
Abstract
Cerebral amyloid angiopathy (CAA) is one of the most common small vessel diseases. With the deposition of amyloid beta within the walls of cerebral blood vessels, CAA can result in damage to blood vessels over time and ultimately intracerebral haemorrhage (ICH). Not only is CAA a leading cause of ICH but CAA is commonly observed in Alzheimer’s disease (AD), yet there are still no effective treatments for CAA. The pathophysiology of CAA has yet to be fully elucidated but we know the perivascular environment is heavily impacted by the deposition of amyloid within blood vessels. In recent years, there has been an increased interest in the role that perivascular clearance may play in the development of the disease. Additionally, the role of the immune system has also come into question, especially regarding CAA-related inflammation. In this review, we aim to discuss the immune impacts of CAA within the perivascular environment, and probe how specific cells within this environment may be involved in the pathogenesis of CAA. Importantly, we also consider how some of these immune cells may be key treatment avenues to explore.
Keywords
Introduction
Introduction to cerebral amyloid angiopathy
As the global population ages, we are faced with new challenges, especially challenges related to brain health. Small vessel disease (SVD), describes diseases which damage small brain blood vessels with these diseases increasing in prevalence as we age.1,2 Damage to these important vessels can lead to cognitive impairment, strokes and even dementia – highlighting the importance of not only increasing our knowledge about the causes of these complex conditions but also focusing on potential treatment avenues.
One of the most common small vessel diseases is cerebral amyloid angiopathy (CAA). CAA is defined as the accumulation of amyloid beta protein (Aβ) within the walls of cerebral blood vessels,3,4 predominantly small cortical and leptomeningeal arteries and arterioles (type 2), although deposition can occur less frequently within capillary walls (type 1). 5 This small vessel disease commonly occurs in elderly individuals, with ~23% (mean age 84.9 years) of the general population having CAA. Additionally, CAA often co-occurs with Alzheimer’s disease (AD), with the presence of CAA observed ranging from 48% 6 to 98% 7 in patients with AD in autopsy studies. Due to this overlap of diseases theoretically any treatments proposed to reduce CAA may also be effective at reducing amyloid plaques in AD and vice versa, highlighting the importance of studying both diseases alone and in tandem.
CAA is associated with the observation of haemorrhagic brain lesions (observed with MRI) in the form of cerebral microbleeds and cortical superficial siderosis. These can coincide with other non-haemorrhagic tissue injuries including white matter hyperintensities and enlarged perivascular spaces. 8 Until post-mortem confirmation, these are among the limited diagnostic features for CAA. CAA can result in clinically devastating intracerebral haemorrhage (ICH) and cognitive impairment. However, there are currently no disease modifying or curative treatments for CAA – highlighting the necessity for a more in-depth understanding of causes, specific diagnostic features and potential treatments of this disease.
Over the years we have gained deeper insights into the cellular changes that occur in CAA, especially regarding how the vasculature is impacted by amyloid deposition. 9 However, one avenue in which there is still a paucity of research is with regards to the role of inflammation, and especially how inflammation impacts cells of the perivascular unit. Although, in recent years there has been an emerging interest in the role of inflammation in the disorder,10–12 which may be due, in part, to an increased interest in a specific subtype of CAA: CAA-related inflammation (CAA-ri). In addition to this, emerging evidence has also shown that some Aβ immunotherapies have resulted in amyloid related imaging abnormalities (ARIA).13,14 Where, in response to the therapy some individuals develop oedema (ARIA-E) or haemorrhages (ARIA-H).15,16 The mechanisms for this have not been fully elucidated yet but, with regards to inflammation, two mechanisms are proposed. One being that the removal of Aβ from the parenchyma becomes deposited within the walls of vessels, increasing the amount of CAA and perivascular inflammation resulting in oedema or microhaemorrhage.17,18 The second mechanism proposes a local immune response due to anti-Aβ antibodies binding to vascular Aβ.19,20 Recently, ARIA-E and CAA-ri were shown to share similar pathological features, suggesting that ARIA-E involves some form of inflammatory response. 21 CAA-ri is a rare, but reversible condition, whereby an acute inflammatory response occurs in response to the build-up of Aβ mainly around arteries and arterioles, resulting in vasogenic oedema, seizures and microhaemorrhages.22–25 High levels of Aβ antibodies have been reported in cerebral spinal fluid (CSF), in the initial stages of CAA-ri, with these levels returning to control levels on remission – highlighting that CAA-ri is likely due to an immune response to vascular Aβ deposition. 19 Yet, recent studies have failed to replicate the above findings, suggesting that detecting Aβ antibodies in the CSF may not be a suitable biomarker for CAA-ri. 26 However, as this subtype of CAA responds to immunosuppressive medication, such as corticosteroids, (which reduce inflammation)27,28 this lends to the hypothesis that CAA may also have an inflammatory profile that could be targeted for treatments.
