
Abstract
The recognition of ferroptosis as a distinct, iron-dependent, yet caspase-independent form of cell death has captivated the cell death research field. This discovery has also rekindled optimism that antioxidant approaches, historically disappointing in clinical trials for stroke and related diseases, may yet achieve therapeutic relevance. However, despite an expanding body of literature, knowledge of ferroptosis has not yet translated into meaningful advances for neurological conditions in humans. Here, we summarize evidence supporting the hypothesis that intracerebral hemorrhage, as a prototypical iron-overload disorder, induces tissue damage and functional impairment through ferroptotic mechanisms. Specifically, erythrocyte lysis releases hemoglobin and its degradation product hemin, promoting lipid peroxidation and antioxidant collapse, culminating in neuronal ferroptosis. We further propose that endogenous homeostatic responses driven by pharmacological selenium supplementation, including a novel selenium-based peptide developed in our laboratory, represent effective strategies to mitigate injury and improve functional recovery in rodent models of hemorrhagic and ischemic stroke. Given reproducible effects across models and laboratories, we argue that anti-ferroptotic interventions, particularly those that limit the toxic effects of redox-active iron and reinforce GPX4-centered defenses, warrant advancement toward human clinical trials.
Introduction
Intracerebral hemorrhage (ICH) is a subtype of stroke that results from the rupture of small penetrating arteries within the brain, causing bleeding into the local parenchyma. Although less common than ischemic stroke, accounting for around 29% of incident strokes worldwide, ICH carries disproportionately poor outcomes. 1 The 30-day mortality is high at ~40%–50%, approximately double that of ischemic stroke, 2 but current management remains largely supportive. While surgical hematoma evacuation has shown potential promise in improving functional outcomes for patients with lobar bleeds, limited benefit is experienced by those with hemorrhage in deep subcortical brain structures, who make up most ICH patients.3,4
ICH pathophysiology is classically described as compromising “primary” and “secondary” injury phases, although these pathogenic processes overlap temporally and spatially. Primary injury occurs immediately: the expanding hematoma directly displaces and mechanically damages tissue, producing mass effect, elevated intracranial pressure, and potentially causing fatal herniation. 5 In contrast, secondary injury evolves over hours to days, as the brain reacts to extravasated neurotoxic blood and cell breakdown products. This reaction initiates neuroinflammation, blood–brain barrier disruption, excitotoxicity, oxidative stress, and regulated cell death pathways. 6
Whereas cell death in the necrotic core of the hematoma is largely unregulated and irreversible, secondary injury within the perihematomal region and beyond is characterized by diverse and overlapping RCD pathways, such as apoptosis, necroptosis, autophagy, and ferroptosis (among others). 7 Among these, ferroptosis, an iron-dependent form of regulated cell death driven by lipid-peroxide accumulation, has emerged as a central contributor to neuronal loss and pathophysiology in ICH, and a promising therapeutic target.
From developmental cell death to therapeutic target
The concept of programmed neuronal death in the nervous system dates back to work by Levi-Montalcini and Hamburger in the 1940s–1950s, who demonstrated that the developing system undergoes massive loss of lumbosacral motor neurons and neurons from spinal ganglia. They hypothesized that the purpose of this massive neuronal loss is to match an initially overabundant neuronal pool to its peripheral targets, thereby sculpting mature neuronal circuits. 8 These seminal findings have spawned whole fields of neuroscientific research but have left the open question of how to connect programmed cell death to normal adulthood, aging and disease. Our lab (and others) have been particularly interested in how oxidative stress induces cell death in aging and in neurological conditions like hemorrhagic stroke, and whether this death is programmed.
Broadly defined, oxidative stress is a shift in redox balance towards oxidation, wherein the production of reactive oxygen and nitrogen species (ROS/RNS) surpasses the buffering capacity of antioxidant systems, driving dysfunction or death. 9 At low or transient levels, oxidative signals (termed “oxidative eustress”) serve as essential physiological cues for processes like synaptic plasticity, immune defense, and cellular adaptation. In contrast, sustained or excessive oxidative stress damages lipids, proteins, and DNA, undermining cellular integrity and triggering degenerative pathways. 10 Despite enthusiasm for targeting “oxidative stress” in stroke and neurodegeneration with antioxidant-based therapeutics, clinical trials have largely disappointed. This likely reflects a lack of consideration of the role of oxidants as signaling molecules, alongside an overly reductionist view of cell death. There are also the considerable challenges of therapeutic timing, dosing, bioavailability, and delivery.
Our lab was the first to demonstrate that oxidative stress could be programmed and require de novo protein synthesis. 11 Cellular survival therefore depends on a cell’s ability to sense the local milieu and activate an appropriate transcriptional response commensurate to the level and type of insult. Initially, the cell activates defense programs, including stress-responsive transcriptional regulation and antioxidant pathways. When these fail, the cell commits to programmed cell death, with the pathway dependent on its context. 12 The first such defined pathways were apoptosis, a tightly regulated caspase-dependent cellular dismantling described in the 1970s, 13 and necrosis, once considered passive but now recognized to include regulated forms such as necroptosis. 14 These discoveries established that cell death is not a singular, accidental process but a collection of orchestrated mechanisms. This landscape has since expanded to include numerous forms of RCD, each characterized by distinct molecular triggers and morphologies, mechanistically distinct yet interconnected through extensive crosstalk. 15 Recognition of this diversity has opened new therapeutic avenues, including those that target ferroptosis.
Ferroptosis in ICH: Definition and evidence
First described by Dixon et al. and Stockwell et al., ferroptosis is a distinct iron-dependent form of regulated cell death characterized by three mechanistic hallmarks: (1) increased availability of redox-active ferrous iron; (2) oxidation of poly-unsaturated phospholipids contained within the cell membrane; and (3) failure of protective antioxidant function that normally detoxifies lipid peroxides, enabling their accumulation. 16 Unlike apoptosis, ferroptosis proceeds in a caspase-independent manner with a distinct morphological signature: shrunken mitochondria with increased membrane density and occasional outer-membrane rupture, lacking apoptotic hallmarks such as chromatin condensation or apoptotic bodies. 17
ICH pathology sequentially triggers each of these hallmarks, providing a mechanistic bridge between the iron-rich perihematomal milieu created by the breakdown of extravasated blood products, and the oxidative death of neurons, as illustrated in Figure 1. 16 This section first establishes that ferroptosis occurs in ICH, then examines each hallmark in detail, considering its biochemical mechanism and evidence for activation after ICH.
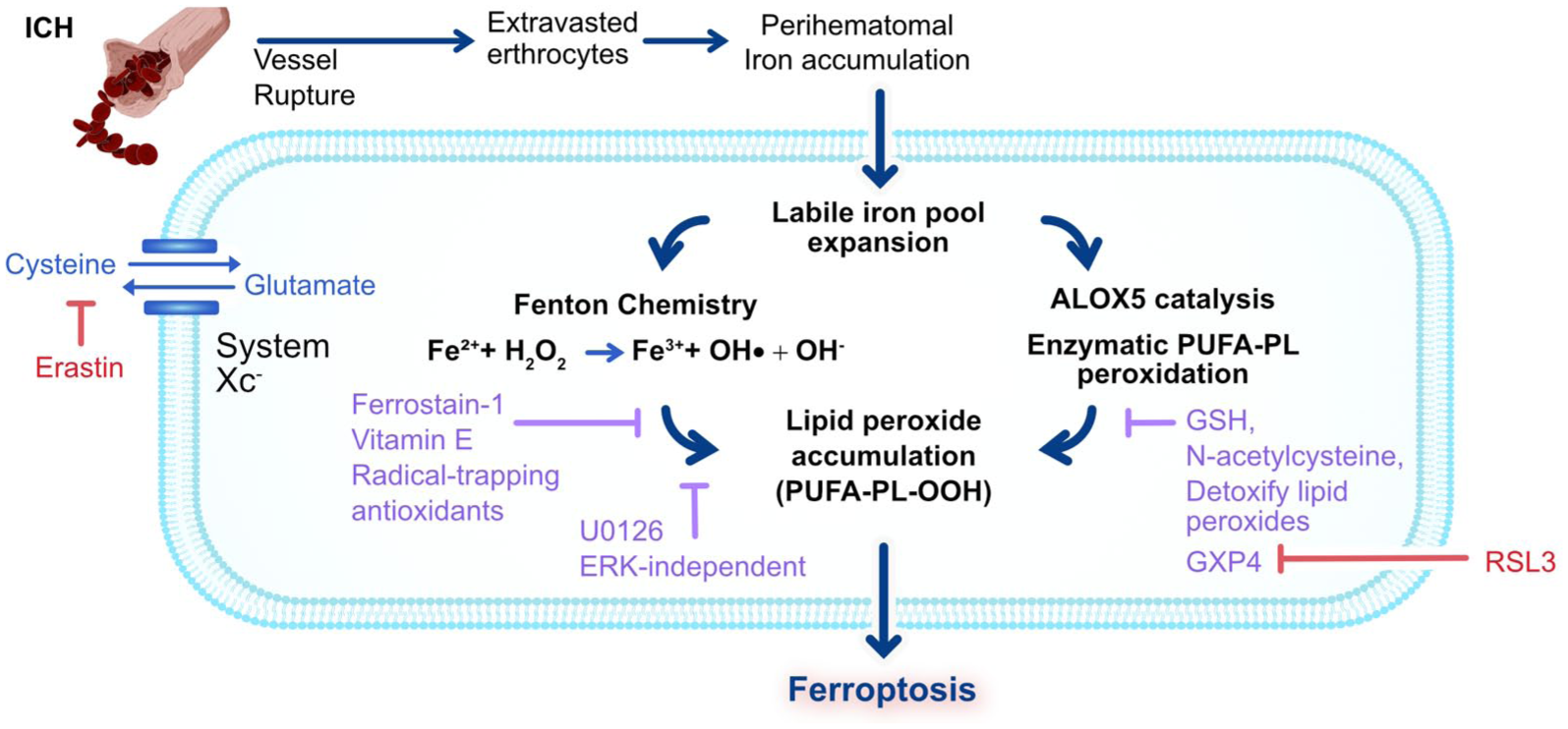
Following vessel rupture, extravasated erythrocytes release heme/iron, expanding the labile iron pool and driving Fenton chemistry that generates ·OH. Together with iron-dependent ALOX catalysis, these reactions promote phospholipid peroxidation (PUFA–PL–OOH), causing membrane damage. Under physiological conditions, cystine import via system Xc− and GSH support GPX4 to detoxify lipid peroxides. After ICH, inadequate antioxidant defence against iron- and lipoxygenase-driven peroxidation leads to ferroptosis. Experimentally, erastin (blocks Xc−) and RSL3 (inhibits GPX4) mimic ICH-induced antioxidant collapse to trigger ferroptosis. Therapeutically, radical-trapping antioxidants (e.g. ferrostatin-1, vitamin E) and the glutathione prodrug, NAC limit lipid peroxidation to rescue cells.
