformed to the National Institutes of Health guidelines for care and use of animals. Male C57Bl/6 20–25-day-old (P20–25, prepubertal) mice (Charles River Laboratory) were used for this study as we recently demonstrated that BDNF mRNA expression is not altered following female juvenile GCI. 15 The average age of CA/CPR was 22.5 ± 0.2 days (n = 64). The average age for sham surgeries was 22.6 ± 0.3 days (n = 65). These mice were weaned and not with dam at the time of experiment. The mice were housed in a standard 12 h light and 12 h dark cycle and had free access to food and water. All experiments in the study adhered to the ARRIVE guidelines for animal experiments. Mice were randomly assigned to experimental groups, and the investigator was blinded through analysis. Twelve mice were excluded from GCI group and two were excluded from the sham group due to premature death.
Cardiac arrest and cardiopulmonary resuscitation
GCI was attained via cardiac arrest and cardiopulmonary resuscitation (CA/CPR) in juvenile mice as previously described.11,12,29 Briefly, male mice were anesthetized using 3% isoflurane and maintained with 2%–2.5% isoflurane in 21% fraction of inspired oxygen (FiO2) via face-mask. Body temperature was maintained at 37 °C using a heat lamp and heating pad while being monitored with temperature probes placed into the left ear canal and rectum. For drug administration, a small incision was made along the right side of the neck, and a PE-10 catheter was inserted into the right internal jugular vein and flushed with heparinized 0.9% normal saline solution and surgical wounds were closed. Animals were endotracheally intubated using a 24G intravenous catheter and connected to a mouse ventilator (Minivent; Hugo Sachs Elektronik, March-Hugstetten, Germany) set to a respiratory rate of 160 breaths/min. Cardiac function was monitored throughout the experiment with electrocardiography (EKG). Anesthesia ceased 1 min prior to the onset of cardiac arrest. Cardiac arrest was induced by turning off the mouse ventilator along with an injection of 30 µL of 0.5 M KCl via the jugular catheter and confirmed by asystole on EKG and absence of spontaneous breathing. The endotracheal tube was disconnected from the ventilator during cardiac arrest, and no spontaneous breathing was observed during asystole. Body warming ceased 1 min prior to cardiac arrest. During cardiac arrest, the pericranial temperature was maintained at 37.5 °C ± 0.5 °C by using a water-filled coil or heat lamp. Peripheral body temperature was allowed to fall spontaneously to 35 °C. Thirty seconds prior to compressions, ventilation through the mouse ventilator was resumed with 100% FiO2 and the respiratory rate was increased to 210 breaths/min. Resuscitation was begun 8 min after the initiation of cardiac arrest by slow injection of 0.2–0.5 mL of calcium chloride epinephrine solution (16 µg epinephrine/mL 0.9% saline; 16 µg calcium chloride/mL 0.9% saline) and chest compressions at a rate of ~300/min. Chest compressions were stopped upon return of spontaneous circulation (ROSC), defined as electrical evidence of cardiac contractions on EKG. If ROSC was not achieved within 3 min of CPR initiation, resuscitation was stopped, and the animal was excluded from the study. Upon the onset of spontaneous respiration, FiO2 was decreased to 50%. When the spontaneous respiratory rate was 30 breaths/min, the ventilator was weaned back to 150 breaths/min over 4 min, and when the animals had at least 60 spontaneous breaths/min, the endotracheal tube was removed. Temperature probes and intravascular catheters were also removed. Mice were single housed for complete recovery. Sham mice received intravascular access and were intubated and subjected to the same FiO2 as above, but saline, rather than KCl was administered. Surgeries were randomized and investigators were blinded through analysis. Fifteen mice were excluded from the study after CA/CPR and one mouse was excluded from the sham group due to premature death.
In vivo drug administration
LY404187 (1 mg/kg) or normal saline vehicle was administered 13 days post GCI or sham surgery via retro-orbital route. Mice were weighed and anesthetized with 3% isoflurane and injected with 0.1 mL of either saline or LY404817 solution. Mice were monitored until anesthesia wore off and righting reflexes were gained. Experiments were performed 24 h after injection.
ELISA
Hippocampal tissue was isolated 14 days after recovery from CA/CPR or sham surgeries. Mice were anesthetized with 3.5%–4% isoflurane. Mice were transcardially perfused with ice-cold (2 °C–5 °C) oxygenated (95% O2/5% CO2) artificial cerebral spinal fluid (aCSF) for 2 min prior to decapitation. Left and right hippocampi were isolated, flash frozen, and store in −80 °C freezer until time of experiment. Unilateral hippocampal tissue was homogenized in neuronal protein extraction reagent (NPER) buffer. Protein extracts were collected and BDNF levels were measured using mouse BDNF ELISA kit (LS-F2404; LifeSpan Biosciences, Seattle, WA, USA) as per manufacture’s instructions. Samples were diluted using sample diluent from kit, then loaded onto a plate and incubated for 90 min at 37 °C. After aspirating liquid from each well, 1× detection antibody solution was added and left to incubate for 1 h at 37 °C. After washing 3×, 1× ABC complex working solution was added and left to incubate for 30 min at 37 °C. After washing 5×, TMB substrate was added and left to incubate for 20 min at 37 °C. Stop solution was added and optical density was measured at 450 nm immediately using SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA, USA). All BDNF concentration values were normalized to total protein content in the sample measured with a Pierce BCA Protein Assay Kit (ThermoFisher Scientific). Note on experimental comparison: ELISA experiments corresponding to Figures 1 to 3 were performed independently on separate plates using different BDNF ELISA kits, each with their own internal standards and controls. Because of this, absolute BDNF values are not directly comparable between experiments. All statistical comparisons were made within individual experiments using internal controls.
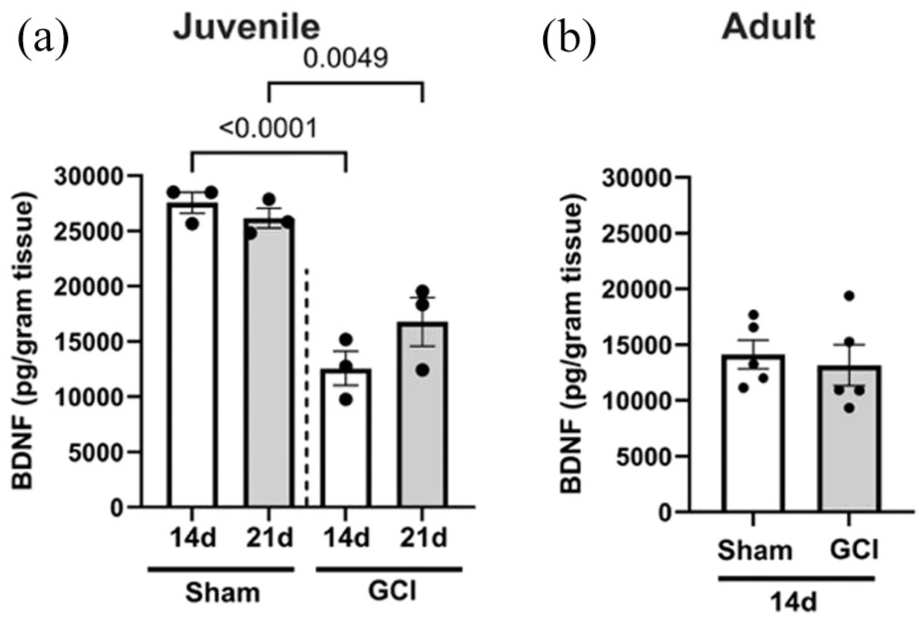
BDNF expression decreases after GCI in juvenile mice. (a) ELISA in juvenile mice 14, 21 days after sham or GCI showing decreased BDNF expression followed by recovery. (b) There is no change in BDNF in adults 14 days after GCI.