Previous reviews have provided thorough examinations of broad vascular pathology and neuroinflammation in CAA, in this review we take a compartment-specific focus, specifically the immune landscape of the arteriolar perivascular space. We integrate endothelial and mural cell dysfunction, basement membrane alterations, immune cell involvement and clinical biomarkers within this perivascular niche to illustrate how progressive vascular Aβ deposition reshapes the local immune composition. With our concentration on the vascular and immune mechanisms within this unique environment, we aim to clarify how vascular changes and inflammatory processes coalesce to drive CAA progression and its inflammatory complications.
Perivascular environments along the cerebrovascular zones
Blood vessels are composed of endothelial cells, surrounded by mural cells such as smooth muscle cells on arteries, arterioles, veins and venules as well as pericytes on capillaries29,30 (Figure 1(a) and (b)). Along all vascular zones, astrocytic endfeet attach to the outer vascular basement membrane separating the vasculature from the brain parenchyma. 31 In between the mural cell layer and astrocytic endfeet is the perivascular space (also known as the Virchow-Robin space) which appears to be most visible on arteries and arterioles in magnetic resonance imaging (MRI) with pathology increasing their prevalence and visibility. 32 With the perivascular space on penetrating arterioles being continuous with the CSF-filled subarachnoid space overlying the brain, this is in line with ultrastructural studies finding empty spaces, presumably once filled with CSF, mainly outside of arteries and arterioles in the healthy human and mouse brain.31,33 The presence of these similar empty or CSF-filled perivascular spaces around capillary vessels have not been observed and therefore appears to be a unique environment mainly around arteries and arterioles. This space is home to a multitude of cells, including immune cells such as perivascular macrophages 34 and fibroblasts 35 (Figure 1(b)). Whereas capillary vessels lack perivascular macrophages and fibroblasts but maintain contact with microglia. 36 The perivascular space around arterioles has been postulated to play a role in the removal of waste from the brain including Aβ via CSF-mediated mechanisms as discussed later,37,38 making it a unique vascular zone for potential CAA therapeutics.
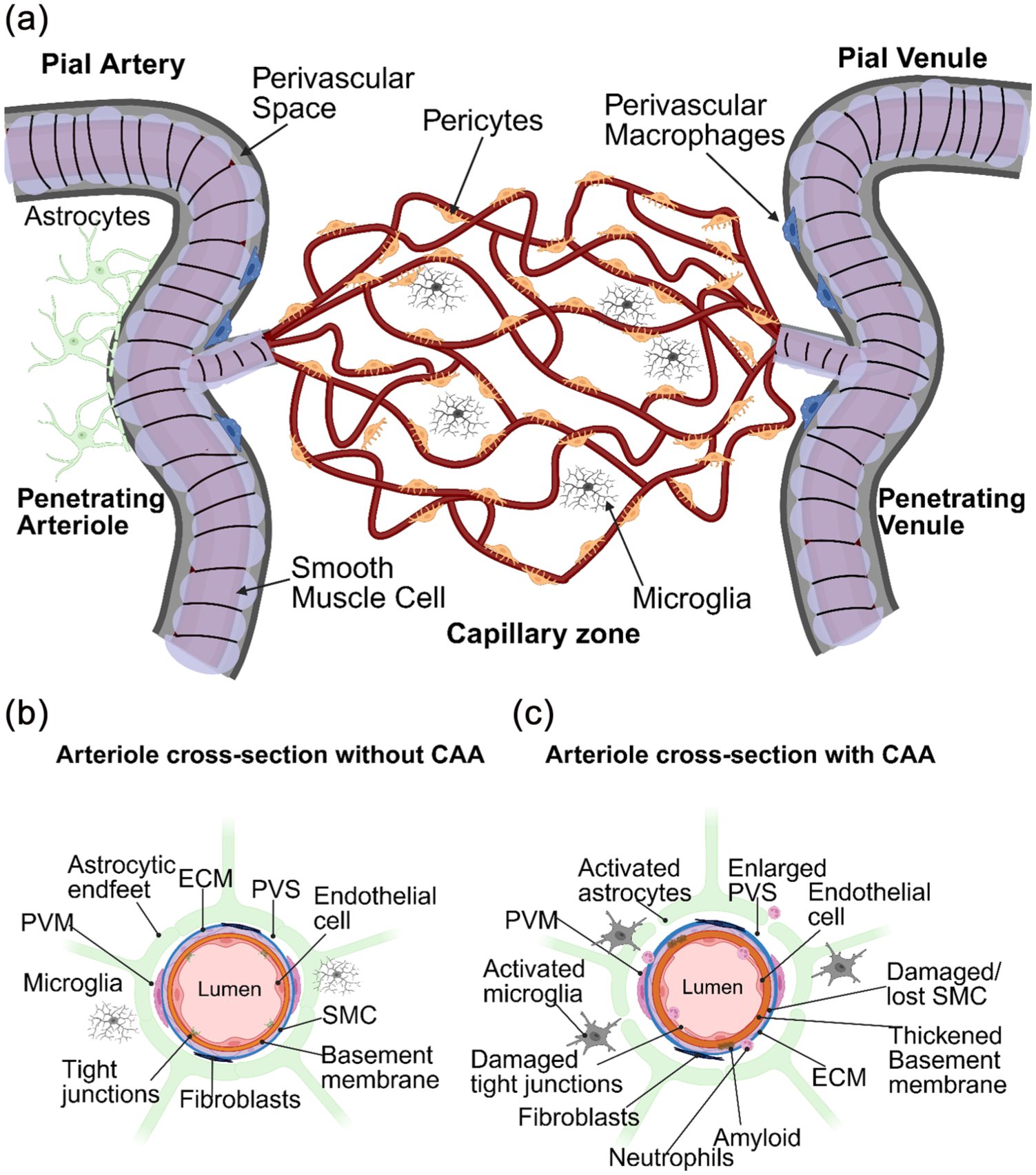