Evidence for ferroptosis following ICH
Both preclinical and clinical evidence support that ferroptosis is an important form of neuronal death after ICH.
Our previous work has shown that the neuronal toxicity observed following ICH shares a common mechanistic basis with ferroptosis. 18 Modelling hemorrhagic injury to cortical neurons with hemoglobin and its breakdown product hemin, we found hemin-induced death in cortical neurons was blocked by established ferroptosis-targeting agents, including ferrostatin-1 (Fer-1), the glutathione prodrug N-acetylcysteine, the iron chelator deferoxamine, and the MAPK inhibitor U0126. Independent work corroborated these findings. Li et al. used the canonical specific inhibitor of ferroptosis, ferrostatin-1 (Fer-1), to prevent neuronal death and reduce iron deposition in hemoglobin-treated organotypic hippocampal slice cultures. 19 In vivo, Fer-1 administration significantly increased neuronal survival, reduced injury volume (quantified from staining of coronal sections) and ameliorated neurological deficits following striatal collagenase ICH. Shrunken mitochondria, and the lack of protection by apoptosis, autophagy, or parthanatos inhibitors supported ferroptosis as the operative pathway. 19 Notably, Fer-1 protection has been tested in clinically relevant paradigms: Chen et al. showed that administration up to 3 h following autologous whole blood injection-induced ICH exerted long-term neuroprotective effects. Neurological deficits and memory function were improved 19, 20, and 21 days after ICH, and post-mortem Nissl staining revealed that brain atrophy was reduced in the Fer-1 treatment group compared to the sham group. 20
Human tissue transcriptomics show a consistent ferroptotic signature in perihematomal regions. Liu et al. re-analyzed the perihematomal tissue gene expression dataset GSE24265 against a curated ferroptosis gene set, identifying 45-ferroptosis linked DEGs, with enrichment of oxidative stress response, TNF signaling, and mitogen-activated protein kinase 1 (MAPK1) pathways, consistent with iron-driven lipid peroxidation after hemorrhage. MAPK1 had been previously implicated as upregulated following ICH in mouse models, in response to iron accumulation and involved in the induction of free radicals following ICH; directionally similar changes were verified through qRT-PCR in an ICH rat model. 21 A similar approach was employed by Diao et al., analyzing the GSE24265 dataset. They identified the novel gene sphingosine kinase 1 (Sphk1) as markedly upregulated. Pharmacologic Sphk1 inhibition, previously shown to enhance BBB integrity in ischemic models, suppressed ferroptosis and reduced secondary injury in mice. In HT22 hippocampal neuroblast cells, Sphk1 blockade rescued glutamate-induced ferroptosis, directly linking Sphk1 to the ferroptotic pathway. 22
While targeting ferroptotic injury mechanisms appears to provide tangible benefit following ICH, it is likely that other forms of RCD occur in parallel, with the potential for crosstalk between different modes of cell death signalling. Although not the focus of this review, this is an area ripe for future investigation.
Iron overload
The initiating hallmark of ferroptosis after ICH is the accumulation of redox-active ferrous iron (Fe2+). 23 Under physiological conditions, iron homeostasis is tightly regulated. Most iron is sequestered within ferritin or compartmentalized in mitochondria and lysosomes, leaving only a small cytosolic labile iron pool. 24 ICH acutely disrupts this balance. Following ICH, sequential erythrocyte lysis releases hemoglobin into the extracellular space (Figure 1). Outside the antioxidant milieu of the erythrocyte, hemoglobin auto-oxidizes to methemoglobin, liberating hemin. 25 Hemin is then degraded by heme-oxygenase (HO-1), yielding carbon monoxide, biliverdin, and, critically, free iron- although the role of heme oxygenase in toxicity is controversial.26–28
Our lab found that Hgb induces cell death in primary neurons in vitro at 1.5 µM. 18 Given that low micromolar extracellular Hb is neurotoxic in primary neurons, even minimal erythrocyte lysis could plausibly raise Hgb to toxic levels. Since hemolysis begins within hours and progresses over the first day after ICH, this likely explains therapeutic windows for anti-ferroptotic agents of 6 h (Figure 2). 19 Continued lysis drives ongoing injury following ICH, supported by intracerebral infusion studies: while infusion of intact erythrocytes causes delayed tissue damage, lysed erythrocytes produce rapid toxicity.29–31 Ultimately, the stepwise breakdown of extravasated erythrocytes results in a prolonged perihematomal excess of redox-active iron. In rodent ICH models, using autologous whole blood infusion into the caudate, non-heme iron rises three-fold within days, and remains elevated for weeks to months. 26 Similarly, collagenase-induced ICH models in rats demonstrate peak iron deposition around 7 days post-injury, with sustained elevation at 14 days (Figure 2). 32 In humans, imaging and postmortem studies demonstrate long-lasting perihematomal iron deposition post-ICH that persists for months and correlates with worse outcomes. 33 It remains unclear whether this iron is in the labile free iron pool or stored in ferritin in professional phagocytes.
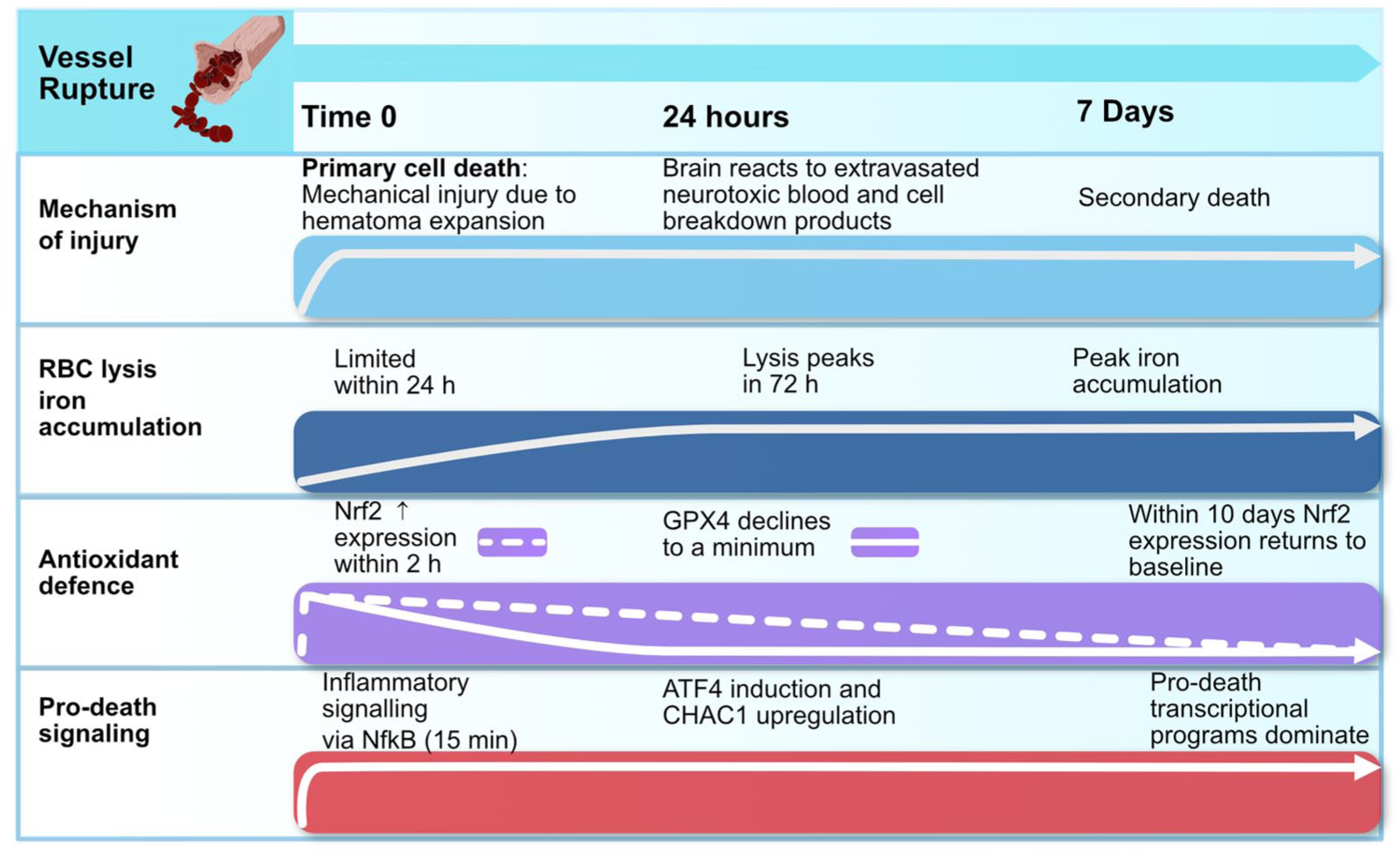
A temporal overview of major ICH injury mechanisms in comparison to known transcriptional changes in antioxidant defence and pro-death programs. Diverse upstream drivers coupled with inadequate endogenous antioxidant defence ultimately converge to precipitate pro-death signalling.
