
Abstract
The hypothalamic–pituitary–adrenal (HPA) axis has long served as a classical model of brain–body communication, illustrating how neuroendocrine signaling maintains physiological homeostasis. While endocrine hormones have traditionally been thought to act on peripheral organs, emerging evidence reveals that several such hormones, including insulin, corticoids, ghrelin, and thyroid hormones, also influence central nervous system (CNS) function. These actions are supported by the presence of corresponding hormone receptors within specific brain regions. However, the mechanisms by which these peripheral signals access the brain remain insufficiently explored, particularly regarding the role of brain barriers, including the blood–brain barrier (BBB) and blood–cerebrospinal fluid barrier (BCSFB). These barriers not only regulate molecular transport into the brain but may also serve as active participants in neuroendocrine communication. This review synthesizes current findings on representative peripheral hormones originating from the pancreas, small intestine, and thyroid gland. We focus on their interactions with brain barriers, identify known transport mechanisms, and discuss how these processes are altered in metabolic and neurological disorders. Understanding the crosstalk between peripheral hormones and brain barriers may provide novel insights into the regulation of CNS function and the pathophysiology of hormone-related brain disorders.
Introduction
The brain continuously monitors peripheral organ function and orchestrates protective responses that serve to maintain systemic homeostasis. 1 Brain barriers, characterized by selective permeability through specialized transporters, restrict nonspecific molecular entry while facilitating tightly regulated bidirectional exchange with peripheral organs. 2 Key barriers include the blood–brain barrier (BBB), separating brain parenchyma from the circulation; the blood–cerebrospinal fluid barrier (BCSFB), formed by the choroid plexus (ChP) epithelium3,4; and the blood–labyrinth barrier (BLB), which regulates access to the inner ear. 5 In addition, specialized circumventricular organs (CVOs) lack a BBB, allowing direct sensing of circulating hormones and bidirectional exchange of mediators with the blood.6,7 Disruption of brain barriers can result in loss of synaptic connectivity and dysregulation of systemic homeostasis. 8 Thus, a comprehensive understanding of brain barrier function is fundamental to elucidating the complex interactions between the brain and peripheral organs.
The hypothalamic–pituitary–adrenal (HPA) axis is a well-characterized neuroendocrine system that links the brain to peripheral endocrine organs. 9 Central production of hormones, including those from the hypothalamus and pituitary gland, influences peripheral target tissues by triggering hormone cascades that regulate processes such as energy balance.10,11 While efferent signaling from the brain to peripheral organs via hormones has been extensively characterized, afferent pathways—through which peripheral hormones signal back to the brain—remain less well understood. Small lipophilic hormones, such as steroid hormones, can readily cross the BBB via transmembrane diffusion without transporters or endocytosis. In contrast, hydrophilic hormones require a specialized transport process, such as receptor-mediated endocytosis, transporters or passage through CVOs, where the BBB is more permissive. Nevertheless, the extent to which peripheral hormones directly affect brain vasculature, pass through brain barriers, or regulate brain functions remains poorly defined. An overview of the interactions between peripheral endocrine organs and major brain barrier interfaces is illustrated in Figure 1.
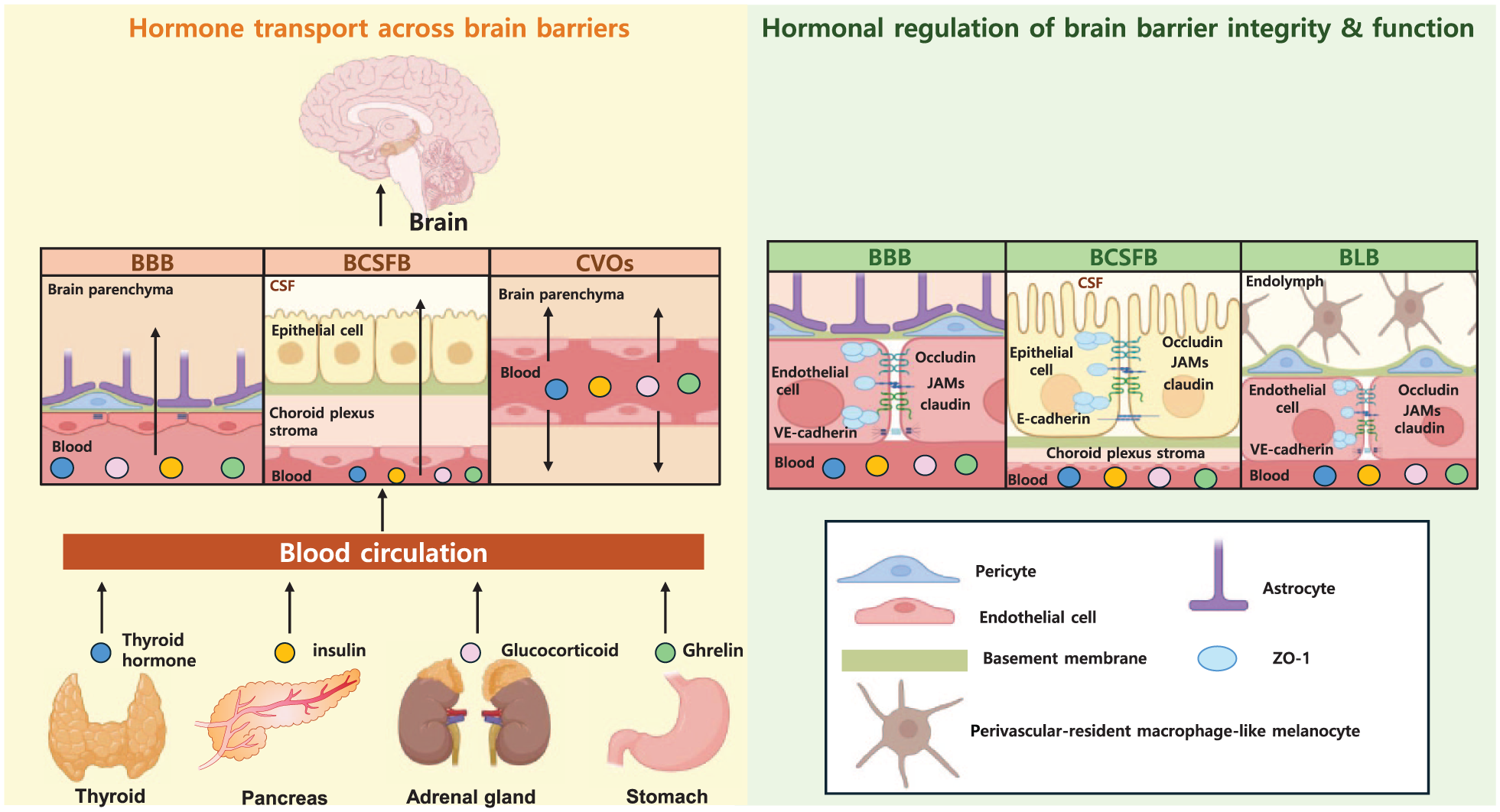
Transport of peripheral hormones across and regulation of brain barriers. Peripheral endocrine organs—including the thyroid gland, pancreas, adrenal gland, and stomach—release circulating hormones (thyroid hormones, insulin, glucocorticoids, and ghrelin). The left panel illustrates routes by which these hormones access the CNS through major brain barrier interfaces: the BBB, BCSFB, and CVOs. Arrows indicate the direction of hormone movement between the circulation and the brain. The right panel highlights hormone-mediated regulation of barrier integrity and function across multiple barrier systems, including the BBB, BCSFB, and BLB.
The primary route through which peripheral molecules access the central nervous system (CNS) is the BBB, composed of three main components: astrocytic endfeet, endothelial cells and pericytes, collectively forming the neurovascular unit. 3 Although hormones typically act by binding to specific receptors, the precise mechanisms by which they traverse the brain barriers—together with the transport routes utilized by individual hormones—are still not fully elucidated. Our recent findings highlight the importance of endothelial energy metabolism in maintaining barrier integrity, with mitochondrial dysfunction causing profound disruption and neurological injury. 12 However, the contribution of peripheral hormones to the structural and functional maintenance of brain barriers, including not only the BBB but also the BCSFB, BLB, and CVOs, remains largely unexplored.
In this review, we examine how four key peripheral hormones—thyroid hormones, insulin glucocorticoids and ghrelin—interact with brain barriers. These hormones provide complementary models for understanding hormone–barrier interactions because they represent distinct endocrine axes and recently it reported that these hormones affect brain barrier integrity. 13 THs and GCs are small lipophilic molecules; however, their CNS access is not solely dependent on passive diffusion but also involves regulated transport, metabolism, and efflux mechanisms.14–16 In contrast, insulin and ghrelin are peptide hormones that require receptor-mediated or carrier-mediated transport processes to traverse brain barriers.17,18 Together, they encompass a broad spectrum of transport modalities, including specific solute carriers (e.g. MCT8 for THs), receptor-mediated transcytosis (insulin), diffusion coupled with efflux regulation (GCs), and peptide transport (ghrelin).19–21 Importantly, these hormones function not only as circulating signals that must cross brain barriers but also as active modulators of barrier structure and function. GCs enhance tight junction integrity, whereas insulin and THs influence endothelial metabolic homeostasis and mitochondrial function.22,23 Originating from the thyroid gland, pancreas, adrenal cortex, and gastrointestinal tract, respectively, these mediators regulate key systemic processes—including stress responses, glucose metabolism, energy balance, and feeding behavior—that are closely linked to vascular and barrier physiology.24–27
Specifically, we focus on how they traverse the BBB, BCSFB, and BLB; how they influence barrier physiology; and how their dysregulation may contribute to CNS disorders. Whereas studies on the BLB and CVOs have provided limited evidence for direct hormone transport, there is research on potential links between these barriers and hormone-associated disorders, which we will explore. This review provides new insights into mechanisms connecting peripheral hormones with brain barrier function. An overview of these hormone–brain barrier interactions is summarized in Figure 1.