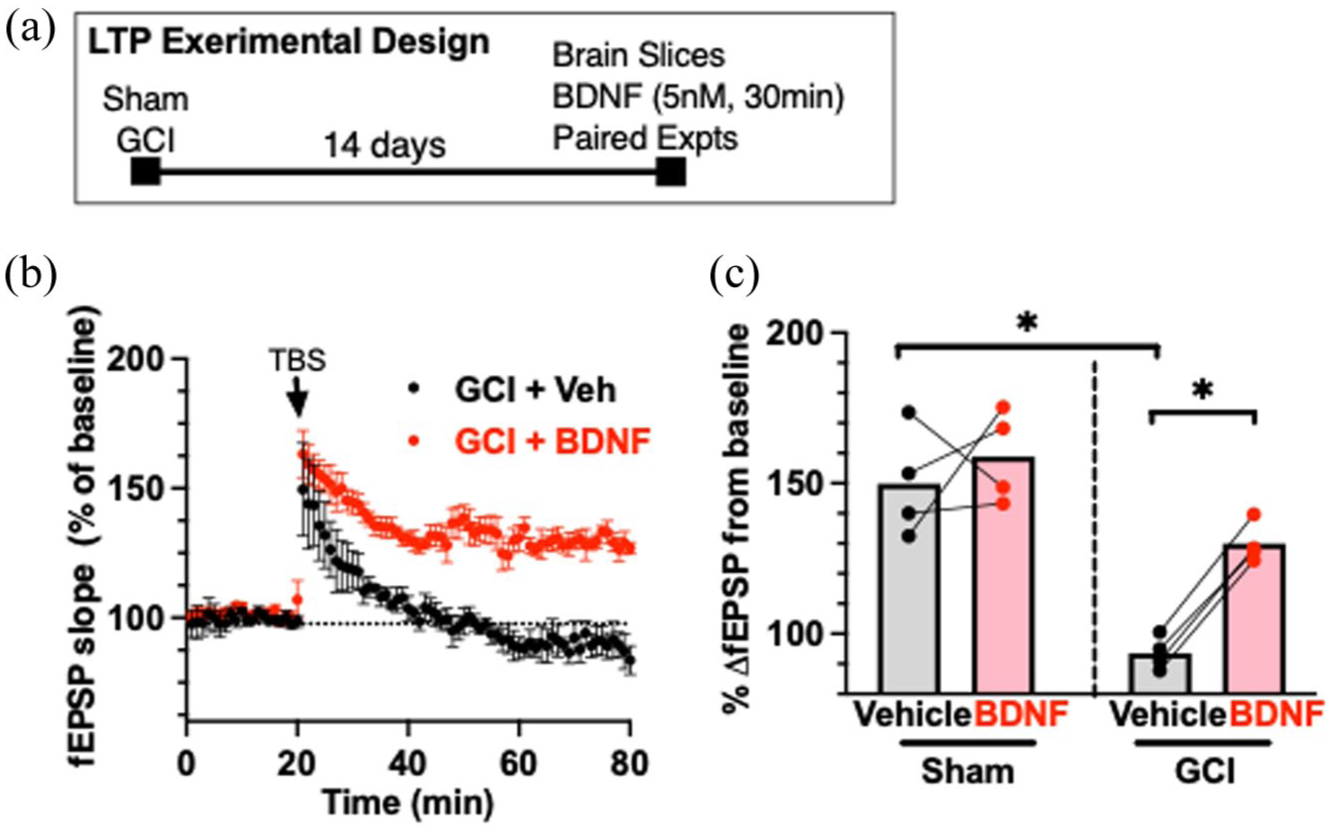
Recombinant BDNF rescues LTP impairment 14 days after juvenile GCI. (a) Experimental design. (b) Time plot of responses to TBS (40 pulses) in paired experiments where slices were treated with vehicle or BDNF. (c) Quantification of LTP experiments when BDNF (5 nM) was applied in paired experiment 14 days after sham or GCI Each dot represents an individual experiment, and the lines indicate the experiments that were paired.
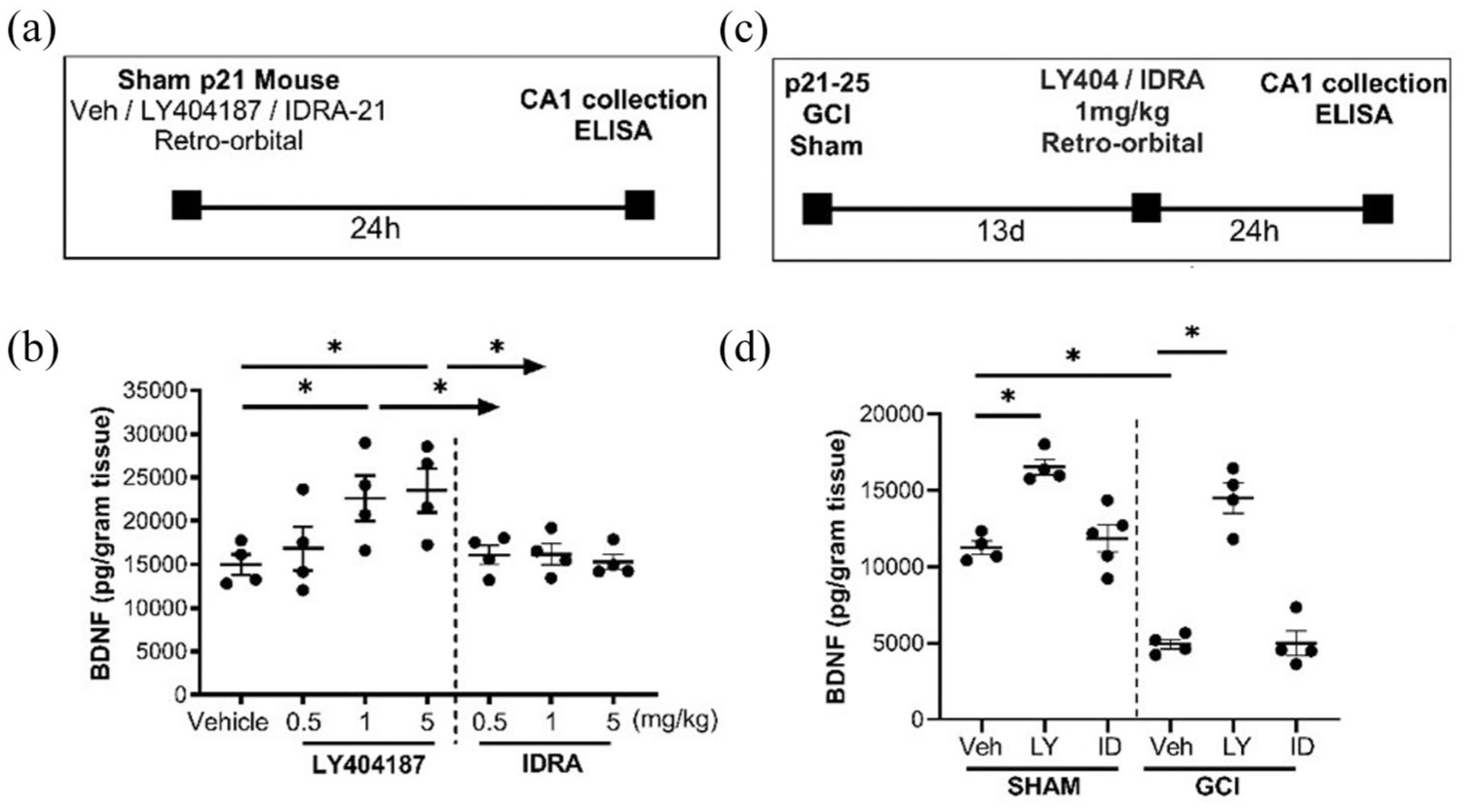
Ampakine effect on BDNF expression. (a) Experimental design for injection of ampakines in sham P21 mice. (b) Quantification of hippocampal BDNF showing dose response of LY404187 and IDRA-21 24 h after injection in sham mice. (c) Experimental design for injection of 1 mg/kg of ampakine 13 days after sham or GCI surgery, followed by collection of (d) quantification of hippocampal BDNF following LY404187 (LY) or IDRA-21 (ID) administration is sham or juvenile GCI mice.
Acute hippocampal slice preparation
Hippocampal slices were prepared at 14 days after recovery from CA/CPR or sham surgeries. Mice were anesthetized with 3.5%–4% isoflurane and transcardially perfused with ice-cold (2 °C–5 °C) oxygenated (95% O2/5% CO2) artificial cerebral spinal fluid (aCSF) for 2 min prior to decapitation. The brains used for electrophysiology were then extracted and placed in ice-cold oxygenated aCSF. The composition of aCSF was the following (mmol/L): 126 NaCl, 2.5 KCl, 25 NaHCO3, 1.3 NaH2PO4, 2.5 CaCl2, 1.2 MgCl2, and 12 glucose (11). Horizontal hippocampal slices (300 µm thick) were cut with a Vibratome 1200 (Leica) and transferred to a holding chamber containing room temperature aCSF for at least 1 h before recording.
Ex vivo drug administration
After hippocampal slice preparation, the brain tissue was transferred to a aCSF. After 1 h in aCSF at room temperature, brain slices were incubated in one of the following drug solutions for an additional time before recording LTP; BDNF (5 nM) for 30 min, LY404187 (25 µM), CX546 (200 µM), IDRA21 (500 µM), and ANA-12 (1 µM) for 2–3 h. All drugs were purchased from Tocris Bioscience (Bristol, UK). Hippocampal slices remained exposed to the drug at 1.5–2 mL/min throughout the recordings.
Electrophysiology
Synaptically evoked field potentials were recorded from hippocampal CA1 slices maintained in a temperature-controlled (31°C ± 0.5 °C) interface chamber and continuously perfused with oxygenated artificial cerebrospinal fluid (aCSF) at 1.5 mL/min. Field excitatory postsynaptic potentials (fEPSPs) were elicited by stimulating the Schaffer collaterals and recorded in the stratum radiatum of CA1. Stimulus intensity was adjusted to evoke fEPSPs at 50% of the maximum slope, with test pulses delivered every 20 s. Paired-pulse responses were recorded using a 50 ms interpulse interval and expressed as the ratio of the second to the first slope. After establishing a stable 20-min baseline, long-term potentiation (LTP) was induced using a theta burst stimulation (TBS) protocol