The capacity for iron to drive secondary injury directly following ICH is supported by studies in rats demonstrating that intrastriatal injections of ferrous iron increased oxidative DNA damage, dendritic atrophy, and progressive tissue loss.34,35 This is also supported by the observed benefit of iron chelators in reducing iron accumulation, neuronal death, brain edema, atrophy, and neurologic deficits.29,36,37 However, not all studies consistently find benefits with iron chelation despite achieving reduction in brain iron levels following ICH, 38 highlighting key issues with chelation therapy, discussed in more detail below.
At the cellular level, extracellular iron enters via transferrin receptor-mediated endocytosis and divalent metal transporter 1 (DMT1), contributing to the cytoplasmic labile iron pool. Fe2+ then drives lipid peroxidation by two routes: as a cofactor for lipoxygenases that peroxidize phospholipids, and non-enzymatically via Fenton chemistry, converting H2O2 to hydroxyl radicals that indiscriminately oxidize lipids, proteins, and DNA.39–41 Both routes converge on the ferroptotic hallmark of lipid peroxidation (Figure 1).
Enzymatic and Non-Enzymatic Lipid peroxidation
Neuronal membranes are particularly susceptible to oxidative damage as they are enriched in polyunsaturated fatty acid-phospholipids, particularly arachidonoyl- and adrenoyl-containing phosphatidylethanolamines. These PUFAs contain bis-allylic hydrogen atoms that are readily abstracted to form lipid radicals.42,43 The primary products, PUFA-phospholipid hydroperoxides (e.g. PUFA–PL–OOH), accumulate to destabilize membranes, dissipate electrochemical gradients, and increase permeability, ultimately precipitating rupture and cell death (Figure 1).44,45 Secondary aldehydes, such as malondialdehyde (MDA), also damage proteins and nucleic acids, propagating oxidative damage and impairing cellular function. Although not unique to ferroptosis, MDA is widely used clinically as a marker of non-enzymatic lipid peroxidation burden, indexing oxidative stress. 46
In ferroptosis, iron drives peroxidation through two complementary pathways: enzymatic oxidation by lipoxygenases (LOX), for which iron is a co-factor, and through catalyzing Fenton chemistry to drive non-enzymatic radical autoxidation. Evidence of lipoxygenase involvement in ferroptosis stems from the observation that pharmacological and genetic inhibition of lipoxygenases can be cytoprotective,42,47 while selective inhibition of other lipid-oxidizing enzymes, including cyclooxygenases (COXs) and cytochrome p450 (CYPs), does not rescue cells from ferroptosis. 48 Consistently, LOX overexpression sensitizes cells to ferroptosis, rescued by both radical-trapping antioxidants and LOX inhibitors (Figure 1).
Notably, inhibition of LOX catalysis does not rescue cells from ferroptosis induced by depletion of the antioxidant enzyme glutathione peroxidase 4 (GPX4), such as through the agent RSL3 (RAS-Selective Lethal 3), while radical-trapping antioxidants can (Figure 1). 49 This suggests a potentially dominant contribution of non-enzymatic autoxidation (e.g. driven by Fenton chemistry) in the execution of the terminal stages of ferroptosis. Accordingly, many free-radical scavengers, including ferrostatin-1 (Fer-1) and liproxstatin-1 (Lip-1), inhibit ferroptosis (Figure 1). 50 In liposomal systems, Fer-1 and Lip-1 act primarily as peroxyl-radical scavengers rather than as LOX inhibitors, suggesting that random radical-chain peroxidation may outweigh LOX-mediated oxidation in some contexts.51,52 However, the physiological relevance of these findings in intact neurons remains to be fully established. 51 Overall, both LOX-driven and Fenton-driven lipid peroxidation converge on the abstraction of a hydrogen atom from PUFA-phospholipids, but their relative contributions remain incompletely characterized. It may be that the balance between LOX and Fenton chemistry shifts depending on the cellular and pathological context in which ferroptosis occurs. This distinction has therapeutic implications, as it dictates whether radical-trapping antioxidants or enzyme-directed inhibitors are more effective, and warrants further study.
In ICH, upstream triggers have been shown to promote both enzymatic and non-enzymatic lipid peroxidation. Hemin intercalates into plasma membranes and sensitizes cells to exogenous oxidants (e.g. H2O2), thereby facilitating lipid peroxidation.53,54 Iron accumulation after erythrocyte lysis may enhance LOX function by altering its subcellular localization. In human macrophages, exposure to extracellular Fe3+ and hemin increases LOX binding to nuclear membranes. 55 Whether this mechanism operates in brain tissue following ICH requires direct experimental confirmation. In neuronal systems, 12-LOX is directly inhibited by glutathione (GSH), an integral part of the cell’s glutathione–GPX4 antioxidant defense against ferroptosis. Glutathione (GSH) depletion has been noted to occur following ICH (discussed at length in the next section). Independently, glutathione depletion has been shown to activate 12-LOX, increasing peroxide generation and cell death, opposed by 12-LOX inhibitors,48,56 although no study yet has provided direct evidence through investigating each putative axis. Therefore, by promoting iron overload and glutathione depletion, ICH provides convergent pressure for LOX-dependent and Fenton-driven peroxidation, exacerbated by pro-oxidant and inflammatory conditions established by neurotoxic blood products.
In vivo models highlight lipid peroxidation burden following hemorrhagic exposure: intrastriatal heme injection induces neuroinflammation and increases brain lipid peroxidation in mice, 57 although ferroptotic death was not directly quantified and so a direct causal link cannot be established. However, intercepting peroxidation is protective in primary ICH models. Our group showed that N-acetylcysteine (NAC), an antioxidant thiol compound, neutralizes ALOX5-derived toxic lipids, with efficacy comparable to chemical or molecular inhibitors (Figure 1). 47 Systemic NAC administration following collagenase ICH in mice reduces hemin-induced ferroptotic neuronal death and improves functional recovery. 47 In a related model of warfarin-treated mice after experimental stroke, 12/15-LOX inhibition lowers lipid peroxidation injury and reduces hemorrhagic transformation. 58 While not primary ICH, this reinforces peroxidation as an active, modifiable driver of hemorrhagic brain injury, potentially supporting strategies that target peroxidation. Translating these insights to human ICH requires both biomarker validation and therapeutic testing.
Correlative evidence suggests that membrane peroxidation is active in patients following ICH. Human biomarker data indicates that serum MDA is elevated at presentation in severe ICH patients versus controls, and further higher in non-survivors than survivors. Baseline MDA independently predicts 30-day mortality after adjusting for confounding factors such as hematoma volume or age. 59 While MDA is not ferroptosis-specific, it is a direct readout of unchecked lipid peroxidation, providing correlative evidence that membrane peroxidation is active in patients following ICH. Therapeutic targeting of lipid peroxidation in human primary ICH is similarly underexplored; it may prove easier to inhibit ROS production than scavenge and neutralize radicals once self-amplifying chain reactions have begun.
Early clinical relevance suggests potential therapeutic relevance of lipid peroxidation. Edaravone, a potent free radical scavenger, improves functional outcomes in patients with aneurysmal subarachnoid hemorrhage (a condition sharing overlapping oxidative injury mechanisms with ICH) 60 but has not yet reduced mortality in ICH trials. As a relatively weak ferroptosis inhibitor, the modest benefit of edaravone hints that more potent radical trapping antioxidants might influence functional outcome, an integrated measure of therapeutic benefit (Ratan Lab, unpublished observations).
Ultimately, lipid peroxidation represents the execution stage of ferroptosis in ICH: excess iron primes and catalyzes the reaction, while PUFA-rich neuronal membranes provide vulnerable substrates. Once initiated, lipid peroxidation is self-propagating, as newly formed lipid radicals abstract hydrogens from adjacent PUFA chains, generating additional peroxides and amplifying oxidative damage across the bilayer. The autocatalytic nature of lipid peroxidation makes the integrity of antioxidant defenses, particularly the GPX4–glutathione system, the final determinant of whether neurons withstand oxidative stress or succumb to ferroptotic death.
Collapse of antioxidant defenses
Cellular defenses against ferroptosis involve multiple levels of endogenous antioxidant systems. Primarily, the GPX4–glutathione axis reduces phospholipid hydroperoxides to alcohols using glutathione, and is supported by System Xc−, which sustains glutathione by importing cystine. In parallel, two GPX4-independent systems, the ferroptosis suppressor protein 1 (FSP1)–coenzyme Q10 (CoQ10) system, and GTP cyclohydrolase 1 (GCH1)–tetrahydrobiopterin pathway, also limit lipid peroxidation. In ferroptosis, these systems fail or are overwhelmed, and the balance between the generation and removal of lipid peroxides tips in favour of lipid peroxide accumulation.
First described by Ursini et al., GPX4 is an antioxidant enzyme that inhibits lipid peroxidation and can degrade H2O2, small-molecule peroxides, complex lipid peroxides, and lipid hydroperoxides, using GSH as a substrate. 61 GPX4 activity requires reduced glutathione (GSH) as an electron donor. GSH synthesis, in turn, depends on the supply of cysteine, imported intracellularly as cystine via the cystine/glutamate antiporter, system Xc−. Inside the cell, cystine is reduced to cysteine and incorporated into GSH. By continuously inhibiting lipid peroxidation in healthy cells, GPX4 serves as a primary enzymatic defense against ferroptosis. 62
The importance of GPX4-mediated protection against ferroptosis is demonstrated by the compounds used to experimentally induce ferroptosis. Following Marcus Conrad’s discovery that genetic inactivation of GPX4 induced a form of non-apoptotic lipid-peroxidation cell death, suppressed by the radical scavenger α-tocopherol, and capable of rescue by overexpression of System Xc−, 63 Dixon et al. proposed the concept of ferroptosis. They found that erastin, by directly inhibiting system Xc−, caused this form of cell death through depletion of cysteine and glutathione, allowing peroxide accumulation and driving ferroptotic death. 16 Directly inhibiting GPX4 (e.g. through chemical inhibition by RSL3, or degradation by FIN56) similarly drives ferroptotic death. 64
In vivo, loss of GPX4 following ICH appears to contribute substantially to secondary brain injury. In autologous whole blood injection models, protein levels of GPX4 decline to a minimum level 24 h post-ICH (Figure 2). 65 A fall in GPX4 expression worsens secondary injury, including blood–brain barrier disruption, edema, inflammation, oxidative stress, and neuronal death. Artificial manipulation of GPX4 levels confirms this as a causative effect: genetic overexpression of GPX4 within the brain relieves secondary injury, whilst pharmacological inhibition or genetic knockdown exacerbates secondary injury, 65 although in this study, ferroptotic death was not characterized sufficiently to draw a direct link between a fall in GPX4 to an increase in ferroptosis.