Overview of brain barrier systems
Brain barrier systems consist of specialized cellular interfaces that regulate molecular exchange between the circulation and the CNS. 3 Although differing in anatomical location, major CNS barriers—including the BBB, BCSFB, and related vascular interfaces—share common structural and functional characteristics. 4
Structurally, the BBB is formed by brain microvascular endothelial cells sealed by tight junction complexes composed of claudins, occludin, and zonula occludens (ZO) proteins. 28 These endothelial cells are supported by pericytes and astrocytic endfeet, together forming the neurovascular unit. 29 The BCSFB, in contrast, is established by tight junction–connected epithelial cells of the choroid plexus, while capillaries at this site are fenestrated. 4 Other interfaces, such as the blood–labyrinth and blood–spinal cord barriers, exhibit similar tight junction-based organization. 30 Functionally, brain barriers restrict paracellular diffusion and exhibit low rates of transcytosis, while expressing selective influx and efflux transport systems, including solute carriers and ATP-binding cassette transporters. 21 Through these mechanisms, they tightly regulate the entry of nutrients, metabolites, immune mediators, and circulating hormones into the CNS. 14
Together, these structural and functional features establish brain barriers as specialized interfaces that tightly regulate molecular exchange between the circulation and the CNS. These properties are central to understanding how peripheral hormones traverse CNS barriers and influence barrier integrity under physiological and pathological conditions.
Thyroid hormones
Recent studies have revealed that thyroid hormones (THs) in the brain maintain neuronal integrity and enhance synaptic activity in adulthood, but are also essential for brain development, including neurogenesis, cell proliferation, and maturation. 31 Beyond their classical endocrine roles, THs also modulate cerebrovascular physiology, linking endothelial metabolism, transporter expression and barrier permeability, thereby sustaining neural homeostasis. 24 During both development and adulthood, the precise orchestration of TH transport across brain barriers—BBB, BCSFB, and BLB—is critical for maintaining regional TH availability and CNS function. 32 However, the molecular routes and regulatory mechanisms governing TH passage through these barriers remain incompletely understood. Recent discoveries combining structural biology and genetic models have begun to uncover how specialized transporters coordinate TH entry into the brain and how their dysfunction contributes to neurodevelopmental and neurodegenerative disorders. 33 This section first discusses physiological mechanisms governing TH entry into the brain and how these mechanisms are altered in disease states, followed by the effects of TH imbalance on brain barrier integrity.
Transport of TH across brain barriers in health and disease
Physiology of TH transport across brain barriers
Figure 2 illustrates that the BBB and BCSFB possess distinct structural and functional properties that fundamentally shape the pathways through which thyroid hormones access the brain. THs depend on specific peptide transporters to cross the BBB. 14 Key transporters identified to date include monocarboxylate transporter 8 (MCT8), organic anion-transporting polypeptide 1C1 (OATP1C1), and L-type amino acid transporters (LATs). Among these, MCT8 is the primary transporter, with THs serving as exclusive substrates. 16 At the BBB, MCT8 expressed in endothelial cells predominantly mediates the uptake of thyroxine (T4) from the circulation. Once inside the CNS, T4 is transported into astrocytes via OATP1C1 and converted to triiodothyronine (T3) by iodothyronine deiodinase 2 (DIO2). T3 is then taken up by neurons through MCT8, where it binds to nuclear receptors and regulates diverse metabolic processes. 15 OATP1C1, selectively expressed in the CNS, is localized mainly to capillary endothelial cells and the choroid plexus (ChP), and preferentially transports T4 and reverse T3. 34 LAT1 and LAT2 also contribute to TH transport across the BBB and BCSFB. 35 LAT1 is expressed in endothelial cells of the BBB and epithelial cells of the ChP, whereas LAT2 is primarily found in neurons and the ChP. 36
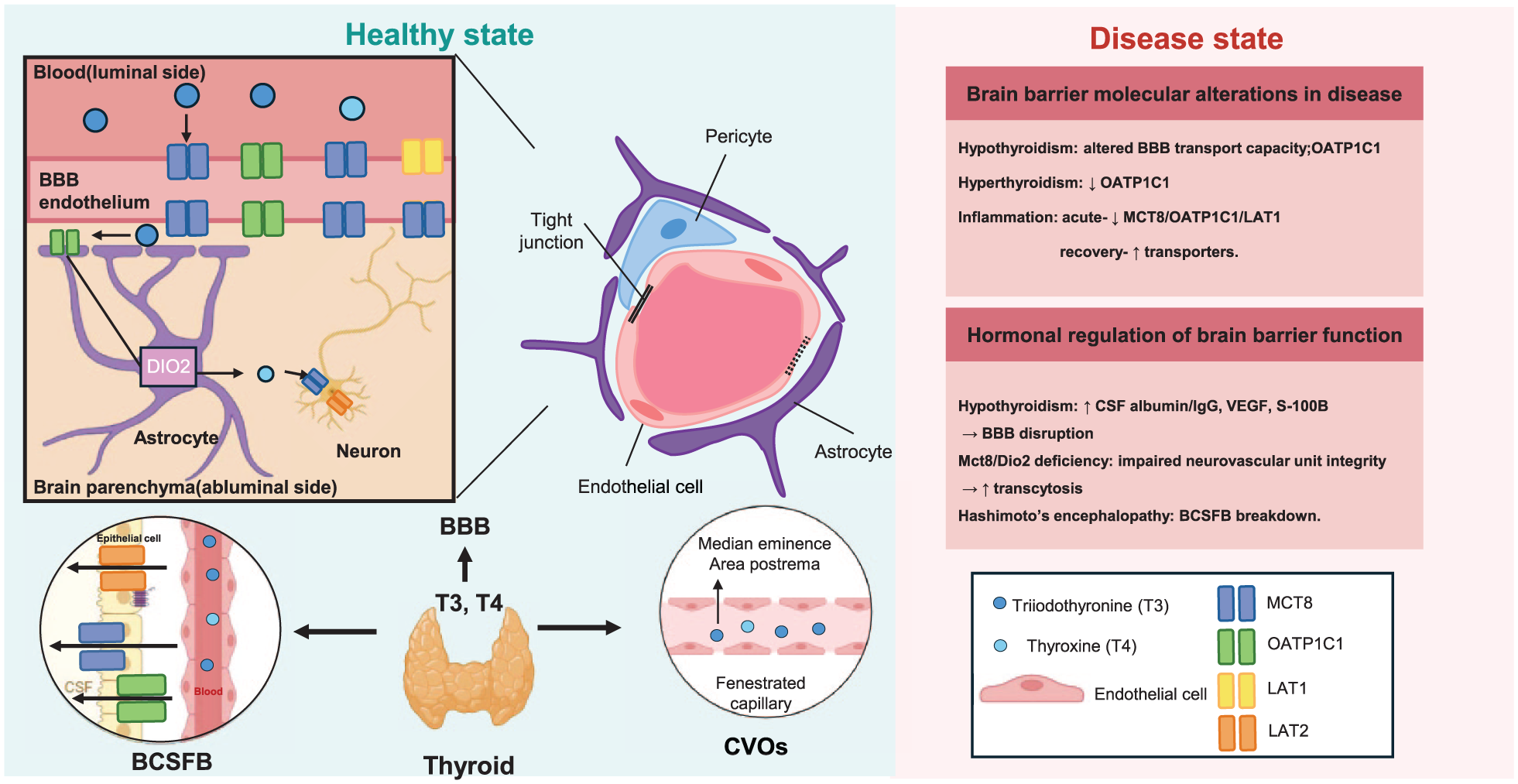
Thyroid hormone transport and regulation of brain barriers. Thyroid hormones (T3 and T4) enter the brain through specialized brain barrier interfaces. At the BBB, transport is mediated by specific membrane transporters expressed in brain endothelial cells, including MCT8, OATP1C1, and LAT1/2. The BCSFB and CVOs provide additional routes for hormone exchange between the circulation, cerebrospinal fluid, and brain. Arrows indicate the direction of hormone movement. The right panel summarizes representative alterations in TH transport and barrier function under disease conditions.
Structural and functional analyses have shown that MCT8 and OATP1C1 are the principal, high-affinity TH transporters at the neurovascular interface, and mutations in these genes cause neurological disorders such as Allan–Herndon–Dudley syndrome. 32 Recent evidence has provided compelling in vivo confirmation of the essential role of endothelial MCT8 and OATP1C1 in mediating TH entry into the brain. In a 2025 study by Alevyzaki et al., 19 mice with conditional knockdown of Mct8 and Oatp1c1 specifically in endothelial cells display region-specific impairments in TH signaling. These animals exhibit reduced brain T3 and T4 levels, diminished expression of TH-responsive genes, and hypomyelination of cortical regions, particularly affecting GABAergic interneuron maturation. However, hippocampal T3 content and TH-regulated transcripts remain relatively preserved compared with that in global double-knockout (DKO) mice. This partial preservation suggests that, in addition to the BBB, alternative MCT8/OATP1C1-dependent routes, such as TH transport through the blood–CSF barrier, may provide a compensatory source of T3 to specific brain regions. Collectively, these findings highlight the roles played by endothelial MCT8 and OATP1C1 as primary pathways for TH entry into the CNS, while recognizing the apparent necessity of additional transport across the BCSFB to sustain complete cerebral TH homeostasis. 19
The BCSFB, located in the ChP, represents a developmentally early, functionally specialized interface that produces cerebral spinal fluid (CSF) and regulates solute entry into the brain. The ChP epithelium forms tight junctions over a fenestrated, highly vascularized stroma, enabling directional transport; during human gestation, features and transporter programs of the ChP emerge before the BBB and coincide with rising T4 exposure (approximately weeks 13–17), consistent with a role in perinatal TH availability.37,38
Besides these canonical barriers, CVOs act as specialized neuroendocrine gateways, allowing hormones to directly enter or exit the CNS. In these areas, the BBB is absent or highly permeable, permitting controlled molecular exchange between the blood and neural tissue. 7 For example, hypothalamic peptide hormones traverse the median eminence (ME), a type of CVO, to enter the hypophyseal portal circulation and act on the anterior pituitary. 16 Through this mechanism, thyrotropin-releasing hormone (TRH) stimulates the secretion of thyroid-stimulating hormone (TSH), completing the hypothalamic–pituitary–thyroid feedback loop. 31
Alterations in TH transport at brain barriers in disease states
Alterations in TH transport across brain barriers under inflammatory conditions and thyroid dysfunction (including hypothyroidism) are summarized in Table 1, together with the relevant experimental findings and references.
Alterations in hormone transport across brain barriers in disease states.