consisting of four pulses at 100 Hz in 30 ms bursts, repeated 10 times with 200 ms interburst intervals. fEPSPs were recorded for 60 min post-TBS. LTP magnitude was quantified as the average fEPSP slope from 50 to 60 min post-TBS, normalized to the 10-min pre-TBS baseline (set to 100%). Signals were amplified (1000×), filtered at 1.0 kHz (Model LP511 AC; Grass Instruments), digitized at 10 kHz, and stored for offline analysis using Clampfit 10.7 (Axon Instruments). The initial slope derivative (dV/dT) was used for analysis. For time course plots, fEPSP slope values were averaged and expressed as a percentage of baseline. Preestablished exclusion criteria included discarding data if LTP increases under control conditions were <120% of baseline. No data met this exclusion criterion.
Behavioral testing
Testing was performed 14 days after sham or GCI surgery. Mice were transported in their home cages to a behavioral suite and allowed to habituate for 1 h prior to testing. Motor activity and function was measured with square open field to assess motor skills and hippocampal dependent memory was testing using contextual fear conditioning (CFC). The same cohorts were used for all three tasks. For all behavior protocols, animals were video-tracked using the ANY-maze system (Stoelting, Wood Dale, IL, USA).
Open field
The open field paradigm was utilized as a motor and anxiety measure. The apparatus consisted of a square open field box with inner and outer zones demarcated with a black square drawn on the floor. Mice were transported in home cages before and after testing. Mice were placed in square open field box for 15 min. Total distance and time spent in the inner and outer zones was measured by video-tracking using ANY-maze software.
Contextual fear conditioning
The contextual fear conditioning (CFC) paradigm was used as a hippocampal-dependent memory task. 30 The apparatus consisted of a chamber with shock grid floors, consisting of 16 stainless steel rods connected to a shock generator (Model H13–15; Colbourn Instruments, Whitehall, PA, USA). Mice were transported in home cages during the training and testing sessions. During day 1, mice were allowed to habituate to the conditioning chamber for 5 min. The following day, during training, mice were allowed to habituate to the conditioning chamber for a 2-min pre-exposure session followed 5-min period consisting of three foot shocks (2 s/1.0 mA electric shock), each separated by 60 s, followed by 2 min without any shocks. Following the shocks, mice were returned to their home cages. Memory was tested 24 h later by placing mice back into the fear conditioning chamber for 5 min. Memory was determined by percentage of freezing behavior, defined as total time freezing over total duration of test. Total time freezing was measured in 10 s intervals across a 5-min testing period video-tracking using ANY-maze, which detects the total episodes of freezing and total amount of time frozen defined as absence of movement except for heartbeat/respiration.