Although GPX4 is clearly a central antioxidant defense, emerging work suggests other glutathione peroxidases may contribute to antioxidant protection. In a recent study, single-cell spatiotemporal transcriptomic mapping around ICH lesions revealed GPX1 upregulation in immune cells. 66 Despite this, the role of GPX1 in ferroptosis suppression has not been clarified, suggesting GPX1 may reflect activation of broader anti-ferroptotic programs involving GPX4, glutathione-s-transferases (GSTs), and FSP1.
Analyses of differential sensitivity to GPX4 inhibitors in cancer cell lines have identified two additional, partially independent safeguards that maintain redox balance: the FSP1–CoQ10 system, and the GCH1–BH4 axis.67–69 In the FSP1–CoQ10 pathway, FSP1 acts as a coenzyme Q oxidoreductase to regenerate reduced CoQ10 (ubiquinol), which traps lipid peroxyl radicals and prevents lipid peroxidation propagation. In the GCH1–BH4 pathway, GCH1 catalyzes the rate-limiting conversion of GTP to tetrahydrobiopterin (BH4). In turn, BH4 directly scavenges lipid peroxyl radicals in a dose-dependent manner, halting self-amplifying phospholipid autoxidation, and acting as a cofactor for its own regeneration. BH4 reinforces the FSP–CoQ10 axis through driving CoQ10 precursor synthesis, changing the amino acids phenylalanine into tyrosine which feeds CoQ10 head-group synthesis. 70 In addition, BH4 remodels the phospholipid bilayer, promoting membrane remodeling toward less peroxidation-prone phospholipids enriched in saturated or monounsaturated fatty acids. 71
In the absence of GPX4, the FSP1–CoQ10 and GCH1–BH4 pathways become key modulators of ferroptosis sensitivity. Using expression cloning in GPX4-null human cancer cells, Doll et al. showed that overexpressing FSP1 rescued cells from GPX4 loss, restoring resistance to ferroptosis inducers by regenerating reduced CoQ10 at membranes. 68 In parallel, Bersuker et al. reported that higher endogenous FSP1 correlates with ferroptosis resistance across hundreds of cancer cell lines, and that manipulating FSP1 levels alters sensitivity to GPX4 inhibitors both in vitro and in xenograft models. 69 However, direct evidence for FSP1–CoQ10, or GCH1–BH4 involvement in ICH remains lacking.
In murine ICH, perihematomal FSP1 is markedly reduced. Dexpramipexole (an R-isomer of the anti-Parkinson drug pramipexole) alleviated perihematomal iron and ROS burden, and suppressed ferroptosis iron and ROS, as assessed by MDA and mitochondrial morphology. Notably, the ameliorative effect on white matter injury depended on GPX4 and FSP1 upregulation, coupling the causative role of collapse of antioxidant systems to ferroptosis within the setting of ICH. 72 Similarly, CoQ10 administration in murine collagenase ICH improved neurological deficits, reduced edema, and limited hematoma in collagenase ICH, likely via NF-κB modulation, although ferroptosis was not directly assessed. 73 These findings highlight the therapeutic potential of boosting parallel antioxidant systems to curb ferroptosis after ICH, while highlighting the need for research to link ferroptosis to these systems in ICH specifically.
Time-dependent transcriptional regulation of ferroptosis in ICH
The preceding section established that ICH triggers the three biochemical hallmarks of ferroptosis: iron overload, lipid peroxidation, and antioxidant collapse. However, ferroptosis represents an attractive therapeutic target precisely because it is not a passive byproduct of iron-mediated oxidative stress, but rather the outcome of regulated metabolic and transcriptional programs. Early evidence for the regulatory control of ferroptosis came from studies demonstrating that inhibitors of transcription and translation (actinomycin D and cycloheximide, respectively) prevented glutamate-induced ferroptosis in neurons, 11 establishing that cell death requires dynamic gene expression.
In ICH, the transcriptional control of ferroptosis is especially relevant because the perihematomal environment evolves over hours to days, progressively challenging neurons with oxidative, inflammatory, and iron-driven stressors. Cells respond actively to these progressive stressors, activating endogenous stress-response pathways that, depending on context and duration, can either protect against or promote ferroptotic death. This section traces the temporal arc of ferroptotic regulation following ICH: from early inflammatory activation (NF-κB, JAK–STAT) through transient antioxidant defenses (Nrf2), to eventual pro-death programming (ATF4).
Early inflammatory transcriptional events after ICH
In the first 24 h following ICH, erythrocyte lysis is relatively limited. 26 Instead, early cytotoxicity derives from blood plasma constituents such as thrombin, coagulation factors, complement, and immunoglobulins released into the parenchyma. Notably, thrombin can directly trigger ferroptotic pathways even before substantial iron release. 74 Concurrently, primary cellular necrosis within the hematoma and perihematomal regions releases damage-associated molecular patterns (DAMPs), which activate resident microglia and infiltrating immune cells through pattern recognition receptors. This initiates inflammatory signaling cascades, especially involving the transcription factors nuclear factor-kappa B (NF-κB) and the Janus kinase/signal transducers and activators of transcription (JAK–STAT) signaling pathway.
These pathways are activated with remarkable rapidity: NF-κB activation is detectable within 15 min following the intrastriatal injection of autologous blood in rats and remains elevated for at least a week (Figure 2). 75 In human ICH tissue, perihematomal NF-κB upregulation correlates with both local cell death and clinical outcome. 76 Functionally, NF-κB and JAK–STAT activation creates a pro-oxidant environment, driving pro-inflammatory cytokine production (e.g. TNF-α, IL-1β), induction of ROS-generating enzymes, and early consumption of the available endogenous antioxidant pool. 77 Additionally, JAK–STAT signaling upregulates hepcidin expression, promoting iron retention, and potentially contributing to later iron overload. 78 This early pro-inflammatory signaling establishes a pro-oxidative environment, likely heightening neuronal susceptibility to ferroptosis once erythrocyte lysis and subsequent iron accumulation begin in earnest. Yet NF-κB signaling is not uniformly pro-death. In some contexts, it induces manganese superoxide dismutase (MnSOD), ferritin heavy chain, and other antioxidant enzymes that transiently buffer oxidative stress, thereby contributing to the resolution of inflammatory responses.78–80 This duality frames NF-κB as a context-dependent redox sensor, where acute activation may confer transient protection, 81 whereas the sustained activation typical of ICH typically amplifies vulnerability to ferroptosis. 82
Adaptive antioxidant responses: Nrf2
Following ICH, neurons and glia mount an endogenous defense by activating an Nrf2-centred antioxidant program. Nuclear factor erythroid 2-related factor 2 (Nrf2, a basic leucine zipper transcription factor) regulates key determinants of ferroptosis sensitivity, positioning it to coordinate a unified anti-ferroptotic response.83,84
Under basal conditions, Nrf2 levels remain low, as it is constitutively tagged for proteasomal degradation by the Keap1–Cul3–RBX1 E3 ubiquitin ligase complex. In the cytosol, the actin-bound protein Keap1 binds Nrf2 and directs it for ubiquitination and proteasomal degradation. 85 In response to electrophilic stress arising from reactive oxygen species, heme, or electrophilic compounds such as sulforaphane, reactive cysteine residues on Keap1 become alkylated or oxidized. This disrupts its interaction with Nrf2, allowing newly synthesized Nrf2 to stabilize and accumulate within the cytoplasm. Stabilized Nrf2 then translocates to the nucleus, where it binds antioxidant response elements (AREs) in target gene promoter regions, inducing a cytoprotective transcriptional program. 86
The Nrf2-driven cytoprotective response limits ferroptosis by modulating susceptibility across its three principal “hallmarks.” First, it reduces redox-active iron accumulation and heme toxicity, upregulating ferritin heavy and light chains (FTH1 and FTL), heme oxygenase-1 (HO-1), and the heme transporter HRG1. Second, it enhances detoxification of lipid peroxidation products, increasing expression of aldo-keto reductases, peroxisome proliferator-activated receptor γ (PPARγ), and glucose-6-phosphate dehydrogenase. This in turn regenerates NADPH to sustain antioxidant enzyme activity. Finally, Nrf2 reinforces the glutathione antioxidant system by promoting GSH synthesis, increasing expression of SLC7A11 (a key system Xc− subunit), glutathione synthetase (GSS), glutathione S-transferases, and GPX4.47,87
Experimental evidence confirms Nrf2’s protective role. Nrf2-deficient neurons are more vulnerable to oxidative stress, a phenotype rescued by re-expression. 88 In rodent ICH models, Nrf2 knockout exacerbates hematoma growth, oxidative injury, and neurological deficits. 89 In vitro, Nrf2 modulation alters sensitivity to ferroptosis inducers such as erastin. 90 Despite this, Nrf2-mediated protection is short-lived. In experimental ICH, Nrf2 expression rises within 2 h, peaks at 24 h, and returns to baseline by day 10 (Figure 2). 91 In ICH patients, perihematomal nuclear Nrf2 levels are increased relative to contralateral tissue but remain lower than in non-ICH controls up to 60 days post-stroke. 92 Together, these findings indicate that Nrf2 activation provides an early but ultimately inadequate buffer against the prolonged oxidative stress of ICH.