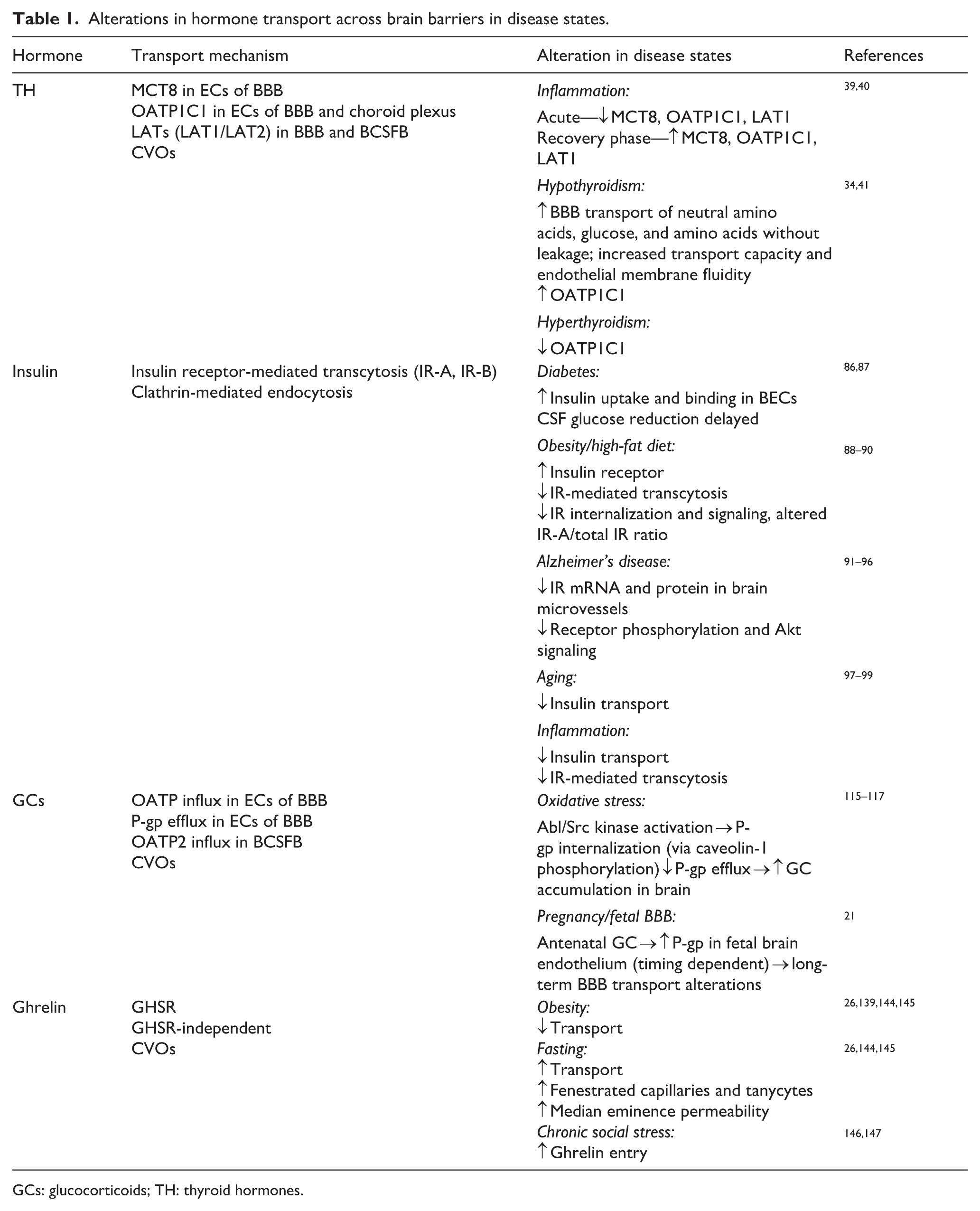
GCs: glucocorticoids; TH: thyroid hormones.
Inflammation
Systemic inflammation can modulate the expression of TH transporters in the BBB. Administration of bacterial lipopolysaccharide (LPS) intraperitoneally to rats or mice induces downregulation of Oatp1c1 and Mct8 mRNA expression in brain endothelial cells (BECs), detected by in situ hybridization 2–9 h after LPS injection. 39 Interestingly, during the recovery phase (24–48 h post-injection), mRNA expression levels were markedly elevated above the level of the control group. Notably, these changes in TH transporter expression were confined to BECs, with no significant alterations in other types of brain cells. Although the mechanisms underlying the rapid downregulation and subsequent upregulation of TH transporters remain incompletely understood, the initial downregulation may result from cytokine or other inflammatory responses triggered by endotoxin exposure. The subsequent upregulation during the recovery period appears to be a compensatory response to reduced TH levels within the brain. 39
Another critical TH transporter, LAT1, was also reported to exhibit marked temporal regulation in response to systemic inflammation. 40 This latter study showed that Lat1 mRNA levels in BECs began to decline as early as 2 h following LPS injection and became undetectable between 4 and 9 h post-injection. During the recovery phase (48 h after LPS administration), Lat1 mRNA levels surpassed those in the control group.
Collectively, these observations show that systemic inflammation resulting from LPS-induced endotoxemia alters the expression of several TH transporters at the BBB. These dynamic changes are likely orchestrated by shared molecular mechanisms activated in response to systemic inflammation (Figure 2).
Hypothyroidism
The expression of TH transporters in the BBB is responsive to TH levels in the blood. OATP1C1 is upregulated at mRNA and protein levels in hypothyroidism and downregulated in a rat model of hyperthyroidism.34,41 This suggests the presence of a compensatory mechanism in the BBB that regulates the expression of TH transporters in response to changes in systemic TH levels and serves to maintain appropriate hormone concentrations within brain tissue. Such adaptive regulation may play a critical role in preserving CNS homeostasis despite systemic thyroid dysfunction. However, it remains unclear whether similar regulatory mechanisms occur in other barriers, such as the BCSFB and the BLB.
Notably, human and organoid-based barrier models reveal species differences (e.g. lower OATP1C1 levels in human BBB microvessels vs rodents), cautioning against direct translation of rodent compensatory patterns to human physiology.32,37 Moreover, endothelial transporter insufficiency per se (e.g. endothelial Mct8/Oatp1c1 loss) can induce a region-specific cerebral TH deficiency, despite normal circulation, underscoring barrier transport failure as a primary disease mechanism rather than a mere consequence of altered serum TH. 19
Hyperthyroidism
In hyperthyroidism, chronic TH excess may saturate or downregulate MCT8 and OATP1C1 at the BBB and choroid plexus, altering the balance between T4 influx, local deiodination, and T3 availability in distinct brain regions. 34 Hyperthyroidism model further suggest changes in endothelial metabolism and neurovascular coupling, raising the possibility that TH overload modulates barrier permeability and regional TH distribution. 42
Effects of TH on brain barrier integrity and function
While Sections 2.1 and 2.2 address mechanisms regulating TH entry into the brain, TH imbalance can also directly influence brain barrier structure and function. Hormone-mediated regulation of brain barrier integrity and function, including the effects of TH on tight junction integrity and barrier permeability, is summarized in Table 2.
Effects of hormones on brain barrier function.

GCs: glucocorticoids; ICH: intracerebral hemorrhage; TH: thyroid hormones.
Tight junction integrity and permeability
A TH imbalance can affect the BBB through two primary mechanisms: first, by compromising the integrity of tight junctions, and second, by modulating the expression and distribution of specific nutrient transporters within brain barriers. 43
Key BBB structural components, including collagen IV, integrin αVβ3, F-actin, claudin-5, and PECAM-1, were reported to be downregulated and disorganized in a rat model of propylthiouracil (PTU)-induced hypothyroidism, together with NHS-biotin leakage into brain tissue, indicating impaired barrier function and compromised BBB integrity.44,45 In a canine model of hypothyroidism, levels of vascular endothelial growth factor (VEGF), a well-known promoter of vascular permeability, and S-100β, a biomarker of astrocytic brain injury, were markedly elevated at 12 and 18 months following iodine-131 treatment, reflecting increased BBB permeability in the hypothyroid state. 23 Furthermore, serum levels of S-100β also showed a delayed increase at 18 months, occurring later than in the CSF. This delay indicates that elevated serum S-100β levels result from changes in BBB permeability, which permit the leakage of S-100β from the brain into the bloodstream. 46
In patients with overt hypothyroidism, CSF levels of albumin and immunoglobulin G (IgG) are elevated, suggesting that reduced TH levels may compromise BBB integrity and alter protein transport. 47 Preclinical studies have also suggested a relationship between TH deficiency and BBB permeability. Studies in Mct8/Dio2-knockout mice, which exhibit TH transporter dysfunction, show significant impairments in neurovascular unit integrity and increased transcytotic flux. 48 Collectively, human and animal studies suggest a potential association between TH disorders and BBB disruption.
Modulation of nutrient transport systems
Altered peripheral TH levels can also modulate BBB transporter systems. For example, in methimazole-induced hypothyroid rats, brain uptake of D-β-hydroxybutyrate (BHB) is significantly reduced compared to controls, whereas the uptake of other nutrients, such as hexoses and basic amino acids, remains unchanged. 49 Because BHB crosses the BBB via the monocarboxylate transporter (MCT) system, which includes the key thyroxine transporter, MCT8, these findings indicate that hypothyroidism may impair MCT function. Importantly, TH replacement was shown to restore BHB uptake, highlighting the reversible impact of a TH imbalance on BBB transport dysfunction.
In severe neonatal hypothyroidism, induced by maternal PTU treatment, BBB transport of glucose and amino acids was shown to markedly increase in the absence of evident barrier leakage, indicating transporter upregulation. 50 Furthermore, T3 treatment alleviated these changes in nutrient transport across the BBB. In a subsequent study using the same rat model, transport capacity was enhanced and endothelial membrane fluidity was increased, suggesting that hypothyroidism modifies BBB kinetics and exposes latent transport pathways. 51 The alterations observed in disease states are also illustrated in Figure 2 in comparison with normal conditions.