Statistical analysis
Analysis was performed by separate investigator from individual performing the experiment in a blinded manner using GraphPad Prism and presented as mean ± standard deviation (SD). Normality was confirmed for all groups using Shapiro–Wilk test. Power analysis prior to study was performed using G*Power software. Based upon previous results, to observe a 40% change in LTP between two groups with a standard deviation of 15 and an alpha error of 5% and a beta error of 80%, a group of four slices per group are required for unpaired experiments and three slices per group for paired experiments. In consideration of biologic variability, no more than two slices per animal used in analysis per condition. For behavior studies, to observe a 30% change in freezing behavior between two groups with a standard deviation of 15%, with an alpha error of 5% and a beta error of 80%, eight animals per group are require for behavior experiments. Statistical analysis of all data was determined using the one-way analysis of variance (ANOVA) and post-hoc Dunnett’s test for comparison of multiple groups compared against the control group. Differences were considered statistically significant with p < 0.05, as indicated by the asterisks.
Results
BDNF levels are decreased after GCI in the juvenile mouse hippocampus
Our prior work demonstrated a reduction in hippocampal BDNF following male juvenile GCI using western blot analysis. 11 To quantify this effect with greater sensitivity, we performed ELISA on homogenized hippocampal tissue collected 14 and 21 days post-surgery from juvenile and adult mice (Figure 1). In juveniles, GCI caused a marked and sustained decrease in BDNF levels, at 14 and 21 days (12,570 ± 2710 pg/g of tissue at 14 days; 16,776 ± 3798 pg/g of tissue at 21 days) compared with age-matched shams (27,552 ± 1641 pg/g tissue at 14 days (p < 0.001); 26,168 ± 1552 pg/g of tissue (p < 0.05)). In contrast, adult mice showed no significant change in BDNF levels 14 days after GCI (14,140 ± 2858 pg/g of tissue in sham vs 13,165 ± 4123 in GCI, p = 0.6755), despite our previous evidence of impaired synaptic plasticity at this time point. 31 These results identify a juvenile-specific, prolonged BDNF deficit after GCI, highlighting a potential age-dependent vulnerability that may be targeted to improve cognitive recovery in pediatric populations.
Exogenous BDNF restores LTP after juvenile GCI
We next tested whether supplementing BDNF could directly reverse the synaptic impairment observed after juvenile GCI. Acute hippocampal slices were prepared 14 days post-injury, and paired experiments from the same animal were treated with either vehicle or recombinant BDNF (5 nM) for 30 min prior to LTP induction. As shown in Figure 2, LTP was decreased following GCI compared to sham after vehicle treatment 14 days after surgery (93% ± 5.5% GCI vs 150% ± 18% sham, n = 4 each, p < 0.05, unpaired t-test). BDNF treatment 14 days after surgery significantly enhanced LTP in GCI slices (93% ± 5.5% vehicle vs 130% ± 6.7% BDNF, n = 4, p < 0.001, paired t-test), restoring it to levels comparable to shams. In contrast, BDNF had no effect on LTP in slices from sham animals (150% ± 18% vehicle vs 159% ± 15% BDNF, n = 4). These findings indicate that the post-GCI plasticity deficit in the juvenile hippocampus is, at least in part, BDNF-dependent, and that restoring BDNF signaling can normalize synaptic function.
Type II ampakines elevate hippocampal BDNF in juvenile mice
After establishing that reduced BDNF expression correlates with impaired long-term potentiation (LTP) in juvenile GCI, we next tested whether type II ampakines could serve as a therapeutic strategy to restore BDNF levels. Type II ampakines have been shown to enhance BDNF expression through sustained AMPA receptor activation, whereas type I ampakines generally do not produce this effect. 24 To directly validate this in our juvenile model, we measured hippocampal BDNF protein levels by ELISA in sham juvenile mice following a single retro-orbital injection of LY404187 (type II ampakine) or IDRA-21 (type I ampakine). Mice received 0.5, 1, or 5 mg/kg of drug, and hippocampi were collected 24 h later. As shown in Figure 3, LY404187 produced a robust dose-dependent increase in BDNF, with significant elevations at both 1 and 5 mg/kg compared with vehicle (14,972 ± 2360 pg/g vehicle vs 22,600 ± 5241 pg/g LY404187 1 mg/kg, n = 4, p = 0.04; 23,487 ± 5082 pg/g LY404187 5 mg/kg, n = 4, p = 0.02). There was no difference between the 1 and 5 mg/kg doses (p = 0.9989), so 1 mg/kg was selected for all subsequent in vivo experiments. By contrast, Figure 3 shows that IDRA-21 produced no change in BDNF levels at any tested dose (all p > 0.99).
We then evaluated whether the effects of LY404187 on BDNF expression persisted in the context of ischemic injury. In line with our earlier findings, vehicle-treated GCI mice exhibited markedly reduced hippocampal BDNF levels 14 days post-injury (Figure 3). Strikingly, administration of LY404187 at 13 days post-GCI (24 h before tissue collection) significantly increased BDNF in both sham animals (11,245 ± 864.4 pg/g vehicle vs 16,541 ± 1018 pg/g LY404187, n = 4, p = 0.0003) and GCI animals (4935 ± 623.6 pg/g vehicle vs 14,508 ± 1985 pg/g LY404187, n = 4, p < 0.0001). In contrast, IDRA-21 again had no effect on BDNF levels in either group (sham: p = 0.9822; GCI: p > 0.99). These results confirm that type II ampakines, but not type I, can robustly elevate hippocampal BDNF in both uninjured and post-ischemic juvenile brains.
Type II ampakines restore LTP and memory after juvenile GCI
We next determined whether the BDNF-enhancing effects of type II ampakines could translate into functional recovery of hippocampal plasticity and memory. Fourteen days after GCI or sham surgery, we assessed CA1 LTP in paired ex vivo slice recordings, applying vehicle or drug to slices from the same animal. Ex vivo slice preparations were used to directly assess the effect of ampakine application in slices from the same animal in a paired fashion, a test that cannot be performed with in vivo administration. LTP in sham animals was unaffected by drug treatment, indicating no ceiling effect under baseline conditions. In contrast, vehicle-treated slices from GCI mice exhibited markedly impaired LTP. Figure 4 demonstrates that treatment with LY404187 (25 μM) restored LTP to near-sham levels (111% ± 12% vehicle vs 156% ± 13% LY404187, n = 4 pairs, p = 0.01), and CX546 (200 μM), another type II ampakine known to increase BDNF,23,24 produced a similar recovery (120% ± 6.2% vehicle vs 167% ± 3.4% CX546, n = 3 pairs, p = 0.004). By contrast, IDRA-21 had no effect on LTP in GCI slices (100% ± 10% vehicle vs 104% ± 12% IDRA-21, n = 4 pairs, p = 0.75).