One possible explanation for the blunted Nrf2 response after ICH is its reciprocal inhibition with NF-κB signaling. The p65 subunit of NF-κB can directly suppress NRf2 transcription by competing for co-activator CBP, and through recruitment of histone deacetylase 3, which promotes local hypoacetylation and reduced Nrf2 expression. Conversely, Nrf2 restrains NF-κB signaling, and so Nrf2 deficiency can enhance NF-κB activity through reducing antioxidant gene expression; Nrf2 products such as HO-1 derived carbon monoxide and bilirubin dampen NF-κB’s nuclear translocation. Importantly, astrocytes are the dominant source of Nrf2 activity in the CNS. Their robust induction of antioxidant programs supplies neurons with glutathione and associated precursors, while neurons themselves express relatively little Nrf2, leaving them heavily reliant on glial support. 83 Given that astrocytic Nrf2 is especially sensitive to NF-κB-mediated suppression, pro-inflammatory activation during ICH may disproportionately blunt this protective glial program, leaving neurons vulnerable to ferroptosis. Although this reciprocal regulation has been demonstrated in other injury models,93–96 it remains to be directly tested in ICH.
Pro-death transcriptional programming in ferroptosis
In the hours to days after ICH, as antioxidant defenses wane, pro-death transcriptional programs increasingly dominate, activating genes that dismantle glutathione metabolism and amplify vulnerability to ferroptosis (Figure 2). These pro-death programs are orchestrated largely by activating transcription factor 4 (ATF4), which integrates oxidative stress signals partly independent of eIF2α phosphorylation. 97 ATF4 is required for the neuronal transcriptional response to oxidative stress, driving a gene program that depletes glutathione metabolism and weakens GPX4-mediated detoxification of lipid hydroperoxides, with the overall effect of amplifying ferroptotic susceptibility. Supporting this, ATF4 knockout abolishes most oxidative stress-induced transcriptional changes: in primary cortical neurons exposed to the glutamate analogue homocysteic acid, 119 transcripts were altered in ATF4+/+ cells but only three in ATF4−/− cells. Loss of ATF4 largely abolished stress-induced gene activation, particularly across categories linked to mitochondrial function, oxidoreductase activity, and amino-acid metabolism. 98
Notably, ATF4 activates a mixed stress-response program, with genes implicated in multiple forms of cell death. Cation transport regulator homolog 1 (CHAC1) is most clearly linked to the induction of ferroptosis, encoding a γ-glutamyl cyclotransferase that degrades glutathione, undermines GPX4 function, increases oxidative stress, and facilitates lipid peroxide accumulation. 99 In parallel, ATF4 can induce classically apoptotic genes such as C/EBP homologous protein (CHOP), which upregulates several pro-apoptotic mediators including PUMA, 100 and TRB3 (Tribbles homolog 3), which promotes apoptosis during ER stress through interference with pro-survival Akt signaling. 101 These signals nonetheless ultimately operate upstream of ferroptotic death. Dixon et al. demonstrated that inhibition of system Xc− with erastin, the canonical ferroptosis inducer, activates an endoplasmic reticulum (ER) stress response, upregulating downstream eIF2α–ATF4 targets including CHOP and ATF3. Ferroptotic death proceeded despite caspase inhibition, confirming its non-apoptotic nature. 102
The importance of ATF4 is reinforced by genetic studies: neurons from ATF4-knockout mice are rendered resistant to ferroptosis induced by homocysteic acid, a functional analog of erastin. Reintroduction of wild-type ATF4 restores their sensitivity, whereas a mutant lacking the DNA-binding domain fails to reverse this effect. 98 This establishes that the DNA-binding activity of ATF4, facilitating its ability to drive transcription of target genes, is essential for ferroptotic execution. In other words, this indicates that ferroptosis is driven by active transcriptional regulation, rather than being a passive consequence of oxidative stress. Consistent with this, the PERK–ATF4 pathway regulates neuronal survival in both in vitro and in vivo ICH models. In hemin-treated neurons, metabolomic analysis revealed upregulation of this pathway during neuronal adaptation to oxidative stress, and PERK inhibition partially reduced ICH volume and improved neurobehavioral performance. 103 In a collagenase model of ICH, the levels of phosphorylated PERK and ATF4 were significantly higher in ICH than in sham tissue, with marked upregulation of ATF4 expression. Notably, the protective effects of the neurotropic factor neuritin (which inhibits neuronal death, improves neurological deficits, and reduces lesion volume and edema in mice) are ERK- and ATF4-dependent; these protective effects were blocked by PERK and Akt signalling pathway inhibitors. 104 Together, these findings indicate that activation of ER stress can convert ATF4 from a pro-ferroptotic transcription factor to one that is anti-ferroptotic, reflecting the context-dependent nature of stress-response signaling. 98
Ferroptosis as a therapeutic target
The preceding sections establish that ferroptosis after ICH represents the failure of transcriptional adaptation rather than passive oxidative injury. Early Nrf2-driven defenses prove insufficient against sustained oxidative pressure from progressive erythrocyte lysis, their effectiveness blunted by reciprocal NF-κB suppression. As oxidative stress intensifies, ATF4 activation commits neurons to a pro-death trajectory, proving maladaptive by actively disabling the glutathione–GPX4 antioxidant axis.
Understanding that ferroptosis is regulated rather than passive reveals multiple therapeutic intervention points (Figure 3). Each hallmark can be targeted: iron chelation limits the accumulation of catalytic ferrous iron that initiates lipid peroxidation, antioxidant reinforcement (selenium, CoQ10) counters membrane peroxidation and restores the GPX4–glutathione axis, and transcriptional modulation prevents the shift from adaptive to maladaptive gene programs (Figure 3). These approaches are complementary rather than competing, allowing broad approaches addressing both the initiation and execution of ferroptotic injury. Each strategy is examined below with attention to mechanistic rationale, existing evidence, and translational challenges.
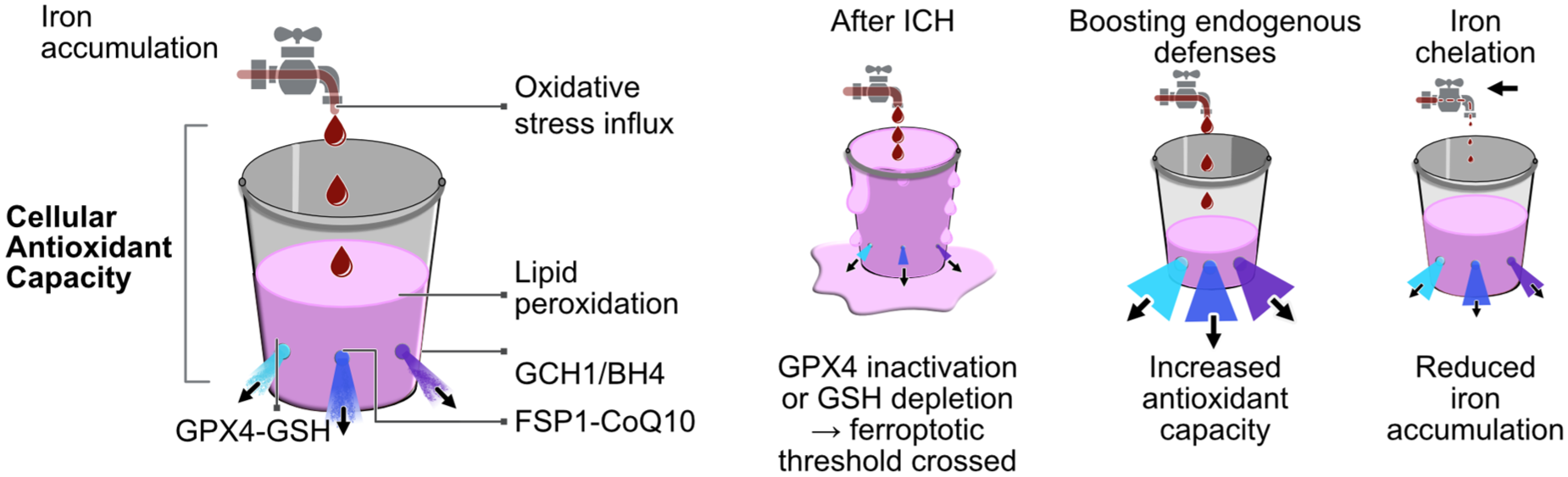
Iron released from lysed erythrocytes acts as the “tap,” driving oxidative influx via Fenton chemistry (Fe2+ + H2O2 → Fe3+ + •OH + OH−). ROS enter the “bucket,” representing oxidative load, while the bucket’s fluid level reflects lipid peroxide accumulation. Antioxidant “drains” depict endogenous detoxification systems (GPX4–GSH, FSP1–CoQ10, GCH1–BH4). After ICH, excess iron and ROS overwhelm these defenses; GPX4 inactivation or GSH depletion raises lipid peroxides beyond antioxidant capacity, triggering ferroptosis (“overflow”). Enhancing antioxidant “outflow” through selenium-driven GPX4 induction or CoQ10 supplementation restores redox balance and prevents ferroptosis. Iron chelation “turns down the tap,” reducing oxidative influx, though residual ROS from inflammatory or mitochondrial sources may sustain lipid peroxidation.
Iron chelation
Iron chelation is an intuitive therapeutic prospect after ICH, as erythrocyte lysis and heme catabolism generate sustained perihematomal pool of redox-active Fe2+, which catalyzes lipid peroxidation via Fenton chemistry and lipoxygenases. Chelators sequester free Fe2+ into chemically inert complexes, unable to participate in Fenton reactions, which can then be cleared into the bloodstream for harmless excretion, 105 meaning that they have the potential to reduce lipid peroxidation burden (Figure 3).