Effects beyond the BBB
Beyond the BBB, other brain barrier structures, including the BCSFB and the BLB, may also be involved in TH disorders. For example, an analysis of CSF in patients with Hashimoto’s encephalopathy revealed albuminocytological dissociation (ACD) in 75%–80% of cases, indicating breakdown of the BCSFB. 52 In addition to TH imbalance, Hashimoto’s encephalopathy is thought to have an autoimmune basis, with antithyroid antibodies and other autoantibodies contributing to vascular and inflammatory injury, and the high frequency of ACD supports the notion that BCSFB dysfunction is a component of its pathophysiology. 53 Although direct evidence for TH effects on the BLB is lacking, THs are critical for auditory system development and function. TH imbalances have been linked to sensorineural hearing loss, likely through disruption of auditory pathway maturation and ion channel expression in the inner ear. 54 Experimentally induced hypothyroidism impairs cochlear development, alters endocochlear potential, and leads to degeneration of hair cells and spiral ganglion neurons, suggesting that TH‑dependent signaling in the cochlea and its microvasculature is essential for maintaining BLB integrity and normal hearing. These findings suggest that, beyond their known effects, TH imbalances may also contribute to auditory dysfunction by compromising the integrity of the BLB.55,56 These findings suggest that, beyond their known effects, TH imbalances may also contribute to auditory dysfunction by compromising the integrity of the BLB. Further investigation is warranted to elucidate the potential link between TH imbalance and BLB integrity, with the aim of preventing auditory complications in patients with TH-related disorders.
Despite these insights, most studies on TH transporter regulation and brain barrier function are outdated, with limited recent mechanistic data. Considering the significant impact of BBB dysfunction on neurodegenerative diseases and mood disorders, future research should focus on elucidating how THs regulate barrier transport and integrity at the molecular level.
Insulin
Insulin, a peptide hormone predominantly secreted by pancreatic β-cells, acts to regulate systemic glucose metabolism and energy homeostasis. 57 Within the CNS, insulin exerts diverse neurobiological effects, notably influencing cerebral glucose utilization and modulating synaptic plasticity—processes essential for higher-order cognitive functions and affective regulation that reflect modulation of neuronal circuitry through the insulin-regulated glutamate transporter, GLUT4.58,59 Most central insulin activity is attributed to pancreatic β-cell-derived hormone delivered to the brain via receptor-mediated transcytosis across brain barriers or by direct activation of insulin receptors (IRs), which are widely expressed on endothelial cells, neurons, and glia throughout multiple brain regions. 60
Transport of insulin across brain barriers in health and disease
Physiology of insulin transport across brain barriers
The IR is a transmembrane glycoprotein that functions as a pivotal receptor tyrosine kinase, mediating intracellular signaling cascades in response to insulin binding. Within the CNS, IRs are expressed on multiple cell types, including neurons, pericytes, astrocytes, and endothelial cells.61,62 Among these, transcriptomic analyses indicate that endothelial cells exhibit the highest IR expression levels in the human brain. 63 Structurally, the IR is a heterotetramer composed of two extracellular α-subunits and two transmembrane β-subunits, the latter possessing intrinsic tyrosine kinase activity essential for downstream signaling. 64 The α-subunit exists in two isoforms, IR-A and IR-B, distinguished by the presence of an additional exon in the IR-B variant. 65 While both isoforms are expressed in astrocytes and endothelial cells, neurons predominantly express the IR-A isoform.66,67 Insulin transport at the BBB is saturable and specific, indicating the involvement of receptor-mediated mechanisms.20,68,69 It has long been assumed that insulin crosses the BBB primarily through receptor-mediated transcytosis involving the IR. Transcytosis typically involves three major steps: (1) receptor-mediated endocytosis at the luminal surface of brain BECs, (2) vesicle transfer across the cytoplasm, and (3) exocytosis at the abluminal surface. 70
However, the specific nature of the endocytic pathway remains controversial. Endocytosis in BECs can occur through clathrin-coated pits, caveolae, or macropinocytotic vesicles. 71 Studies suggest that the mechanism of insulin endocytosis may vary depending on vascular bed and anatomical region. For example, clathrin-mediated endocytosis predominates in the microvasculature, whereas caveolin-1-dependent pathways are more prominent in macrovascular regions, such as the aorta.72,73 This regional heterogeneity suggests that insulin trafficking mechanisms vary across brain regions and physiological conditions. A study using isolated mouse brain microvessels reported that clathrin-mediated mechanisms are primarily responsible for insulin binding at the luminal surface, whereas caveolin-mediated endocytosis may play a role in insulin transport in specific brain regions, such as the hypothalamus. 18 Furthermore, genetic and pharmacologic inhibition of caveolin-1 impairs insulin signaling in BECs,74,75 whereas clathrin-mediated uptake is disrupted by hyperinsulinemia, which reduces clathrin-coated pit formation and insulin receptor internalization. 76
Despite extensive evidence for endocytosis, it remains unclear whether the internalized vesicles actually complete transcytosis and deliver insulin into the brain parenchyma. Recent in vivo studies using IR-deficient endothelial cells (EndoIR-KO) and pharmacological IR antagonists (S961) have shown that insulin can still cross the BBB in the absence of IR signaling, challenging the concept that IR-mediated transcytosis is the exclusive delivery pathway.77,78 These observations have prompted the hypothesis that insulin also utilizes non-IR-mediated transport pathways across the BBB, though the identity and functional relevance of such mechanisms remain poorly defined. Consistent with this possibility, a surface biotinylation-based proteomic study in a human cerebral microvascular endothelial cell line (hCMEC/D3) that readily detected transferrin receptor internalization (positive control) failed to detect internalization of the IR. While this does not exclude a role for IR-mediated transport in vivo, it underscores potential limitations of IR involvement in BBB insulin uptake and highlights the need to further explore alternative transport mechanisms. 79
Transport through the BCSFB is considerably slower and less efficient than that across the BBB, underscoring the dominant route of the latter in insulin entry into the CNS. Insulin transport into the brain occurs primarily across the BBB, with only a limited contribution by the BCSFB.80,81 Blood-borne radiolabeled insulin has also been shown to bind to CVOs, such as the ME and area postrema,82–84 supporting limited signaling at these specialized sites, but providing little evidence that this represents a major route for insulin entry into the brain parenchyma. Interestingly, a recent study has identified IRs in tanycytes of the hypothalamic arcuate nucleus—a CVO—where they appear to gate feeding-state-dependent regulation of Agouti-related protein (AgRP)-expressing neurons. 85 Importantly, in vivo studies using radiolabeled insulin coupled with electrophoresis-based detection techniques have confirmed that intact insulin crosses the BBB without undergoing degradation, providing direct evidence for functional trans-barrier transport. 18
Taken together, the current evidence indicates that multiple mechanisms contribute to insulin entry into the brain. Although evidence supports a contribution of IR-mediated transcytosis, insulin has also been detected independently of IR signaling, pointing to alternative, yet unidentified, pathways. 18 Identifying these alternative transporters remains critical for elucidating the regulation of brain insulin entry in health and disease. These physiological mechanisms are summarized in Figure 3.
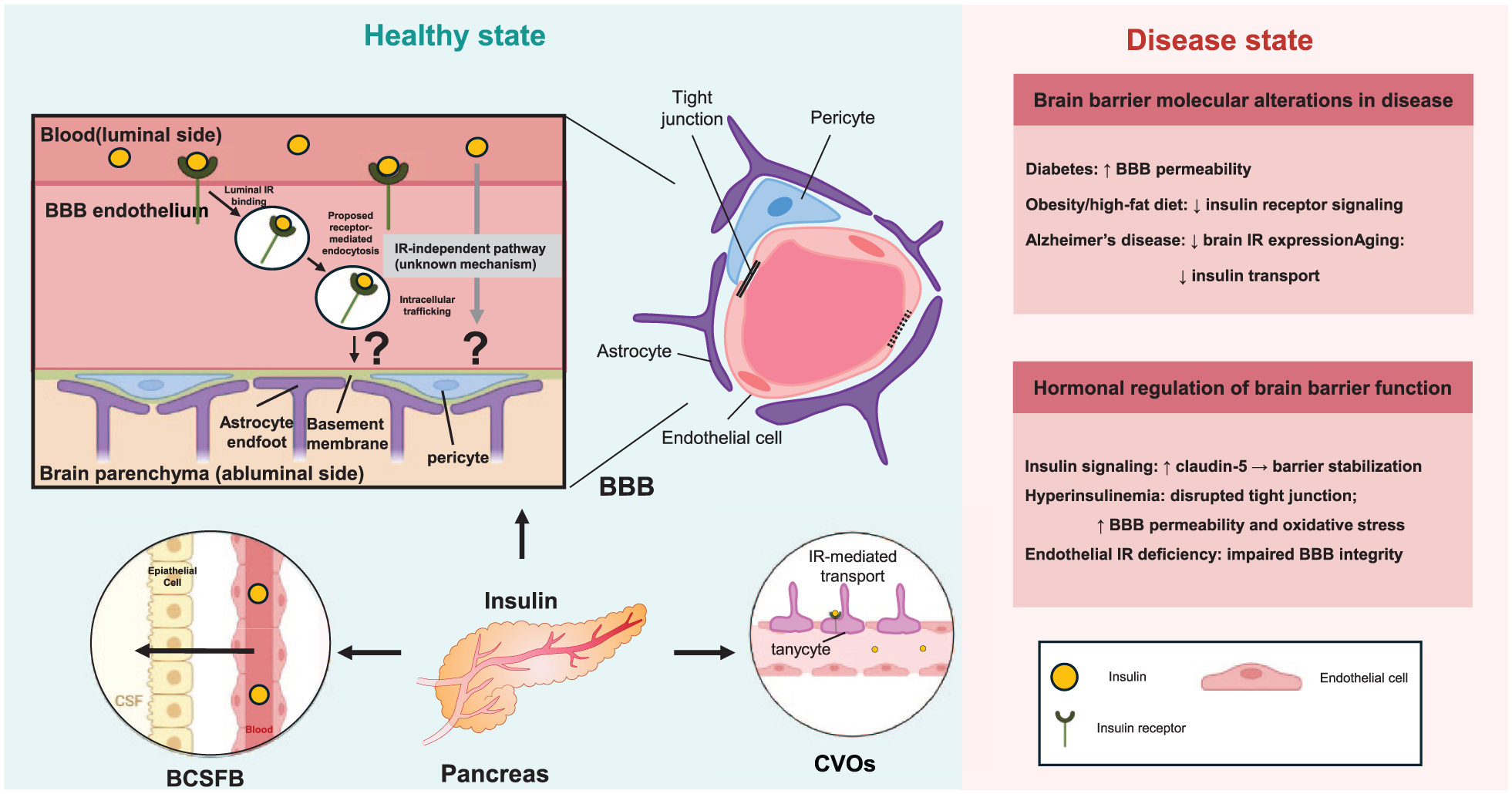
Insulin transport and regulation of brain barriers. Insulin produced by the pancreas reaches the central nervous system through interactions with brain barrier interfaces. At the BBB, insulin binds to IRs expressed on brain endothelial cells and undergoes receptor-associated uptake and intracellular trafficking. Arrows in the BBB panel illustrate proposed transport routes and highlight the uncertainty regarding insulin delivery into the brain parenchyma. In addition to the BBB, insulin can interact with other barrier interfaces, including the BCSFB and CVOs. The right panel summarizes representative alterations in insulin transport and brain barrier function in disease states such as diabetes, obesity, aging, and Alzheimer’s disease.