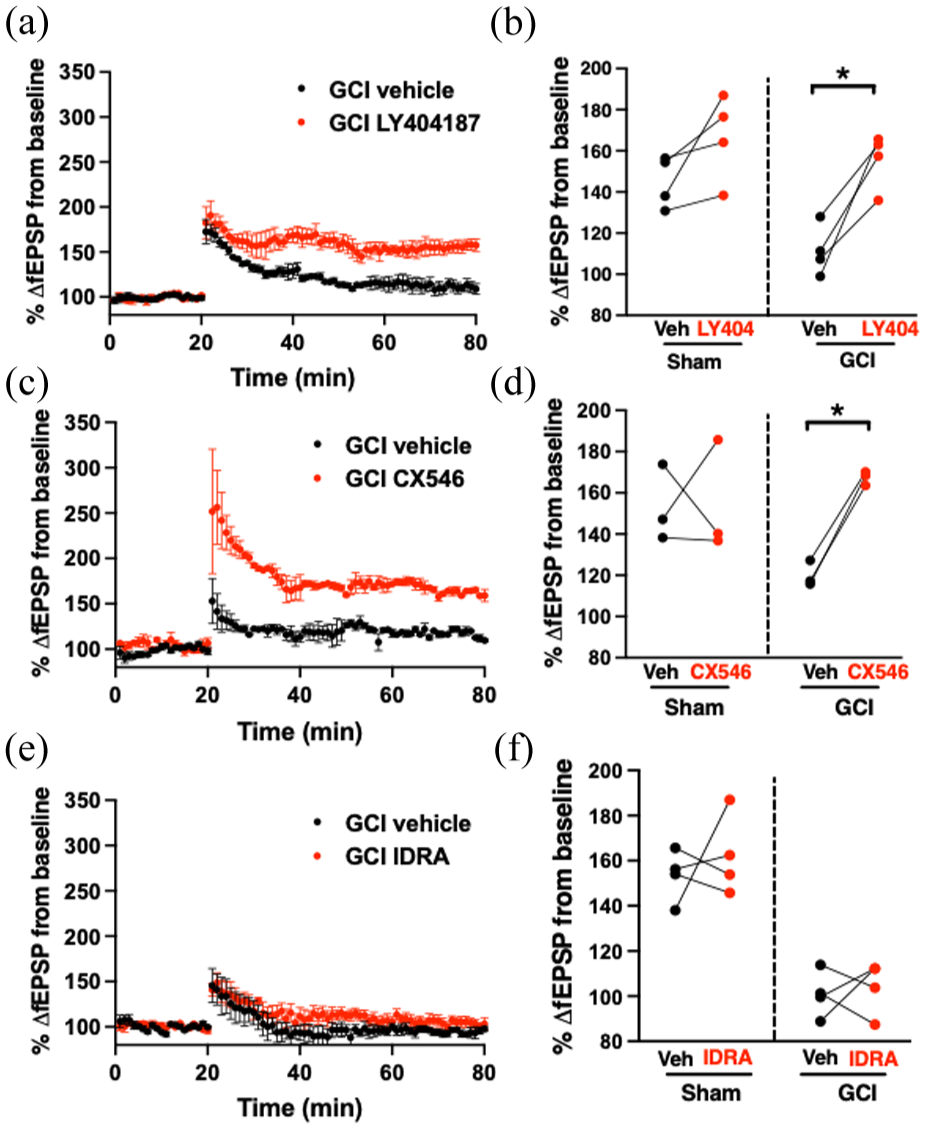
(a) Time plot of LTP experiments in slices exposed to vehicle or LY404187. (b) Quantification of LTP experiments when LY404187 was applied in paired experiment 14 days after sham or GCI. (c) Time plot of LTP experiments in slices exposed to vehicle or CX546. (d) Quantification of LTP experiments when CX546 was applied in paired experiment 14 days after sham or GCI. (e) Time plot of LTP experiments in slices exposed to vehicle or IDRA-21. (f) Quantification of LTP experiments when IDRA-21 was applied in paired experiment 14 days after sham or GCI. Each dot represents an individual experiment, and the lines indicate the experiments that were paired.
To test whether these synaptic improvements extend to behavior, LY404187 (1 mg/kg) was administered in vivo 13 days post-GCI, and contextual fear conditioning was performed 24 h later. Consistent with prior reports,11,12 GCI in juvenile mice significantly reduced freezing (indicating impaired memory) compared with sham animals (26% ± 7.3% GCI vehicle, n = 8 vs 54% ± 19% sham vehicle, n = 8, p = 0.01; Figure 5). Delayed LY404187 administration restored freezing to sham levels (54% ± 13%, n = 7, p = 0.01 vs GCI vehicle) without affecting sham animals (64% ± 22%, n = 8). Open field testing confirmed that differences in freezing were not due to motor deficits: GCI vehicle animals traveled farther than all other groups, and no differences were observed in time spent in the inner zone. These findings demonstrate that type II ampakines can reverse both electrophysiological and behavioral deficits when administered well beyond the acute injury phase.
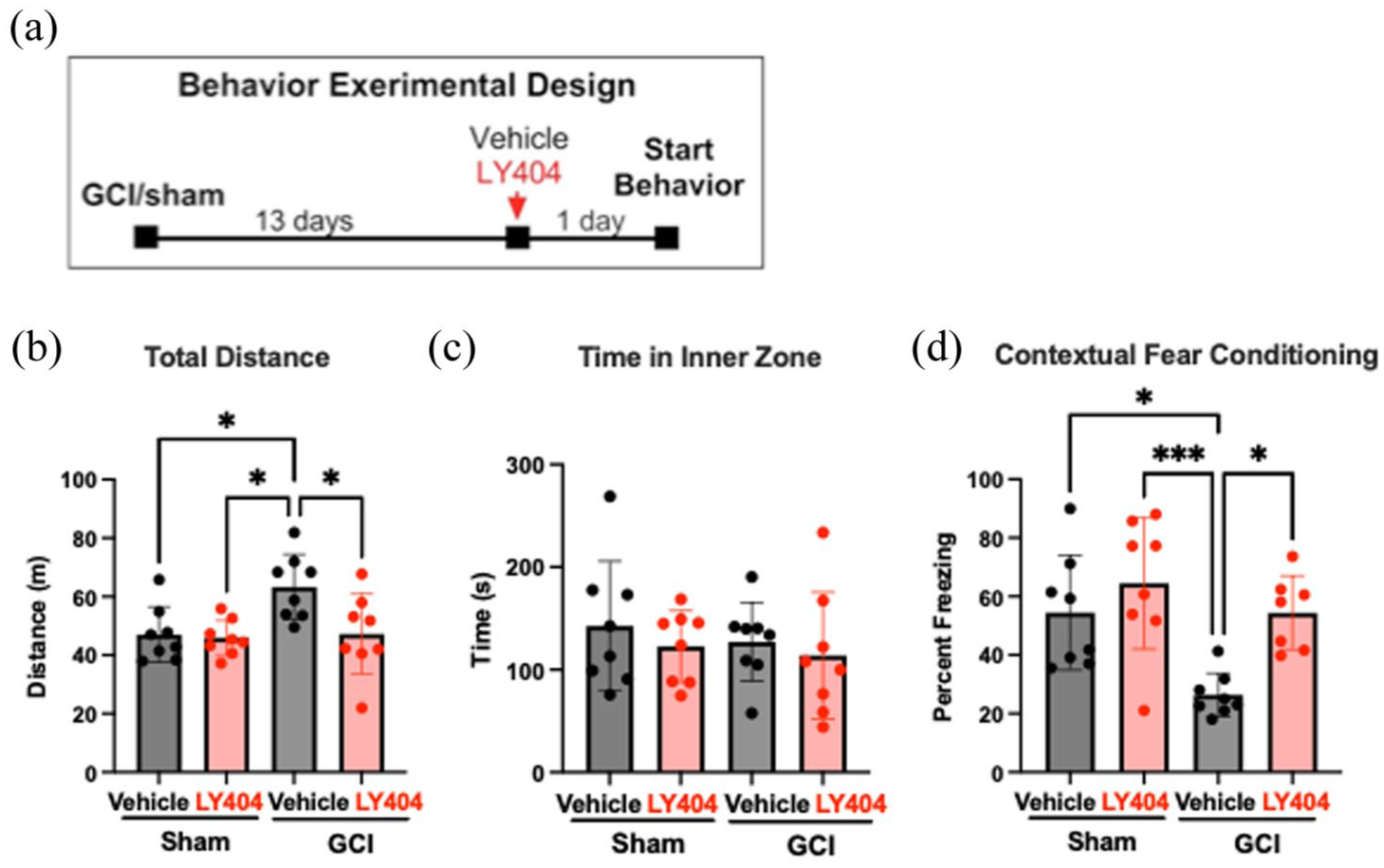
(a) Experimental design for behavioral testing. (b) Quantification of total distance traveled by mouse in square open field apparatus in 15 min, revealing increased distance traveled by GCI vehicle group compared to sham vehicle (p = 0.0016), sham Ly404187 (p < 0.001), and GCI LY404187 (p = 0.0141). (c) No statistical difference between any groups when