Iron chelation has demonstrated benefit in numerous preclinical ICH models. The classical chelator deferoxamine (DFO) consistently lowers perihematomal iron, suppresses lipid peroxidation, and improves histological and behavioural outcomes including edema, tissue atrophy, and neurological function.36,37 For example, systemic DFO after autologous whole blood ICH in rats reduces brain iron burden, attenuates MDA/4-HNE accumulation (marking oxidative stress), limits edema and neuronal death, and enhances neurobehavioral recovery. 37 However, protection by DFO is inconsistent: in a collagenase-induced ICH model, DFO reduced brain iron, but failed to reduce neurological damage and improve long-term prognosis. 38 Conversely, in another study using the same model, DFO reduced iron accumulation, ROS production, microglial activation, and neuronal loss, yielding modest functional improvement, yet without reducing injury volume or edema. 33
Despite some encouraging preclinical evidence, DFO has not demonstrated clear clinical efficacy. The initial HI-DEF trial, testing higher doses of DFO (up to 63 mg/kg/day over 5 days) was terminated early due to toxicity, specifically an increased incidence of acute respiratory distress syndrome. 106 This established that higher exposures of DFO are not tolerable in acute ICH, prompting the design of the modified i-DEF trial, using lower doses and shorter duration. The phase II i-DEF trial confirmed that 3-day DFO administration (32 mg/kg/day) post-ICH is safe but failed to improve 90-day functional outcomes versus placebo, precluding phase III advancement. 107 However, subsequent post-hoc analyses have revealed a more complex picture of DFO’s effects. A 2022 analysis found that DFO-treated patients showed improvement in modified Rankin scores, denoting an improvement in functional disability, specifically between 3 and 6 months post-stroke. 108 This might highlight delayed therapeutic benefit not captured by the primary 90-day endpoint in i-DEF. More recently, an analysis demonstrated that DFO was associated with 2.3-fold higher odds of early major neurological improvement (EMNI) during the first week post-ICH, and this early improvement was independently associated with favorable long-term functional outcomes at both 90 and 180 days. 109 While these post-hoc findings are intriguing, they are exploratory by nature, unable to establish causation in the same way as pre-specified primary endpoints. The delayed benefits observed could reflect statistical noise, patient selection effects, or genuine therapeutic mechanisms operating on extended timescales. Nonetheless, the consistency of benefits across multiple post-hoc analyses, combined with the biological plausibility of delayed iron-mediated injury, has reinvigorated clinical interest in DFO. These findings also highlight the importance of extended follow-up periods in ICH trials, and highlight that EMNI could serve as a valuable early biomarker in future ICH trials.
The disconnect between preclinical and clinical outcomes reflects pharmacokinetic and biological challenges. For one, DFO has poor oral bioavailability, rapid systemic clearance, and limited blood–brain barrier permeability, making it difficult to sustain therapeutic concentrations in the brain parenchyma. This is further confounded by the progressive nature of iron accumulation following ICH, as erythrocytes are lysed gradually over days. Given this extended timeline of iron release, the brief infusion protocols used in clinical trials (3–5 days) may be insufficient to meaningfully impact iron-mediated injury throughout the critical period of secondary damage.
Alternative chelators have been explored to overcome the pharmacokinetic limitations of DFO, isolating delivery hurdles from potential therapeutic benefit. Deferiprone is an orally bioavailable, blood–brain barrier–permeant chelator that lowers brain iron, yet translational success in chronic neurodegeneration has been mixed-to-negative. For example, in Parkinson’s disease, the FAIRPARK-II trial showed that deferiprone (30 mg/kg/day) successfully reduced substantia nigra iron but did not improve motor outcomes; numerical trends suggested potential worsening in some motor assessments, though the clinical significance remains debated. 110 Similarly, in a randomized trial in amyloid-confirmed early Alzheimer’s disease, deferiprone decreased hippocampal iron but was associated with accelerated cognitive decline on some executive function measures. 111 These findings indicate that adequate CNS exposure and iron reduction do not necessarily translate into neuroprotection, underscoring the context-dependence of iron chelation strategies and the importance of iron in normal physiology of the brain.
Notably, the selectivity of iron chelation, towards either redox-active ferrous (Fe2+) versus ferric (Fe3+) iron, may influence efficacy. Wang et al. compared deferiprone with clioquinol, a Fe2+/Cu2+ chelator, in experimental collagenase-induced rat ICH models. While both reduced brain iron levels, clioquinol provided superior neuroprotection, attenuated brain edema and ROS production, and improved neurological outcome, unlike deferiprone. 112 One possible mechanistic explanation is that clioquinol preferentially chelates labile ferrous iron (Fe2+), the redox-active species that directly catalyzes Fenton chemistry and lipid peroxidation, whereas deferiprone may have greater affinity for Fe3+, which can be recycled back to Fe2+ within the reducing cellular environment, allowing Fenton chemistry to continue. Additionally, the two chelators exerted opposing effects on iron transporters: clioquinol increased ferroportin expression without increasing DMT1, whereas deferiprone increased DMT1 without increasing ferroportin, differences consistent with divergent impacts on iron export versus import pathways.
While extending chelation may sustain effective parenchymal concentrations, it would likely amplify adverse effects. DFO exhibits dose-dependent toxicity, including visual and auditory neurotoxicity with chronic exposure, and acute gastrointestinal symptoms such as nausea, diarrhea, and abdominal pain. 105 More fundamentally, iron is physiologically indispensable, critical for neurotransmitter synthesis, myelination, mitochondrial respiration, and DNA replication. 113 Prolonged indiscriminate chelation may disrupt fundamental neurological processes, potentially exacerbating cognitive deficits in neurologically vulnerable stroke survivors. Thus, iron represents an intersection of physiology and pathology, rendering nonspecific chelation a potentially blunt therapeutic approach in ICH. Targeted delivery systems, such as nanoparticle-conjugated chelators with BBB permeability, 114 may overcome some challenges, but by the time chelation is feasible, downstream oxidative cascades may already be self-propagating, rendering intervention “too little, too late.”
The protective effects of iron chelation extend beyond simple metal sequestration: chelators also modulate oxygen-sensing pathways that activate adaptive stress responses. 115 Our work supports this observation by identifying the underlying mechanism: DFO and related chelators inhibit ferroptosis in vitro by targeting hypoxia-inducible factor (HIF) prolyl hydroxylases (PHDs). 116 Under normoxic conditions, HIF PHDs use iron as a cofactor to hydroxylate HIF-1α, marking it for proteasomal degradation.117,118 Chelation inhibits PHD activity, stabilizing HIF-1α and promoting the transcription of genes involved in angiogenesis, metabolism, and cytoprotection.115,119 These findings suggest that target-selective iron chelation and PHD inhibition, rather than bulk iron removal per se, may mediate the central protective mechanism of DFO. Supporting this idea, the small-molecule PHD inhibitor adaptaquin, a branched analog of clioquinol, provided neuroprotection in rodent ICH models without the limitations of chelation. Adaptaquin reduced neuronal death and neurological deficits without any detectable increase in HIF-1α levels or activation of canonical HIF target genes. Instead, adaptaquin reduced the expression of ATF4-dependent pro-death transcriptional programs induced by oxidative stress, indicating that PHD inhibition can modulate redox-sensitive gene networks independently of canonical HIF signaling. 113 Looking forward, selective PHD inhibitors may isolate the protective effects of iron chelation from its significant drawbacks, preserving iron’s physiological functions while limiting its pathological role after ICH. At the same time, research into ferroptosis has also revealed a host of iron-independent targets that will be discussed below. These may offer the best opportunity for treatments with potentially broader windows of therapeutic efficacy.
Augmenting endogenous anti-ferroptotic defenses
While iron overload may trigger ferroptosis, focusing exclusively chelation risks missing the transcriptional regulation and antioxidant failure that define ferroptotic death. A more salient therapeutic strategy may be to target and augment pre-existing intrinsic protective responses mounted by neurons and glia in response to ferroptotic stress (Figure 3). As discussed earlier in this review, neurons deploy an initial transcriptional adaptive program in response to ferroptotic stress, which is ultimately insufficient to protect cells from death as sustained erythrocyte lysis progresses (Figure 3). Acutely, cells upregulate classical antioxidants pathways such as Nrf2, but this response is transient, becoming overwhelmed under sustained oxidative stress. Notably, our laboratory found that exposure to pro-ferroptotic stimuli, such as hemin or glutamate analogues, triggers a distinct transcriptional response in both cultured neurons and murine ICH models. Rather than inducing canonical antioxidants like catalase or superoxide dismutase, the dominant adaptation is the selective upregulation of selenoenzymes, including GPX4, thioredoxin reductase 1 (TXNRD1), glutathione peroxidase 3 (GPX3), and selenoprotein P (SelP).120,121
Evidence for the protective nature of GPX4 induction
As discussed previously, GPX4-mediated detoxification of lipid hydroperoxides is a central anti-ferroptotic safeguard. To confirm that GPX4 induction represents a protective response, we used a conditional protein-stabilization system to control GPX4 levels following injury onset. Enhancing GPX4 expression conferred robust neuroprotection 4–8 h following ferroptotic insult with hemin or glutamate. Similarly, artificially elevating GPX4 levels before hemorrhage reduced neuronal death and resulted in nearly complete functional recovery after ICH by day 14 in behavioral tests of sensory or spatial neglect. 120 These findings suggest that reinforcing selenoenzyme expression could augment an otherwise submaximal antioxidant response following ICH (Figure 3), sustaining lipid hydroperoxide detoxification and preventing the redox collapse that commits cells to ferroptosis.
Consistent with this rationale, selenium supplementation dose-dependently inhibited ferroptosis induced by hemin or homocysteic acid, triggering a protective transcriptional program that upregulated GPX4, selenoprotein K (which mitigates ER stress), and genes conferring resistance to excitotoxicity. These effects extended in vivo: intracerebroventricular injection of selenium 2 h following striatal ICH in mice reduced neuronal death and improved functional recovery, without shrinking hematoma volume, indicating mitigation specifically of secondary injury processes.