Alterations in insulin transport at the brain barriers in disease states
Pathological conditions differentially regulate insulin transport across the BBB, either enhancing or impairing its delivery to the brain parenchyma. Disease-associated alterations in insulin transport across brain barriers are summarized in Table 1. These alterations likely reflect disease-specific changes in transporter expression, receptor activity, and/or barrier integrity. The following examples illustrate the diverse regulatory patterns observed in metabolic, neurodegenerative, aging-related, and inflammation-related disorders.
Metabolic disorders: Diabetes and obesity
Disturbances in glucose and lipid metabolism reshape insulin transport across the BBB, underscoring the dynamic interplay between peripheral metabolic state and central insulin signaling. In streptozotocin-induced diabetes, BECs exhibit markedly increased insulin uptake and binding, consistent with a compensatory response to hypoinsulinemia.86,87 Moreover, insulin transport across the BBB is markedly reduced in canine and murine models of diet-induced obesity, accompanied by elevated plasma insulin levels. This impairment appears to be driven by chronic hyperinsulinemia and excess adiposity, which blunted receptor-mediated insulin transcytosis. Notably, such transport deficits are reversible through metabolic interventions, including fasting or triglyceride supplementation.88,89 In high-fat-diet-induced hyperinsulinemia models, hippocampal microvessels display increased total IR abundance but reduced internalization and signaling, alongside a lower IR-A/total IR ratio—features indicative of receptor desensitization and impaired insulin responsiveness. 90 Collectively, these findings reveal that systemic metabolic context—whether defined by insulin deficiency or chronic hyperinsulinemia—critically determines BBB insulin transport capacity and signaling fidelity.
Neurodegenerative disorders: Alzheimer’s disease
Analyses of human postmortem brain tissue from Alzheimer’s disease (AD) patients have revealed marked reductions in IR expression at both mRNA and protein levels, particularly within brain microvessels. 91 Functional analyses further demonstrated attenuated insulin signaling, reflected in decreased insulin-stimulated receptor phosphorylation and reduced activation of downstream protein kinase B (PKB/Akt). 92 These deficits are reported to be accompanied by altered subcellular distribution of the IR, with decreased membrane localization and increased cytosolic sequestration, suggesting impaired receptor trafficking dynamics. 93 Consistent with this, single-cell RNA sequencing of human AD brain tissue has shown reduced expression of IR-related genes in BECs. 94 Complementary studies in AD mouse models also recapitulate similar receptor dysfunction and signaling deficits, with decreased insulin delivery to the brain, together with increased Aβ accumulation and cognitive dysfunction, indicating that the impairment in insulin signaling resulting from altered IR-mediated insulin transport is associated with increased amyloid and tau pathology and accelerated cognitive decline.95,96
Aging and inflammation
Aging exerts heterogeneous effects on BBB insulin transport. For instance, whereas senescence-accelerated postnatal day 8 (P8) mice show no significant changes in transport efficiency, aged C57BL/6J mice exhibit a marked decline, 97 highlighting the influence of genetic background on age-related BBB function and insulin delivery. Similarly, systemic inflammation induced by LPS significantly reduces insulin transport across the BBB, despite concomitant increases in paracellular permeability to albumin. 98 This dissociation suggests a selective impairment of receptor-mediated insulin transcytosis under inflammatory conditions, 99 emphasizing that overall barrier permeability can decouple from the activity of specific transport pathways.
Collectively, these observations indicate that BBB insulin transport is a dynamic process, modulated by metabolic state, neurodegenerative pathology, aging, and systemic inflammation. Chronic pathological conditions, such as metabolic syndrome and Alzheimer’s disease, induce molecular and functional changes in cerebrovascular IRs that may compromise brain insulin signaling and nutrient delivery. Elucidating the mechanisms underlying these disease-specific alterations will be crucial for understanding their contribution to CNS dysfunction and for developing targeted therapeutic strategies.
Effects of insulin on brain barrier integrity and function
In addition to governing its own entry into the CNS, insulin signaling within endothelial and barrier-associated cells actively modulates barrier integrity and transport capacity. The effects of hormones on brain barrier integrity and transport function, including insulin-mediated regulation, are summarized in Table 2 together with the relevant references.
Regulation of tight junction integrity
Insulin exerts multifaceted effects on brain barrier function, extending beyond its classical metabolic role in modulating BBB structure, endothelial signaling, and nutrient transport. IR signaling is a key determinant of BBB integrity: upon insulin binding to the IR extracellular α-subunit, the β-subunit undergoes autophosphorylation, activating downstream pathways such as Akt and the mitogen-activated protein kinase (MAPK) cascade. 100 Knockdown of IRs in hCMEC/D3 cells results in reduced expression of the tight junction proteins, claudin-5, occludin, and zonula occludens-1 (ZO-1), and glucose transporter 1 (GLUT1); increased expression of inflammatory adhesion molecules, including intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1); and impaired barrier integrity. 101 Conversely, insulin stimulation of BECs protects BBB function by activating the IR/AKT2/forkhead box protein O1 (FOXO1) signaling pathway, which promotes claudin-5 expression and enhances barrier integrity. 102 However, pathological conditions can reverse these protective effects. Chronic hyperinsulinemia disrupts tight junctions, increases permeability, and promotes neurovascular complications102,103; an insulin deficiency, as observed in streptozotocin-induced diabetes, also similarly elevates BBB permeability and reduces or mislocalizes tight junction proteins. 104 This barrier disruption is linked to elevated matrix metalloproteinase-2 (MMP-2) and MMP-9 activity, as supported by partial restoration of BBB integrity following pharmacologic inhibition of these enzymes. 105 In addition, prolonged hyperinsulinemia promotes oxidative stress within the cerebrovascular endothelium, further compromising IR signaling and barrier function. This combination of receptor desensitization and reactive oxygen species (ROS) accumulation may exacerbate BBB dysfunction in metabolic disease states. 90 Furthermore, chronic hyperinsulinemia impairs endothelial nitric oxide synthase (eNOS) signaling by reducing insulin-stimulated phosphorylation at Ser1177, a key activation site, thereby diminishing eNOS activity and contributing to cerebrovascular endothelial dysfunction. 74
Modulation of nutrient transport systems
In addition to structural and metabolic regulation, insulin regulates nutrient transport at the BBB. 18 Early studies demonstrated that insulin enhances the transport of neutral amino acids, such as tyrosine and tryptophan, by modulating competitive substrate availability,106,107 and increases the blood-to-brain delivery of molecules including leptin and azidothymidine via facilitative transporters.86,108 IR knockdown selectively reduces GLUT1 expression, whereas LAT1 and transferrin receptor levels remain unchanged, indicating that IR-mediated insulin transport modulates specific transporter networks rather than broadly altering nutrient uptake. 101 Interestingly, despite these regulatory effects on transporter expression, glucose uptake into the brain parenchyma remains largely insulin-independent and is primarily mediated by GLUT1. 109 EndoIR-KO studies confirm that, although insulin signaling is delayed in brain regions such as the hypothalamus and hippocampus, overall brain glucose uptake is unaffected. 110 This suggests that insulin dynamically regulates BBB metabolic capacity and transporter expression without being essential for basal glucose delivery.
Effects beyond the BBB
Overall, studies of insulin’s actions at brain barriers have largely focused on the BBB, with comparatively little attention given to the BCSFB, BLB, or CVOs. It is well established that chronic hyperinsulinemia disrupts tight junctions, potentiates inflammatory signaling, promotes MMP-mediated protein degradation, and induces oxidative stress; these processes are likely to similarly compromise the BCSFB, which relies on junctional proteins to maintain barrier integrity.80,87,104 Moreover, diabetes has been shown to impair the BLB through hyperglycemia and insulin resistance, resulting in microenvironmental perturbations and sensory dysfunction.111,112 Taken together, these findings suggest that, beyond its classical metabolic roles, insulin not only traverses brain barriers but also exerts significant influence on their maintenance, offering a broader perspective on its role in brain barrier physiology (Figure 3).
Glucocorticoids
Glucocorticoids (GCs), produced by the adrenal cortex, regulate stress response and energy metabolism under the control of the HPA axis. 113 In mammals, the principal endogenous GCs include cortisol (predominant in humans) and corticosterone (the major GC in rodents). For GCs to play their negative feedback role in this axis, they must be able to access brain areas (e.g. the hypothalamus); this, in turn, requires that GCs traverse blood–CNS barriers, which involves complex processes. 114
Transport of glucocorticoids across brain barriers in health and disease
Physiology of GC transport across brain barriers
As highly lipophilic steroid hormones, GCs have traditionally been presumed to enter the CNS via passive diffusion. 110 However, accumulating evidence indicates that GCs not only diffuse into the CNS, but also interact with several transport systems at the BBB and ChP—including influx transporters, such as organic anion transporting polypeptide (OATP), and the efflux transporter, P-glycoprotein (P-gp)—in both mice and humans.109,111–114 By comparing hormone distribution in wild-type and Abcb1a-deficient mice (which lack the gene encoding the efflux transporter P-glycoprotein at the BBB and are commonly used to investigate transporter-mediated drug and hormone transport across the BBB) after systemically administering cortisol and corticosterone, Mason et al. demonstrated that P-gp impedes the entry of cortisol, but not corticosterone, into the mouse brain. 114 In contrast, Uhr et al. reported that both cortisol and corticosterone penetration are modulated by P-gp, based on studies in mice lacking both Abcb1a and Abcb1b. 112 Additionally, these mice exhibit chronically reduced corticosterone levels and altered stress-related behaviors, indicating the relevance of P-gp in glucocorticoid homeostasis and HPA axis regulation. 115
GC transport also occurs in the pituitary gland, a CVO and a critical component of the HPA axis.109,113 In situ brain perfusion studies with radiolabeled corticosterone and cortisol in mice have demonstrated the presence of influx mechanisms—including OATP2-mediated transport—that contribute to GC accumulation in the pituitary gland. 109 Notably, no significant differences in corticosterone or cortisol distribution were observed in the pituitary gland between Abcb1a/b−/− and wild-type mice, suggesting that the efflux transporter P-gp does not influence GC transport at this site.109,113 These results highlight that the facilitative influx mechanism has the potential role in modulating GC levels and regulating the HPA axis.