comparing total time spent in the inner zone of the square open field. (d) Quantification of total time freezing out of 5 min during CFC testing, revealing decreased freezing behavior in GCI vehicle group compared to sham vehicle (p = 0.0207) and restored freezing behavior with administration of LY404187 at 13 days post GCI (p = 0.0376).
TrkB antagonist blocks LTP recovery
Finally, we tested whether the restorative effects of LY404187 depend on BDNF–TrkB signaling. Our previous work showed that BDNF–TrkB binding is essential for reversing LTP and memory deficits after juvenile GCI. 11 Here, we applied ANA-12, a selective TrkB antagonist, in three-way paired experiments (vehicle, LY404187, and LY404187 + ANA-12) using slices from the same mouse 14 days post-GCI. As shown in Figure 6, LY404187 alone restored LTP (112.1% ± 6.65% vehicle vs 159.6% ± 18.87% LY404187, n = 3, p = 0.02), whereas co-administration with ANA-12 abolished this recovery (118.1% ± 19.08% LY404187 + ANA-12, n = 3, p = 0.04 vs LY404187 alone). TrkB blockade also reduced LTP in sham animals (149% ± 12% vehicle vs 123% ± 6.2% ANA-12, n = 3, p = 0.05), underscoring the importance of endogenous BDNF–TrkB signaling in maintaining normal plasticity. Together, these results demonstrate that LY404187 restores synaptic and cognitive function after juvenile GCI via a BDNF/TrkB-dependent mechanism.
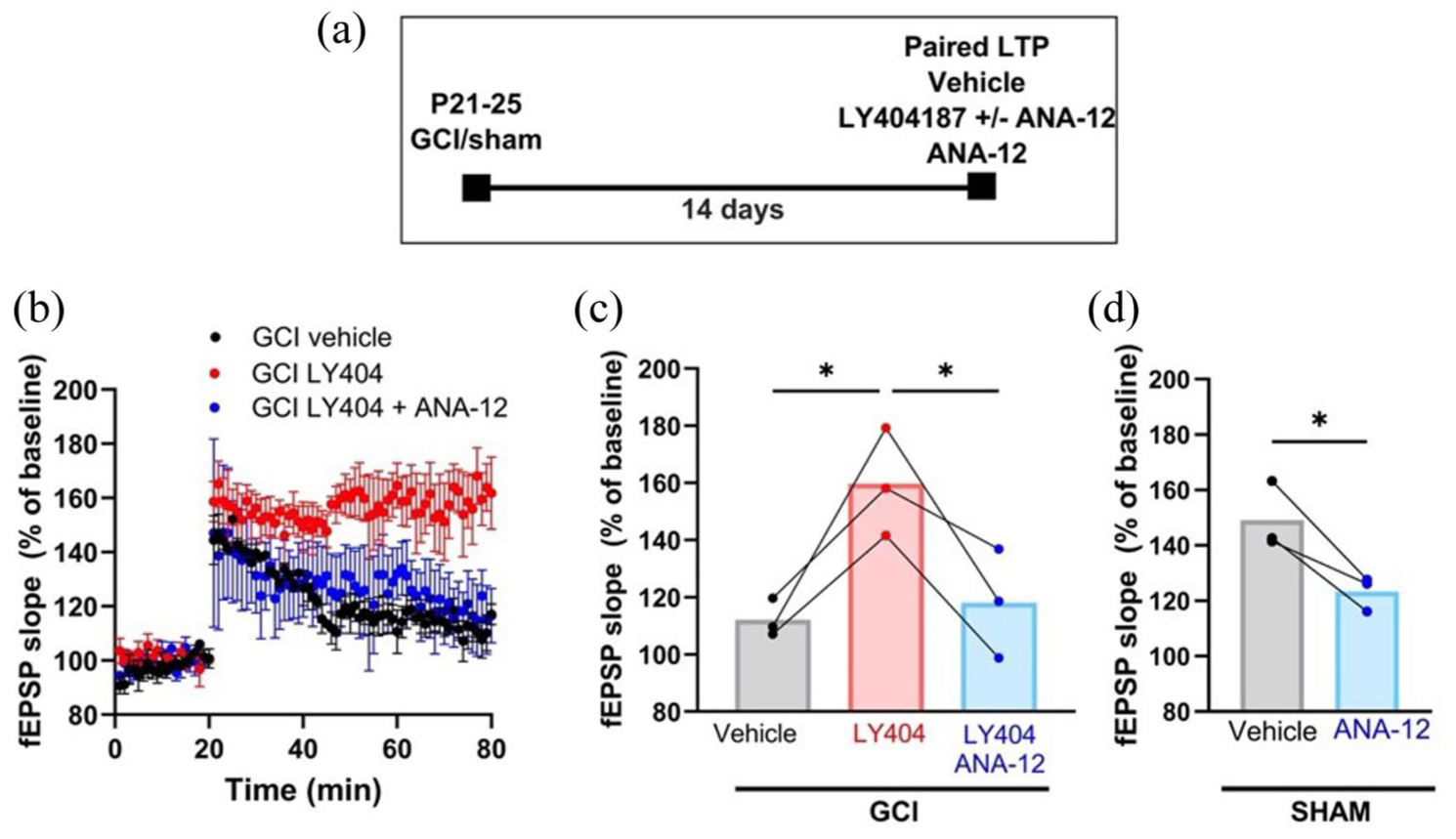
LY404187 rescue of LTP after GCI is attenuated with TrkB receptor antagonist. (a) Experimental design showing delayed administration of drug or vehicle. (b) Time plots of EPSP slope in mice 14 days after GCI treated in three-way paired experiments with vehicle, LY404187, and LY404187 + ANA-12. (c) Paired experiments 14 days after GCI showing the rescue of LTP provided by LY404187 is blocked by ANA-12. (d) Quantification of LTP with ANA-12 compared to vehicle in paired sham experiments. Each dot represents an individual experiment, and the lines indicate the experiments that were paired using slices from the same mouse.