An illustrative schematic of the brain vasculature with a focus on how the perivascular environment is impacted by CAA: (a) the brain is highly vascularised by a web of vessels, where blood is perfused into the brain by penetrating arterioles branching off pial arteries. Arterioles elaborate into a capillary network where blood ultimately drains out of penetrating venules and pial veins. SMCs reside on arterioles with pericytes on capillaries. Astrocytes surround the entire vascular tree with endfeet, helping to create prominent PVS mainly around arterioles. The main immune cells interacting with the brain vasculature are PVM in the PVS as well as microglia within the brain parenchyma, (b) a cross-section of an arteriole detailing the perivascular environment in a healthy state, and (c) a cross-section of an arteriole demonstrating how the perivascular environment is affected by CAA. In the higher grades of CAA, amyloid is deposited within the walls of blood vessels, SMCs are damaged/lost, tight junctions are altered and weakened, the PVS becomes enlarged and immune responses are heightened. The ECM is also altered. In addition to activated microglia and astrocytes, neutrophils can be observed sticking to blood vessels and migrating into the parenchyma.
A healthy perivascular environment is integral to brain health. However, this environment is heavily impacted by Aβ that accumulates within blood vessel walls in CAA where type 1 CAA refers to capillary involvement and type 2 CAA indicates Aβ accumulation on arteries and arterioles. For type 2 CAA, this results in the eventual loss of smooth muscle cells altering their ability to contract and dilate properly (Figure 1(c)).12,39 Furthermore, vasomotion, a spontaneous, low frequency oscillation of arteries and arterioles thought to be important for brain clearance mechanisms by driving the movement of CSF, has also been shown to decline in the APP23 AD/CAA mouse model. Altered vasomotion in these mice correlates with initial Aβ deposition (and most closely with age), as well as precedes smooth muscle cell loss and cerebral microbleeds. 39 It is also well documented that individuals with suspected CAA have impaired vascular reactivity.40–42 Likely also contributing to this, vascular basement membranes are also damaged, thickened, and/or unorganised in composition in addition to enlargement of the perivascular space43,44 (Figure 1(c)). Overtime, arteries and arterioles become so fragile that they bleed, resulting in potentially devastating ICH. 45 In contrast, along capillary vessels, Aβ has been shown to induce a pericyte-mediated constriction within the capillary network, influence basement membrane changes and is associated with capillary rarefaction.46–50 While some perivascular pathologies may be shared among arteries, arterioles and capillaries, some are unique and this may be attributed to their differing immune interactions along these zones as discussed later.