Additionally, GC transport occurs across another CNS interface: the BCSFB. Similar to the case in CVOs, both corticosterone and cortisol are taken up into the ChP via a saturable influx mechanism involving multiple transporters, including OATP2. 109 In humans, systemically administered cortisol has been shown to cross the blood into the CSF, indicating that cortisol can traverse the BCSFB. 116
Taken together, these observations indicate that, not only are GCs able to enter the CNS via passive diffusion, their access is also actively regulated by transporter systems, including OATPs and P-gp, at the BBB, CVOs, pituitary, and BCSFB. This allows precise control over steroid distribution under physiological conditions and suggests potential relevance in pathological states, with implications for brain barrier function and CNS homeostasis (Figure 4).
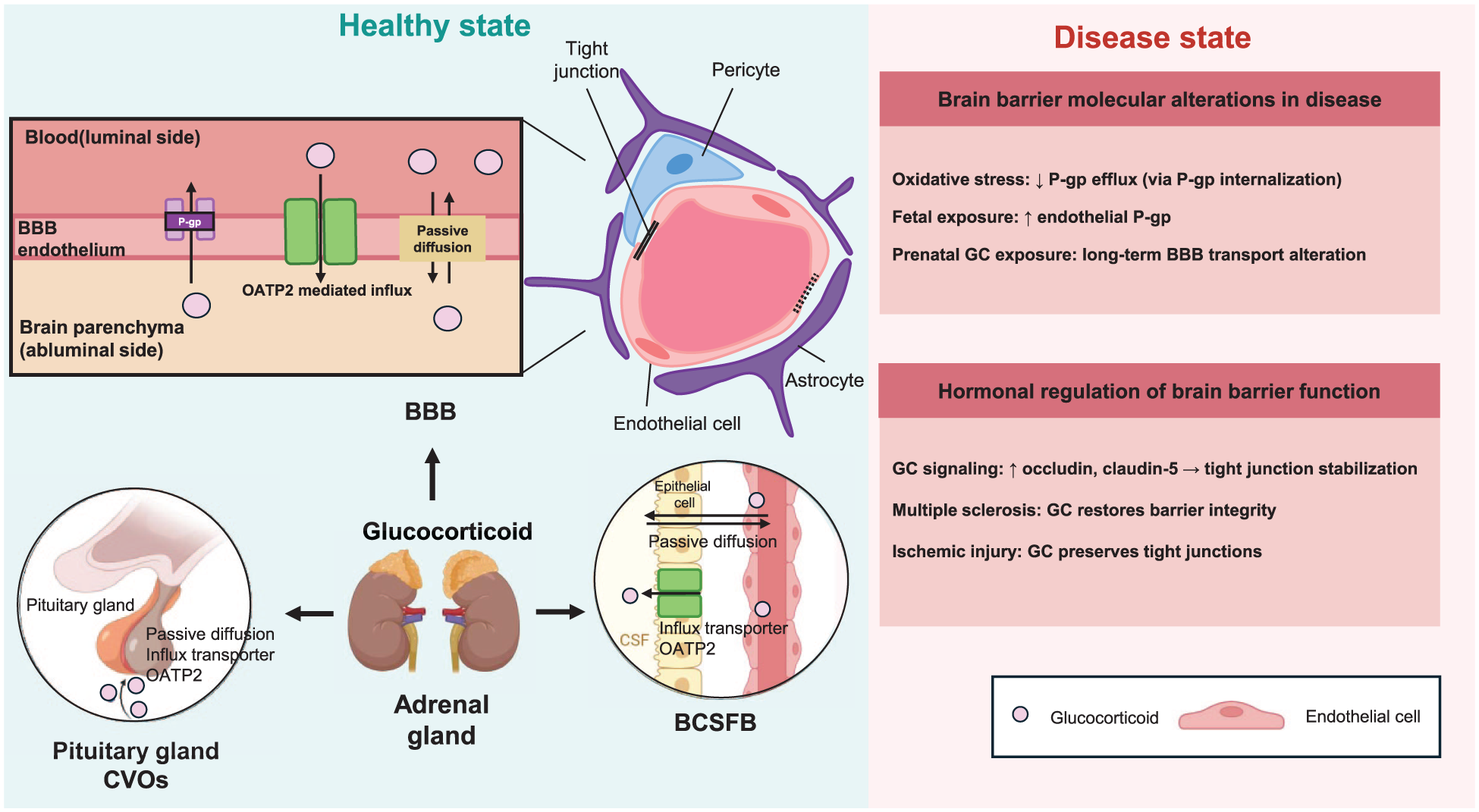
GC transport and regulation of brain barriers. GCs, produced by the adrenal glands, access the central nervous system through multiple brain barrier interfaces. At the BBB, GC entry occurs primarily through passive diffusion but can also be modulated by transporter systems expressed in brain endothelial cells, including OATPs and the efflux transporter P-gp. Arrows in the BBB panel indicate the proposed transport route between blood and brain parenchyma. In addition to the BBB, GCs can interact with other barrier interfaces, including the BCSFB and CVOs, contributing to hormone exchange between the circulation, CSF, and brain. The right panel summarizes representative alterations in GC transport and brain barrier function under pathological or developmental conditions.
Alterations in GC transport at the brain barriers in disease
GC transport into the CNS is altered under pathological conditions. Disease- and development-associated alterations in GC transport across brain barriers are summarized in Table 1. Under pathological conditions associated with acute oxidative stress, such as ischemia–reperfusion injury and inflammatory insults, exposure to reactive oxygen species (e.g. H2O2) activates Abl and Src kinases at the BBB. These kinases phosphorylate caveolin‑1 on tyrosine‑14, driving internalization of P‑gp together with caveolin‑1 and rapidly decreasing P‑gp‑mediated cortisol efflux, thereby increasing GC accumulation in the brain. 115 Given the widespread clinical use of GCs, these finding emphasize the importance of understanding how pathological conditions modify GC transport across the BBB.
Developmental regulation of GC transport at brain barriers
During physiological development, glucocorticoid exposure can modulate BBB properties, particularly in late gestation. Antenatal GC administration in pregnancies at risk of preterm delivery promotes lung maturation but also affects fetal brain microvessels, where cortisol and synthetic GCs such as dexamethasone increase P‑gp expression and activity in a gestational age‑dependent manner.116,117 In guinea pig BECs, GC treatment in late gestation and early postnatal life upregulates Abcb1 mRNA and P‑gp protein, likely through changes in vesicular trafficking (e.g. enhanced Rab‑dependent recycling), leading to a persistent increase in efflux capacity at the developing BBB. 116 This sustained elevation of P‑gp may reduce fetal and neonatal brain exposure to both endogenous GCs and xenobiotics, with potential long‑term consequences for neurodevelopment and drug response. 21 These observations indicate that GC transport capacity is dynamically regulated during both disease and development, with implications for CNS exposure and barrier function.
Local metabolism of GC at brain barriers
Beyond transporter-mediated mechanisms, glucocorticoid exposure at brain barriers is also regulated by local steroid metabolism. The enzymes 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1) and 11β-HSD2 interconvert active cortisol and inactive cortisone, thereby modulating the availability of biologically active glucocorticoids at the neurovascular interface. 118 11β-HSD1 primarily functions as an 11β-reductase that regenerates cortisol from cortisone and amplifies intracellular glucocorticoid signaling. 119 This enzyme is widely expressed in forebrain regions such as the cortex and hippocampus and is also present in glial cells including microglia, where inflammatory activation can further upregulate 11β-HSD1 expression and enhance local cortisol regeneration. 120
In contrast, 11β-HSD2 catalyzes the oxidative inactivation of cortisol to cortisone and is highly expressed during brain development, where it functions as a protective metabolic barrier limiting excessive glucocorticoid exposure to developing neural cells. 121 At brain barrier interfaces such as the BBB and BCSFB, differential expression of 11β-HSD isoforms is thought to regulate local cortisol–cortisone conversion and thereby influence the fraction of active glucocorticoid that reaches the CNS parenchyma. 122
Through this enzymatic interconversion, brain barriers may not only influence the physical passage of glucocorticoids but also modulate the proportion of hormone that remains biologically active after entering the CNS. This metabolic layer of regulation therefore likely complements passive diffusion and transporter-mediated pathways in shaping glucocorticoid exposure within the brain, potentially influencing brain barrier physiology and function.