Discussion
Our findings confirm and extend previous work showing that juvenile global cerebral ischemia (GCI) leads to persistent synaptic dysfunction, characterized by impaired hippocampal long-term potentiation (LTP) and deficits in learning and memory.11,12 These results reinforce the concept that ischemic brain injury in the juvenile period produces distinct and enduring disruptions in circuit function, even when overt neuronal death has subsided. A key advance of the present study is the demonstration that type II ampakines, administered at a delayed time point well beyond the established window of ischemia-related neuronal death (~3 days post-injury), 29 can restore hippocampal function. This expands the therapeutic scope beyond acute neuroprotection to include neurorestoration; targeting the surviving but functionally compromised neuronal network.
Historically, GCI research has focused on acute neuroprotective strategies, with therapeutic hypothermia emerging as the most widely adopted intervention. While effective in neonates and adults,6,7 large randomized controlled trials in pediatric patients post-cardiac arrest have shown little or no improvement in survival or neurological outcomes,8,9 underscoring a persistent gap in treatment options for older infants and children. The juvenile brain’s response to GCI remains comparatively understudied, 10 and the lack of age-specific mechanistic insight has hindered innovation in therapeutic development. Given the limited efficacy of acute neuroprotective strategies, we propose synaptorestoration (restoring synaptic function in surviving neurons) as a promising complementary approach. Using ELISA, we demonstrate that post-GCI hippocampal dysfunction is associated with reduced brain-derived neurotrophic factor (BDNF) expression for up to 21 days after injury, consistent with earlier work showing that BDNF levels are reduced at 7 days post-injury but recover by 30 days. 11 Apparent differences in absolute BDNF concentrations between Figures 1 and 3 reflect independent ELISA experiments performed on separate plates with distinct standards and controls; thus, comparisons were made within, not across, experiments. The persistence of low BDNF over several weeks defines a unique therapeutic window. In juvenile mice injured at postnatal days (P) 21–25, this period extends to at least P42, which may correspond to nearly a decade of neurodevelopment in a child injured at 3 years of age. 16 This prolonged vulnerability—and opportunity—suggests that interventions targeting synaptic plasticity could meaningfully improve long-term function.
Mechanistically, ischemic injury triggers glutamate excitotoxicity during reperfusion, when excess glutamate release overstimulates AMPA and NMDA receptors, leading to calcium overload, mitochondrial dysfunction, and early neuronal death.32–34 Because AMPA receptor activation at early time points can exacerbate injury, we deliberately delayed ampakine administration to time points well beyond the excitotoxic window. Nevertheless, glutamatergic dysregulation does not resolve immediately. In rodent models, post-ischemic extracellular glutamate rises and transporter dysfunction have been reported and implicated in delayed neuronal injury assessed at 7 days. 35 Reviews and primary studies of glutamate transporter expression also document impaired EAAT/GLT-1 expression at ~7 days after ischemia, a mechanism that would promote prolonged extracellular glutamate accumulation.36,37 To avoid confounding effects of ongoing glutamate toxicity, we assessed the impact of ampakines at 14 days post-injury, a time point chosen to be outside this subacute excitotoxic window. At later stages, excitotoxicity wanes, and instead synaptic inactivity, diminished glutamatergic signaling, and reduced neurotrophic support become the dominant contributors to persistent circuit dysfunction. 38 Reperfusion injury also triggers oxidative stress and inflammation, 39 activating IKK/NF-κB signaling. 40 Sustained NF-κB activity can downregulate BDNF expression, 41 providing a plausible link between post-ischemic inflammation, neurotrophic factor depletion, and impaired synaptic plasticity. Future studies dissecting this pathway in the juvenile brain could identify synergistic targets for combination therapy—for example, pairing ampakines with anti-inflammatory or antioxidant agents to preserve BDNF signaling.
Our age-specific comparisons reveal a significant post-GCI decrease in BDNF juveniles but not in adults, suggesting that hippocampal vulnerability and repair mechanisms differ across development. In the adult brain, sustained hippocampal impairment after GCI is unlikely to be driven by persistent reductions in BDNF. Instead, several other multiple mechanisms have been implicated in attenuating synaptic plasticity after GCI, including the effects of sex steroids,42,43 reduced activity of small conductance Ca2+-activated potassium channels, 44 altered autonomous Ca2+/calmodulin-dependent protein kinase II (CamKII), 45 tonic activation of alpha 5 GABA-A receptors, 46 and prolonged activation of transient receptor membrane potential 2 (TRPM2) channels.31,47 Although some studies suggest that BDNF expression may be reduced 7 days after cardiac arrest in adult rats, 48 our data suggest that such reductions are transient and not directly linked to ongoing deficits in synaptic plasticity. By contrast, in the juvenile brain, both our results and previous work 11 indicate that post-ischemic BDNF attenuation, particularly in males, is a key determinant of impaired hippocampal plasticity. Because synaptic remodeling, pruning, and network refinement remain highly active during this developmental period, depletion of neurotrophic support may have a disproportionately large impact on long-term circuit function and cognitive recovery. In adults, on the other hand, injury outcomes are more likely dominated by structural damage and the disruption of established synaptic networks. These fundamental developmental differences underscore the importance of age-specific mechanisms in shaping hippocampal vulnerability and highlight BDNF as a particularly relevant therapeutic target in the juvenile brain.
The juvenile model offers distinct advantages, as the animals are prepubescent at the time of injury,29,49 minimizing the confounding effects of circulating sex steroids on ischemic outcomes. 49 Nevertheless, we and others have reported sex-specific differences in hippocampal vulnerability and recovery mechanisms following juvenile GCI. In the current study, we focused on male mice, as we have previously demonstrated that hippocampal BDNF expression is selectively reduced in males, but not females, after juvenile GCI. 15 These findings are consistent with growing evidence that recovery from brain injury during development is sexually dimorphic and shaped by distinct molecular pathways. For example, in a parallel model of juvenile GCI, we recently demonstrated that LTP recovery in juvenile females is delayed and mediated by estrogen receptor alpha (ERα) activation, independent of BDNF signaling. 15 Importantly, our work has also demonstrated that delayed TRPM2 inhibition can reverse hippocampal impairments in both juvenile sexes. 12 TRPM2 channels are redox-sensitive, calcium-permeable channels implicated in post-ischemic oxidative stress. Notably, TRPM2 channel inhibition has been reported to increase BDNF expression in models of diabetes-induced cognitive decline, 50 suggesting a mechanistic link between TRPM2 modulation and neurotrophic factor repletion. Together, these findings underscore the importance of incorporating both age- and sex-specific considerations into preclinical models to elucidate mechanisms of injury and recovery and to guide the development of targeted neurorestorative therapies.