The activation of such a broad, multi-pathway transcriptional program suggests therapeutic utility of selenium beyond ICH, encompassing both hemorrhagic and ischemic cerebrovascular injury. Supporting this, our laboratory found that selenium-driven GPX4 induction improved outcomes not only after experimental ICH but also following middle cerebral artery occlusion. 120
Overcoming toxicity challenges of Se
Selenium therapy, aimed at inducing GPX4 and reinforcing endogenous antioxidant defenses, holds broad therapeutic promise for inducing GPX4 and reinforcing endogenous antioxidant defenses, but clinical translation faces significant challenges related to its narrow therapeutic index and chemical reactivity. Multiple selenium compounds have been successfully administered in vivo with neuroprotective efficacy in models of ischemia-reperfusion injury and other neurological conditions, indicating that CNS delivery can be achieved. 121 However, the remaining challenge lies in the narrow therapeutic window imposed by selenocompound reactivity, making it difficult to achieve therapeutic CNS concentrations while avoiding systemic toxicity. Selenium exhibits a parabolic dose-response curve: low concentrations may be sub-therapeutic, failing to induce adequate selenoenzyme expression, while overdose generates pro-oxidant selenide species that damage proteins and lipids. 122 Current administration routes compound these difficulties: intracerebroventricular delivery, while effective in rodent studies, would require invasive catheter placement in humans, introducing infection risk and logistical barriers incompatible with acute stroke care. Conversely, nutritional selenium is preferentially targeted to the brain and testes (in men) in conditional deficiency. Given the parabolic dose-response relationship of Se supplementation, this raises concerns about the safety of achieving therapeutic CNS concentrations before peripheral toxicity occurs. 120
To address these barriers, our laboratory developed Tat-SelPep, a blood–brain-barrier-penetrant fusion of the HIV-Tat transduction domain with selenoprotein P, the principal carrier that circulates in the body to distribute selenium. This peptide-based delivery approach circumvents selenium toxicity through several mechanisms. Unlike free selenium supplements that release reactive selenide species (Se2−) capable of oxidizing cellular thiols and disrupting redox homeostasis, Tat-SelPep delivers selenium as selenocysteine residues within a stable protein scaffold. This mimics the natural selenium transport function of selenoprotein P, buffering reactive selenium and releasing it in a controlled fashion via receptor-mediated cellular uptake. Further, The Tat transduction domain enhances blood–brain barrier penetration, allowing lower systemic doses to achieve therapeutic brain concentrations, thereby widening the therapeutic index. 120 This controlled, protein-based delivery maintains selenium bioavailability while minimizing off-target oxidative damage.
In vitro, Tat-SelPep prevents hemin-induced ferroptosis, upregulating GPX4 and the broader selenoproteome, and has a wide therapeutic window. In vivo, administration intraperitoneally up to 6 h following ICH in mice enhanced behavioral recovery. 120 Clinically, this is meaningful as most spontaneous ICH cases present several hours after symptom onset, 123 and door-to-first medication times still lag behind ischemic stroke (median ~55 min). 124 By extending the therapeutic window, Tat-SelPep could significantly increase the proportion of patients who might benefit from antioxidant intervention, addressing a key unmet need in acute ICH care.
Beyond Tat-SelPep, other developments in targeted selenium delivery are emerging. Bovine serum albumin–stabilized selenium nanoparticles (BSA–SeNPs) offer a delivery scaffold that improves selenium stability and pharmacokinetics, enables controlled release, and can enhance brain exposure (e.g., via albumin-mediated transcytosis). In murine ICH, BSA–SeNPs protect hippocampal neurons from damage, reduce neurological deficits, and activate the Nrf2–GPX4 axis, thereby inhibiting lipid peroxidation and ferroptosis. 125 Collectively, these advances in formulation address the exposure-toxicity trade-off by improving selenium stability, controllable release, and therapeutic window, thereby improving the feasibility of selenium as a therapeutic in ICH. This is especially important as growing evidence indicates that selenium’s therapeutic value extends beyond its role in selenoprotein synthesis, potentially with immediate selenoprotein-independent antioxidant effects.
Selenium shows dual-phase protection dependent on ubiquinol
Recent research reveals selenium confers biphasic protection against ferroptosis via two temporally separate mechanisms. First, it induces a cytoprotective transcriptional program, upregulating selenoenzymes such as GPX4 that enhance antioxidant capacity. Second, it exerts rapid selenoprotein-independent activity: intracellular selenide is used by mitochondrial SQOR (sulfide:quinone oxidoreductase) to reduce ubiquinone to ubiquinol, a potent lipid radical–trapping antioxidant, that halts peroxidation chain reactions. This acute protection persists in GPX4-knockout cells and in PSTK-deficient cells, which cannot synthesize selenoproteins. By driving ubiquinol production, selenium naturally intersects with the FSP1–CoQ10 axis, another radical-trapping antioxidant pathway important in membrane integrity. This dual action supports selenium as a therapeutic in ICH, providing rapid membrane protection immediately after hemorrhage and sustained reinforcement of antioxidant defenses during ongoing erythrocyte lysis.
The FSP1–CoQ10 antioxidant system relies on adequate ubiquinol supply. CoQ10 deficiency has been reported in some neurodegenerative diseases such as Parkinson’s, 126 suggesting CoQ10 supplementation may confer neuroprotection in these patients. 127 However, CoQ10 delivery is challenging given CoQ10’s lipophilicity, poor intravenous suitability, and rapid hepatic and renal clearance. Tet1-targeted CoQ10 liposomes improve neuronal uptake: in hemoglobin-stimulated neurons, they enhanced FSP1 activity, suppressed ferroptosis, lowered cellular iron and oxidative stress, and rescued mitochondrial morphology. In vivo, in a subarachnoid hemorrhage model, CoQ10–Tet1–Lipos reduced MDA accumulation, neuronal damage, and neurological deficits. 128
Antioxidant reinforcement is an attractive therapeutic strategy in ICH as it amplifies endogenous yet insufficient stress responses, shifting redox balance away from ferroptotic collapse. By boosting cellular antioxidant capacity, this approach can restore equilibrium between pro-oxidant and antioxidant forces, pulling the system back from the ferroptotic “tipping point.” Conceptually, antioxidant reinforcement pairs naturally with interventions targeting upstream ferroptotic triggers, such as iron overload: chelation reduces the catalytic iron burden that initiates lipid peroxidation, while antioxidant enhancement fortifies the defenses that suppress its propagation. Hence, selenium- and CoQ10-based strategies intercept ferroptosis at multiple inflection points. Together, these principles motivate combinatorial therapies that target both the initiators and executors of ferroptosis.
Future directions: Cell-type specificity, regulated cell death crosstalk, and combinatorial therapies
As established in preceding sections, ferroptosis is a critical contributor to secondary brain injury after ICH, with multiple therapeutic targets including reducing iron accumulation, boosting antioxidant defences, and transcriptional modulation, demonstrating reproducible neuroprotection across preclinical ICH models. However, translating these insights into effective clinical interventions has been limited, as evidenced by the failure of deferoxamine trials, despite robust preclinical signals, and the absence of any FDA-approved pharmacological therapy for ICH beyond supportive care. The translational gap might reflect the complexity of ICH pathophysiology inadequately addressed by single-target approaches: the perihematomal environment involves cells beyond neurons, which current research focuses on, with distinct ferroptotic vulnerabilities and contributions to injury. Further, ferroptosis operates alongside and interacts with other regulated cell death pathways, with shared activation and common signaling pathways.
This section explores key areas and gaps in current understanding shaping future ferroptosis research and therapeutic development for ICH, considering cellular heterogeneity, crosstalk among death pathways, and the integration of these insights into rational combinatorial approaches.
Cell type specificity and therapeutic targets in ferroptosis following ICH
While ferroptosis has been predominantly studied in neurons after ICH, accumulating evidence reveals distinct cell-type specificity, with different ferroptotic vulnerabilities and contributions to secondary injury. Notably, comparing ferroptosis sensitivity across primary cortical astrocytes, microglia, and neurons in both monoculture and tri-culture systems, Jiao et al. found that neurons were the least sensitive, and neuronal resistance increased further in tri-culture. Hence, intercellular interactions may be protective following ICH, highlighting the importance of coculture for physiologically relevant modelling, and considering other cell types for therapeutic intervention. 129
Microglia were most susceptible in both primary and co-culture settings, replicating findings from human iPSC-derived tri-cultures, while neurons were least sensitive. 129 As the brain’s resident phagocytes, microglia are recruited to the perihematomal region following ICH, where they phagocytose erythrocytes and clear hematoma debris, becoming heavily iron-laden in the process. Consistent with this role, microglia exhibit robust ferroptosis-associated transcriptional responses and heightened sensitivity to iron overload, potentially amplifying neuronal degeneration under ferroptotic stress.129,130 However, microglial ferroptosis depends on phenotypic plasticity: M1-polarized pro-inflammatory microglia appear more vulnerable, while M2 polarization confers enhanced ferroptosis resistance and neuroprotection. Huang et al. demonstrated that the canonical ferroptosis inhibitor ferrostatin-1 biased microglia toward an M2-like phenotype, enhancing phagocytic function, reducing inflammation, and promoting hematoma absorption in vivo, ultimately improving neurological function. 131 Beyond inhibiting microglial ferroptosis, it may be beneficial to bias microglial phenotype toward M2 to enhance clearance capacity and resilience.
Astrocytes, the most abundant glial cell type, demonstrate bidirectional roles in ferroptosis after ICH. On one hand, astrocytes are vulnerable to hemoglobin breakdown products: hemin triggers rapid ROS generation and activates ERK1/2 signaling. 132 This in turn depletes intracellular glutathione and engages regulated death programs including necroptosis. 133 Conversely, astrocytes mount robust Nrf2-mediated antioxidant transcriptional programs following ICH, are important in iron handling through ferritin storage, heme metabolism (including HO-1 expression), ferroportin-mediated iron export. Additionally, they support neuronal glutathione availability via precursor provision. 134 Recent studies suggest additional beneficial roles for astrocyte-derived molecules. Fan et al. showed in an Alzheimer’s disease model that astrocytic lactoferrin overexpression reduced neuronal ferroptosis. This occurred through two mechanisms: limiting neuronal iron accumulation via chelation and preserving neuronal GPX4 by preventing chaperone-mediated autophagy-dependent degradation. 135 While demonstrated in Alzheimer’s models rather than ICH, this positions astrocyte-targeted strategies as promising therapeutic targets for further investigation.