Effects of GCs on brain barriers integrity
Beyond influencing their own CNS access, GCs directly modulate barrier structure and function. Hormone-mediated regulation of brain barrier integrity and function, including the effects of GCs, is summarized in Table 2. Studies using in vitro models of the BBB have reported that GCs increase transendothelial electrical resistance (TEER), indicating enhanced barrier integrity.123–127 This improvement is associated with cytoskeletal rearrangements that alter cell-cell contacts and morphology. 123 Tight junction proteins are key targets of GC signaling: occludin is directly upregulated via glucocorticoid receptor (GR) in BBB endothelium, and claudin-5 expression is also increased at both RNA and protein levels by GC signaling.124–127 Additional mechanisms, such as modulation of plasmalogen levels and acceleration of adjacent junction formation, further contribute to reinforcement of the BBB by GCs. 126 Glucocorticoids may also influence brain barrier physiology through regulation of the neurovascular unit (NVU). Chronic glucocorticoid signaling has been shown to impair astrocyte–vascular communication by reducing inwardly rectifying K+ channel (Kir2.1)-mediated vasodilation in parenchymal arterioles, thereby disrupting neurovascular coupling in a glucocorticoid receptor-dependent manner. 122
In multiple sclerosis (MS), a chronic inflammatory disorder of the CNS, GCs such as dexamethasone and methylprednisolone remain the standard of care for the management of acute exacerbations. In vitro studies using mouse brain microvascular endothelial cells exposed to MS patient serum showed reduced TEER and downregulated tight junction proteins, including claudin-5 and occludin—effects that were partially reversed by GC administration. 22 Treatment of cEND cells, an immortalized mouse cerebral endothelial cell line, with serum from MS patients induced upregulation of MMP-9, an effect that was significantly reduced by GC treatment, which also enhanced the expression of tissue inhibitor of metalloproteinases-1 (TIMP-1), mitigating changes in the expression of the microvascular integrin α1 subunit and further reducing MMP-9 levels.22,128 These findings indicate that GC therapy preserves BBB integrity in MS by modulating tight junction proteins and matrix-remodeling enzymes, thereby reinforcing its therapeutic efficacy. GC therapy has also been shown to restore BLB integrity in models of hearing loss, highlighting the role of vascular regulation in GC-mediated tissue protection. 129 Disruption of junctional proteins in the BLB increases permeability and can contribute to hearing impairment. Experimental studies have shown that GC treatment upregulates the expression of tight junction proteins, including occludin and claudin-5, thereby reinforcing BLB integrity and mitigating hearing loss. 130 This restoration of barrier function underscores the potential of GC therapy in preserving cochlear vascular integrity and offers new insights into the mechanisms linking GC action with BLB maintenance.
In contrast, GC treatment can be ineffective or even detrimental in acute ischemic stroke. Oxygen and glucose deprivation reduced tight junction protein expression and TEER in cEND cells, and GC was ineffective in reversing these effects in the setting of hypoxia owing to proteasome-mediated degradation of the GC receptor. 131 The effects of glucocorticoids on brain barrier structure and function in disease states are summarized in Figure 4.
Taken together, these observations indicate that GCs regulate barrier integrity across multiple contexts: they protect the barrier in chronic inflammatory and sensory disorders, can be limited in acute ischemic injury, and modulate barrier function during development. These findings highlight the need for further studies to elucidate the precise mechanisms of GC action and optimize therapeutic strategies.
Ghrelin
Ghrelin, a 28-amino acid peptide, that is, mainly released by the stomach, has an orexigenic effect in both humans and rodents. 132 Within the brain, ghrelin functions as a neuroendocrine signal that regulates feeding-related behaviors and maintains energy homeostasis. 133 Because circulating ghrelin acts on hypothalamic and extrahypothalamic targets, its physiological effects depend on regulated access to the brain through multiple barrier interfaces.
Transport of ghrelin across brain barriers in health and disease
Physiology of ghrelin transport across brain barriers
Ghrelin can cross the BBB of the mouse brain in both periphery-to-brain and brain-to-periphery directions through saturable transport mechanisms, 134 which have been studied in both directions using radio-labeled human ghrelin. 135 Ghrelin exerts its effects by binding to growth hormone secretagogue receptor-1a (GHSR-1a), which is abundantly expressed in the CNS. 136 However, studies using radiolabeled ghrelin have shown that BBB transport rates do not differ between wild-type and GHSR-null mice, suggesting that ghrelin crosses the BBB through a GHSR-independent mechanism. 134
Ghrelin can also enter the brain via CVOs, which lack a conventional BBB and possess fenestrated capillaries. 17 Using real-time in vivo imaging, Schaeffer et al. demonstrated that intravenously (i.v.)-administered fluorescently labeled ghrelin (F-ghrelin) rapidly and passively extravasates through the fenestrated vasculature of the ME, a representative CVO. 26 These fenestrated capillaries project toward the arcuate nucleus (ARC) of the ventromedial hypothalamus, facilitating rapid diffusion of ghrelin and mediating orexigenic effects.26,137–140 After i.v. injection, F-ghrelin was observed in ME tanycytes, specialized ependymal cells lining the floor of the third ventricle that regulate molecular exchange between the blood, CSF, and hypothalamus, indicating the involvement of these cells in transporting ghrelin into the brain.137,139 In vitro studies of tanycytes obtained from neonatal rats demonstrated that these cells internalize fluorescently labeled ghrelin via clathrin-mediated endocytosis. 141 Ghrelin transport has also been observed in the area postrema, another CVO with fenestrated capillaries, although higher peripheral concentrations of F-ghrelin were required for detectable uptake in this region.137,138
In addition, ghrelin can be transported through the BCSFB, composed of the ChP and hypothalamic tanycytes, to reach the brain via the CSF. 140 Peripherally administered F-ghrelin is detected in epithelial cells of the ChP and is selectively internalized into the CSF, predominantly in a GHSR-dependent manner, although GHSR-independent uptake has also been observed. 142 Once in the CSF, ghrelin diffuses rapidly and passively into periventricular brain regions to exert its central effects.140,142 Within the CSF, F-ghrelin is subsequently internalized by hypothalamic tanycytes via clathrin-mediated endocytosis, contributing to a reduction in CSF ghrelin levels and shortening the duration of its central effects. 141
Collectively, these findings indicate that ghrelin transport into the brain involves multiple brain barrier systems: across the BBB via GHSR-independent mechanisms, through CVOs via tanycytes and clathrin-mediated endocytosis, and at the BCSFB via epithelial cell uptake. This suggests an integral role of brain barriers in regulating ghrelin access to the CNS. Building on this understanding, further investigations into how pathological conditions affect brain barrier function may provide new insights into the mechanisms linking ghrelin signaling with barrier integrity and CNS homeostasis (Figure 5).
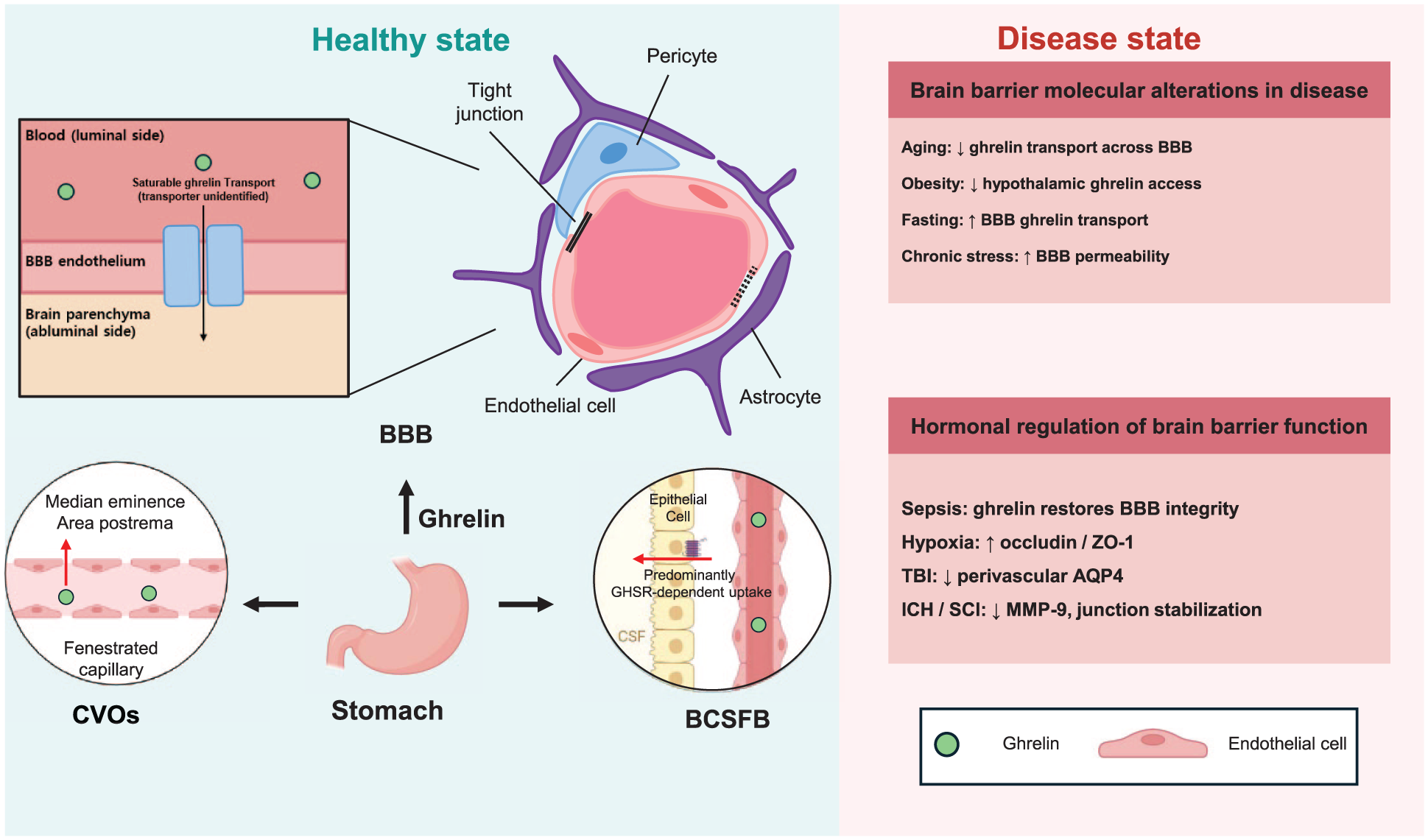
Ghrelin transport and regulation of brain barriers. Ghrelin, a peptide hormone primarily produced in the stomach, can access the central nervous system through multiple brain barrier interfaces. At the BBB, ghrelin crosses the endothelium through a saturable transport mechanism, although the molecular identity of the transport system remains unclear. Arrows in the BBB panel indicate the proposed transport route into the brain parenchyma. In addition to the BBB, ghrelin can enter the brain through CVOs, which contain fenestrated capillaries, and through the BCSFB at the choroid plexus. These routes allow circulating ghrelin to reach the CSF and hypothalamic regions. The right panel summarizes representative alterations in ghrelin transport and brain barrier function under physiological or pathological conditions, including aging, obesity, fasting, and brain injury.
Alterations in ghrelin transport at brain barriers in disease states
Metabolic and stress-related conditions can alter ghrelin transport across brain barriers. Disease- and condition-associated alterations in ghrelin transport across brain barriers are summarized in Table 1.