Our ex vivo experiments further support the role of BDNF in recovery. Application of recombinant BDNF restored LTP in hippocampal slices from juvenile GCI mice but had no effect in sham controls. These results are consistent with the established role of BDNF in supporting CA1 synaptic potentiation 51 and suggest that post-ischemic BDNF deficits directly contribute to the observed plasticity impairments. However, exogenous BDNF is not clinically viable due to its instability in plasma and inability to cross the blood–brain barrier.20,21 Thus, pharmacologic agents that enhance endogenous BDNF production represent a more practical translational strategy.
Type II ampakines are positive allosteric modulators of AMPA receptors that, unlike type I ampakines, both slow deactivation and reduce desensitization, prolonging excitatory postsynaptic currents and enhancing dendritic depolarization. 22 This activity promotes calcium-dependent transcription of BDNF. 24 Previous reports have demonstrated that ampakines restore motor pathways in an adult stroke model,25,26 but this is the first study to examine the effects of ampakines on hippocampal synaptic plasticity in a GCI model. In our experiments, LY404187 increased hippocampal BDNF in a dose-dependent manner in juvenile mice reversed LTP and behavioral deficits when administered 14 days post-GCI. The use of a second, chemically distinct type II ampakine, CX546, an ampakine known to increase BDNF,23,24 showing reversal of LTP impairments strengthens the robustness and translational potential of these findings. Importantly, we co-administered LY404187 and the TrkB antagonist ANA-12, blocking LTP recovery, and suggesting that ampakine-mediated restoration depends on BDNF/TrkB signaling. Previous reports suggest that in vivo ANA-12 attenuates freezing in contextual fear conditioning, 52 thus performing the experiments using in vivo administration of LY404187 and ANA-12 to test whether BDNF/TrkB interaction on the LY404187-improved LTP impairments may have significant confounders. However, the LTP experiments provide an important proof of principle that the effects of LY404187 in synaptic recovery are reliant on TrkB receptor availability. From a translational perspective, these results suggest that type II ampakines could provide a targeted means of enhancing synaptic plasticity during a prolonged period of post-injury vulnerability in pediatric patients. The broad therapeutic window identified here increases feasibility for clinical implementation, especially in real-world settings where treatment delays are common. Furthermore, while we did not perform specific cortical motor testing, we did not observe changes in motor activity in open field testing, suggesting that improvements in memory and learning are not secondary to nonspecific motor effects.
Using two distinct type II ampakines, LY404187 and CX546, strengthens the translational relevance of type II ampakines as therapeutic agents for restoring synaptic function following GCI in the juvenile brain. Importantly, administration of LY404187 showed similar effects in reversing LTP deficits and reversal of hippocampal-dependent memory and learning behavioral impairments, increasing the translational appeal of this strategy. Co-administration of a TrkB antagonist (ANA-12) with LY404187 blocked recovery of LTP, underscoring the critical role of BDNF/TrkB signaling in the neurological improvements observed with ampakine treatment. Together, these findings emphasize the therapeutic potential of type II ampakines as a therapeutic strategy to improve neurological function in pediatric GCI populations.
Our data demonstrate that a single dose of a type II ampakine can restore hippocampal plasticity when tested 24 h after in vivo administration. Prior studies provide mechanistic context for this effect: in hippocampal slice cultures, a single ampakine exposure rapidly increased BDNF mRNA within hours, and maintained elevated protein levels for at least 48 h after treatment, 24 while repeated or “spaced” dosing paradigms sustained BDNF elevations for up to 5 days. 23 In vivo, systemic administration of CX546 also increased hippocampal BDNF expression in adult rodents, though the detailed time course of sustained elevations is less well established compared to slice culture experiments. 24 Together, these data suggest that repeated dosing every 2–3 days may be required to maintain elevated BDNF until endogenous levels recover. 11 Importantly, type II ampakines do not alter AMPA receptor expression. 53 Because LTP maintenance depends on rapid AMPA receptor trafficking to the postsynaptic density to strengthen synaptic transmission 54 the absence of receptor expression changes indicates that the restorative effects observed here are most likely mediated by increased BDNF. While our findings with ANA-12 demonstrate that ampakine-mediated rescue of LTP requires TrkB activation, they do not prove that this effect is exclusively BDNF-dependent. Prior studies have shown that type II ampakines selectively increase BDNF expression in hippocampal and cortical neurons, and we confirmed that LY404187 elevates hippocampal BDNF in vivo. Along with our finding that exogenous BDNF restores LTP after juvenile GCI, these data strongly support BDNF as the primary TrkB ligand mediating recovery. Nevertheless, other AMPA receptor-mediated signaling pathways could also contribute, and future studies employing BDNF-specific knockdown or neutralization approaches will be needed to establish exclusivity. Together, these considerations will be critical in designing translational strategies for patient care.
In conclusion, our study demonstrates that delayed administration of type II ampakines restores synaptic and cognitive function after juvenile GCI via a BDNF/TrkB-dependent mechanism. These results shift the therapeutic focus from preventing early neuronal death to reactivating latent plasticity in surviving neurons. Given the devastating and long-lasting cognitive consequences of pediatric GCI, therapies that enhance adaptive plasticity could profoundly impact developmental trajectories. Future work should prioritize: (1) testing ampakines in large-animal and nonhuman primate models to refine dosing and safety profiles; (2) investigating combination therapies that simultaneously address inflammation, oxidative stress, and neurotrophic support; and (3) exploring biomarkers to identify patients most likely to benefit from delayed neurorestorative intervention. By defining both a mechanistic pathway and a broad post-injury window for intervention, this work lays the foundation for a new class of age-specific, plasticity-targeted treatments in pediatric ischemic brain injury.
Footnotes
Author contributions
Jamie E Henry: acquisition of data, analysis and interpretation of data, drafting of manuscript. April A Fineberg: acquisition of data, analysis and interpretation of data, drafting of manuscript. Tanner B McVey: acquisition of data, analysis and interpretation of data, drafting of manuscript. Erika L Tiemeier: acquisition of data, analysis and interpretation of data. James E Orfila: acquisition of data, analysis and interpretation of data, critical revision. Paco S Herson: study conception and design, analysis and interpretation of data, critical revision. Robert M Dietz: study conception and design, acquisition of data, analysis and interpretation of data, critical revision.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: 1. Boettcher Foundation Webb-Waring Biomedical Research Award 2. NIH NINDS 1R21 NS128784.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.