Oligodendrocytes may represent the earliest and most vulnerable target following ICH. Using single-cell and spatial transcriptomics in a rat autologous whole blood ICH model, Gu et al. found that ferroptosis was the most enriched programmed cell death pathway and predominantly affected mature oligodendrocytes, occurring hyperacutely and peaking at 24 h post-injury. 136 This selective vulnerability likely stems from oligodendrocytes’ enrichment in polyunsaturated fatty acid-containing lipids essential for myelin synthesis, coupled with limited antioxidant capacity compared to other glial cells. 137 Oligodendrocyte ferroptosis likely drives substantial white matter injury after ICH, suggesting that oligodendrocyte-targeted anti-ferroptotic therapies may specifically preserve white matter integrity, improving long-term functional outcomes.
Crosstalk between regulated cell death pathways
Ferroptosis does not operate in isolation following ICH. Multiple regulated cell death pathways, including apoptosis, necroptosis, and autophagy, are activated simultaneously, share overlapping triggers and signaling pathways. Understanding these interactions is critical for developing effective therapeutic strategies that fully address secondary injury.
The inflammatory milieu surrounding the hematoma, for instance, drives multiple death pathways through common mechanisms. Heme- and DAMP-mediated activation of Toll-like receptors on immune and glial cell amplifies NF-κB signaling, which simultaneously promotes cytokine release, enhances iron retention through hepcidin upregulation, and generates ROS, ultimately creating conditions that bias neurons toward both ferroptotic and necroptotic death. Oxidative stress-induced ROS and lipid peroxidation following ICH can induce apoptosis, while sustained superoxide generation may switch cell fate from apoptotic to necroptotic programs.138,139 Consistent with this, our ultrastructural analysis following experimental ICH confirms coexistence of ferroptotic (shrunken mitochondria), autophagic (double-membrane vesicles), apoptotic (chromatin condensation), and necrotic features in perihematomal neurons, demonstrating these pathways likely operate in parallel. 18
The relationship between autophagy and ferroptosis adds particular complexity to ICH pathophysiology. Autophagy, a lysosomal degradative pathway that sequesters and degrades cytoplasmic cargo, including damaged organelles and misfolded proteins, is engaged by hemorrhagic injury. He et al. demonstrated that intracerebral infusion of autologous blood or ferrous iron into the rat striatum activated autophagy, evidenced by LC3-II accumulation, cathepsin D upregulation, and increased autophagosome formation. Notably, iron chelation with deferoxamine attenuated these changes, linking iron accumulation, an early and sustained feature of ICH, to autophagy activation in perihematomal tissue. 140
Mechanistically, autophagy can potentiate ferroptosis through ferritinophagy, the NCOA4-mediated selective autophagic degradation of ferritin, enabling expansion of the cytosolic labile iron pool. In vitro, cystine deprivation or pharmacological system Xc− inhibition with erastin (conditions mimicking GSH depletion after ICH) induces ferritinophagy, resulting in ferritin loss, increased labile iron, and accelerated lipid peroxidation culminating in ferroptotic death. 140 Consistent with a causal role for autophagy in ferroptotic death, inhibition of core autophagy machinery (e.g. Atg5/Atg7 knockdown or knockout) suppresses erastin-induced ferroptosis, preserving ferritin, and limiting iron-dependent lipid peroxidation. 141
In ICH, where progressive iron loading due to erythrocyte lysis coincides with impaired cystine import and glutathione depletion, autophagy-driven ferritin degradation likely becomes maladaptive, liberating redox-active iron precisely when antioxidant defenses are overwhelmed. Supporting this, Shen et al. demonstrated that autophagy inhibition through pretreatment with the autophagy inhibitor 3-methyladenine reduced brain edema and cell death following collagenase-induced ICH in mice, whereas rapamycin, an autophagy activator, produced the opposite pattern, implicating autophagy activation as maladaptive in this setting. Mechanistically, 3-methyladenine’s protective effects were associated with suppression of NF-κB signalling, cytokine production, and apoptosis markers, consistent with a coupled autophagy–inflammation–cell death axis. 141 While this study did not directly quantify ferroptosis, the reduction in iron-driven oxidative injury and the known role NF-κB in regulating both autophagy and ferroptotic pathways suggests that autophagy modulation might amplify or dampen multiple pathways of cell death simultaneously, depending on cargo selectivity and injury stage. Direct mechanistic attribution based off this study still requires some caution, given the broad effects of 3-methyladenine, potentially modulating PI3K-dependent signaling independently of autophagy, and similarly rapamycin directly targets mTOR, involved in regulating more broadly fundamental cell processes from autophagy to protein synthesis.142,143
Further, autophagy’s role may be context-dependent. While ferritinophagy may exacerbate ferroptotic susceptibility through iron mobilisation, other cargo-selective autophagic programs, like mitophagy, can be protective by removing ROS-generating mitochondria and limiting cellular stress. 144 This duality suggests that rather than indiscriminate autophagy suppression, more selective therapeutic approaches should target NCOA4-dependent ferritinophagy specifically. This would reduce iron liberation while preserving protective autophagy functions such as mitophagy. However, this strategy remains to be tested in hemorrhagic injury.
Future directions in ICH therapeutics: Combinatorial and precision therapies
The extensive crosstalk among regulated cell death pathways reveals shared biochemical vulnerabilities that can be exploited therapeutically. Interventions targeting shared nodes, such as oxidative stress, iron overload, or inflammatory signaling, may provide broader neuroprotection than pathway-specific inhibitors by simultaneously attenuating ferroptosis, apoptosis, and necrosis.
Antioxidant reinforcement exemplifies this principle. Oxidative stress and mitochondrial dysfunction represent the intersection of multiple death pathways. 145 By reinforcing redox homeostasis, antioxidants act at this convergence point to blunt ferroptotic lipid peroxidation, apoptotic cytochrome c release, and necroptotic ROS amplification. Within this framework, selenium supplementation has a dual effect, providing rapid lipid radical scavenging through SQOR-mediated ubiquinol production while inducing longer-term selenoenzyme expression (GPX4, TXNRD1) that reinforces antioxidant capacity to deal with prolonged insult. Pairing selenium with targeted CoQ10 delivery enhances this protection as CoQ10 increases the pool of reducible ubiquinone, supporting FSP1-mediated radical scavenging at membranes and mitochondrial electron transport chain function to address both lipid peroxidation and mitochondrial ROS generation.
In parallel, iron-targeted interventions address an established upstream trigger of several death pathways. Selective iron chelation to reduce labile iron accumulation as erythrocyte lysis progresses both blocks both enzymatic ROS generation (upstream of ferroptosis, apoptosis, and necroptosis) and non-enzymatic Fenton chemistry driving ferroptotic lipid peroxidation. Together, these complementary strategies could address both the initiation (iron-catalyzed lipid peroxidation) and execution (failure of antioxidant defenses) phases of ferroptosis. By addressing common drivers (oxidative stress, iron, inflammation) in concert, combinatorial therapeutics blunt several death programs simultaneously, yielding more robust and durable neuroprotection within the complex post-hemorrhagic microenvironment.
Similarly, the cellular heterogeneity of vulnerability to ferroptosis highlights that optimal therapeutic strategies may require cell-type specific targeting. Targeting of specific cell types could be beneficial based on the time-point of intervention. Oligodendrocytes appear to undergo ferroptosis earliest and most extensively, with peak death at 24 h and implications for white matter injury. Early intervention, if clinically possible (i.e. in patients with immediate or near-immediate presentation), may preserve these myelin-producing cells. Accordingly, rapid therapeutics such as radical-trapping antioxidants or selenium-based therapies might offer direct oligodendrocyte protection. Similarly, therapies targeting microglia should consider their dual role as both ferroptosis-susceptible cells and inflammatory modulators. The observation that ferrostatin-1 shifts microglia toward M2 polarization while providing cytoprotection suggests that anti-ferroptotic agents could modulate the inflammatory response to protect cells beyond directly preventing cell death. A potential area of investigation is the combination of ferroptosis inhibitors with M2-promoting agents, potentially synergistically enhancing hematoma clearance while limiting inflammation. In a similar vein, the role of astrocytes providing metabolic support, for example, through the provision of glutathione precursors suggests that enhancing astrocytic Nrf2 activity, for example, through relieving NF-κB suppression, or pharmacological activators, might indirectly protect neurons. The finding that astrocytic lactoferrin overexpression reduces neuronal ferroptosis supports this possibility, although whether strengthening astrocyte-neuron networks provides benefit in ICH models remains to be tested. 135 This is potentially promising, however, given the enhanced neuronal resistance observed in tri-culture systems.
Conclusion
Converging lines of evidence indicate that ferroptosis is a valuable in vitro model of cell death for identifying therapeutic targets in rodent models of intracerebral hemorrhage. Ferroptosis is a regulated, iron-dependent, form of necrosis (necroptosis) that may have evolved as a mechanism enabling cells to generate pro-ferroptotic lipids (e.g. 5 hydroperoxyeicosatetraenoic acids) and downstream immunomodulatory lipids, including leukotriene B4, which recruit immune cells to clear cellular debris. Future studies will focus on emerging signaling and transcriptional targets that could neutralize diverse pathological stimuli and modes of cell death. Novel selenium-based peptides represent a promising therapeutic strategy, as they can activate a suite of protective genes with local, cellular, and systemic effects aimed at preserving neuronal function following brain hemorrhage.
Footnotes
Acknowledgements
We acknowledge all the members of the Ratan Laboratory over the past 31 years that have contributed to understanding of ferroptosis after ICH, and our collaborators from the Sansing and Aronowski Labs. Graphic artwork was done by Laura Gilmartin.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from The Dr. Miriam and Sheldon G Adelson Medical Research Foundation, The Burke Foundation, The Cure Alzheimer’s Fund, and the NIH under grant number 1R01NS140765.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Rajiv R Ratan serves on the Scientific Boards for Alevian and Neuronasal and holds equity interest in Neuronasal. Neuronasal is developing intranasal N-acetylcysteine, an antiferroptotic agent, for treatment of Parkinson’s disease and traumatic brain injury.