Metabolic states: Obesity and fasting
Ghrelin transport across brain barriers is differentially regulated in different pathological conditions, particularly those related to metabolic status and energy balance. 143 Studies measuring the unidirectional influx rates of systemically administered, radiolabeled ghrelin have shown that the capacity to transport peripheral ghrelin into the brain is reduced in obese and aged mice.26,144 This impairment is associated with an inverse relationship between body weight and BBB permeability to ghrelin. 145 In addition to adult obesity, neonatal overnutrition has been reported to attenuate ghrelin transport into the mediobasal hypothalamus, including the ARC. 139
In contrast, brain perfusion studies with radiolabeled ghrelin have demonstrated that fasting enhances ghrelin transport across the BBB—effects opposite those observed in obesity. 145 Reduced blood glucose levels during fasting alter the structure of fenestrated capillaries and tanycytes, improving access of metabolic substrates to the ARC. 26 Furthermore, Langlet et al. showed that VEGF-A expression in tanycytes and VEGF accumulation in the hypothalamic ME modulate these barrier properties. 144 Elevated hypothalamic VEGF levels during fasting activate VEGF receptors 1 and 2 in ME endothelial cells, promoting microvessel permeability and reorganization of tight junction proteins in the ME and ARC. 144
Chronic social stress
In mice exposed to chronic social defeat stress, ghrelin entry is increased not only in the hypothalamus but also in extrahypothalamic regions, including the dentate gyrus, ventral tegmental area, and nucleus accumbens.146,147 This increased in BBB permeability is related to morphological changes in astrocytes in the mediobasal hypothalamus. 146
Taken together, these findings indicate that ghrelin accessibility to the brain is not fixed, but instead is modulated by metabolic and stress conditions through structural and molecular remodeling of brain barriers (Figure 5).
Effects of ghrelin on brain barriers integrity
In addition to its well-characterized orexigenic effects, ghrelin has recently been shown to exert neuroprotective functions, including maintenance of BBB integrity, within the CNS. 148 Hormone-mediated regulation of brain barrier integrity and function, including the effects of ghrelin, is summarized in Table 2. In a mouse model of cecal ligation and puncture (CLP)-induced sepsis, BBB integrity was shown to be significantly compromised compared with sham-operated controls. However, administration of ghrelin preserved BBB integrity, as indicated by reduced Evans blue dye extravasation. This protective effect was abolished by LY294002, a PI3K inhibitor, indicating that activation of the PI3K/Akt signaling pathway is involved in ghrelin-mediated preservation of BBB integrity. 149 Systemic hypoxia has also been shown to affect the permeability of the BBB to ghrelin. 145 In rat models, both acute and chronic systemic hypoxia lead to reduced expression of tight junction proteins, including occludin and ZO-1, which are critical for maintaining BBB integrity. 150 Systemic administration of ghrelin during hypoxic conditions was found to upregulate ZO-1 expression under acute hypoxia and increase the expression of both occludin and ZO-1 under chronic hypoxia. 151 In addition, ghrelin prevents BBB disruption after post-traumatic brain injury by decreasing perivascular aquaporin-4 (AQP4). 152 These findings suggest that ghrelin exerts a protective effect on BBB integrity.
In addition to modulating junctional protein expression, ghrelin has also been reported to exert direct protective effects on vascular endothelial cells. In cultured endothelial cells, ghrelin suppresses advanced glycation end product-induced apoptosis through activation of GHSR1a and downstream nitric oxide (NO)/cyclic guanosine monophosphate (cGMP) signaling pathways. 153 Similarly, ghrelin has been shown to ameliorate homocysteine-induced endothelial dysfunction by increasing endothelial nitric oxide synthase (eNOS) expression and reducing oxidative stress in a GHSR-dependent manner. 154 In addition, ghrelin has been reported to attenuate cerebral microvascular leakage by regulating inflammatory responses and endothelial apoptosis, potentially through a p38 MAPK–JNK-dependent signaling pathway. 155 These endothelial-protective actions may contribute to stabilization of the BBB and preservation of NVU function under pathological conditions.
Recent studies using an intracerebral hemorrhage (ICH) animal model have demonstrated that the reduction in junction proteins in the intestinal barrier can be reversed by administration of the brain-gut peptide ghrelin. 156 This finding highlights protective roles of ghrelin beyond the BBB, extending to multiple barrier systems. Although direct investigations of ghrelin on the BCSFB remain limited, evidence from spinal cord injury models indicates that ghrelin can recover junction protein expression in the blood–spinal cord barrier (BSCB) and suppresses MMP-9 expression, thereby preventing infiltration of blood cells. 157 The ability of GHSR1 antagonists to block these effects supports the conclusion that ghrelin signaling via GHSR1, which is expressed at the BCSFB, may contribute to barrier maintenance under inflammatory conditions.
Collectively, these findings suggest that ghrelin can cross not only classical brain barriers such as the BBB and BCSFB but also CVOs, where barriers are naturally leaky. Ghrelin transport appears to be dynamically regulated under metabolic or stress conditions that accompany barrier remodeling. Moreover, several studies have reported that ghrelin can restore the expression of junctional proteins critical for barrier integrity, implying a neuroprotective role in damaged brain barriers. However, comprehensive and mechanistic investigations are required to clarify the precise effects and underlying pathways of ghrelin across different disease models associated with barrier dysfunction (Figure 5).
Conclusions
Brain barriers, including the BBB, BCSFB, BLB, and CVOs, are not just passive physical barriers; they function as dynamic checkpoints for maintaining systemic homeostasis. The transport of peripheral hormones across these brain barriers is tightly regulated and plays a pivotal role in maintaining CNS homeostasis. THs, insulin, GCs, and ghrelin all rely on distinct, often saturable, transport mechanisms to access the brain, and these pathways are selectively modulated under both physiological and pathological conditions (Table 1).
In particular, TH transport via MCT8, OATP1C1, and LAT1 is critically involved in neurodevelopment and adult brain function, whereas systemic inflammation and thyroid dysfunction can disrupt both transporter expression and BBB integrity. Similarly, insulin transport across the BBB remains a subject of ongoing debate, with evidence suggesting receptor-mediated transcytosis as well as receptor-independent mechanisms, which are differentially affected by aging, metabolic diseases, and neurodegeneration. GCs, traditionally considered to diffuse passively into the CNS, also interact with influx and efflux transporters such as OATP2 and P-gp—interactions that can be significantly altered by oxidative stress and exposure to therapeutic. Ghrelin demonstrates both saturable BBB transport and passive diffusion through circumventricular organs and the BCSFB, with transport efficiency fluctuating according to nutritional and stress-related states.
Importantly, beyond transport, these hormones modulate the structural and functional integrity of brain barriers (BBB, BCSFB, BLB; Table 2). A TH deficiency compromises tight junction composition and permeability; insulin deficiencies and hyperinsulinemia disrupt barrier properties and nutrient transport; GCs exert both protective and detrimental effects, depending on disease context; and ghrelin exerts neuroprotective actions that maintain barrier integrity under systemic insults such as sepsis and hypoxia.
These findings collectively underscore the complexity of hormone–brain-barrier interactions. A deeper understanding of the molecular mechanisms governing transport and barrier modulation will be essential for developing therapeutic interventions targeting neuroendocrine dysfunction and neurodegenerative diseases. Future research should focus on identifying novel transporters, elucidating disease-specific regulatory pathways, and clarifying how systemic metabolic and inflammatory states dynamically reshape hormone access to the brain.
Footnotes
List of abbreviations
AD, Alzheimer’s disease
ARC, Arcuate nucleus
ATP, Adenosine triphosphate
BBB, Blood–brain barrier
BCSFB, Blood–cerebrospinal fluid barrier
BECs, Brain endothelial cells
BHB, D-beta-hydroxybutyrate
BLB, Blood–labyrinth barrier
BUI, Brain uptake index
cEND, Mouse cerebral microvascular endothelial cell
CLP, Cecal ligation and puncture
CNS, Central nervous system
CSF, Cerebrospinal fluid
CVOs, Circumventricular organs
DAB, Diaminobenzidine
EndoIRKO, Endothelial-specific insulin receptor knockout mice
F-actin, Filamentous actin
F-ghrelin, Fluorescently labeled ghrelin
GCs, Glucocorticoids
GHSR, Growth hormone secretagogue receptor
GLUT1, Glucose transporter 1
GR, Glucocorticoid receptor
hCMEC/D3, Human cerebral microvascular endothelial cell line
HPA, Hypothalamic–pituitary–adrenal
ICAM-1, Intercellular adhesion molecule-1
IgG, Immunoglobulin G
IR, Insulin receptor
IV, Intravenous
LAT1, L-type amino acid transporter 1
LPS, Lipopolysaccharide
MAPK, Mitogen-activated protein kinase cascade
MCT, Monocarboxylic acid transporter
MCT8, Monocarboxylate transporter 8
ME, Median eminence
MS, Multiple sclerosis
OATP1C1, Organic anion-transporting polypeptide 1C1
PECAM-1, Platelet endothelial cell adhesion molecule-1
PKB/Akt, Protein kinase B
PTU, Propylthiouracil
TEER, Transendothelial electrical resistance
TH, Thyroid hormones
TIMP-1, Tissue inhibitor of metalloproteinases-1
TRH, Thyrotropin-releasing hormone
TSH, Thyroid-stimulating hormone
T3, Triiodothyronine
T4, Thyroxine
VCAM-1, Vascular cell adhesion molecule-1
VEGF, Vascular endothelial growth factor
ZO-1, Zonula occludens-1
Author contributions
The project was conceptualized by JYH, and YEK, DYK, MJL, SK, and SYP drafted the manuscript and prepared the figures and table. JYH, WC, and YEK corrected and commented on the first draft of the manuscript. JHA, CHP, JJ, Y-HP, and MKY provided corrections of the manuscript. DYK, MJL, JYH, and YEK revised the manuscript. JYH, YEK, and WC contributed to discussion, review, and editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT; RS-2022-NR070073, RS-2024-00406568, RS-2021-NR061617), Basic Science Research Program funded by the Ministry of Education (RS-2023-00237665, RS-2024-00463129), the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (RS-2022-KH130308, RS-2020-KH088690, RS-2025-24536373) and by research fund of Chungnam National University.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.