Cells of the perivascular environment are exposed to increasing accumulation of Aβ over time, yet we still know relatively little about how this impacts all the perivascular cells, including macrophages, especially early on in disease. The arteriolar perivascular space is difficult to study in vivo due to its relatively small size (<3 μm). On the contrary, there is a wealth of information about the impact of CAA on overall brain health from imaging studies. For example, we can observe haemorrhages, white matter damage and cortical superficial siderosis on brain scans using MRI, which has given us an unparalleled understanding into what CAA can do to brain tissue at the macroscale.8,51 However, there is a lack of detailed knowledge at the cellular level in the arteriolar perivascular environment as obtaining post-mortem tissue from humans during early disease stages is very limited. Some studies have been able to circumvent this barrier by correlating the severity of Aβ deposition with other vascular pathologies like blood–brain barrier (BBB) leakage and perivascular inflammation. 12 Mouse models have also been integral in studying the various stages of disease, allowing for more mechanistic focusses. 52 Leveraging these types of studies will ultimately be informative about the pathophysiology of CAA and what mechanisms are involved in the transition to CAA-ri and/or ARIA. Understanding these gaps in knowledge could be crucial in the identification of potential therapeutic targets and focussing on the perivascular environment will likely be key in these goals.
Perivascular involvement in Aβ clearance and accumulation
Amyloid precursor protein (APP) is expressed by all major cell types in the brain including, neurons, glia and vascular cell types. APP can be processed by β- and γ-secretases to generate Aβ with neurons demonstrating the most dominant source.53,54 In the healthy brain Aβ is cleared through cellular uptake, enzymatic digestion, fluid-mediated clearance mechanisms or across the vasculature into the blood. There are numerous points where these mechanisms can fail resulting in the aberrant accumulation of Aβ in the brain. 55 It is also possible that cell populations with normally low production of APP and Aβ become dysregulated with age or trauma and contribute to an increased load of Aβ that the brain can no longer manage. It is unclear what the initiating mechanisms are but likely there is an imbalance in production and clearance that becomes the tipping point. It is also possible that increased Aβ production by vascular cell types, possibly coupled with poor Aβ removal, is the major contributor to vascular accumulation of Aβ. A possibility for this was recently shown with pericytes but so far smooth muscle cell involvement is unknown. 56 More work is needed to study the individual cell populations and their potential contributions of Aβ accumulation throughout the brain.
There are a number of ways that Aβ is removed from the brain, including those that involve perivascular cells and the perivascular space. This involves transport of waste across the BBB, cellular uptake and enzymatic-degradation and Aβ can also be cleared via the exchange of interstitial fluid (ISF) and CSF along perivascular spaces. One of the leading hypotheses in the field is that the initiating mechanisms for vascular Aβ accumulation in CAA is due to impairment of perivascular waste clearance mechanisms.37,38 It is important to note that it is unclear to what degree CAA plays in AD pathology and whether it is an initiating factor, it is generally agreed upon that CAA likely exacerbates parenchymal Aβ accumulation given the numerous mechanisms the brain vasculature and the perivascular space utilise in the removal of Aβ from the brain. 4 Thus, it is postulated that improving perivascular clearance of Aβ could be a target for therapeutics in CAA and AD, 57 therefore it is crucial to understand perivascular clearance mechanisms.
Along all vascular zones, transportation of Aβ across the brain vasculature relies heavily on the endothelium where localisation of the low-density lipoprotein receptor-related protein 1 (LRP1) on the abluminal side of the vasculature binds Aβ and transports it out of the brain via the bloodstream. 58 Other Aβ-binding proteins like ApoE assist in LRP1-mediated removal of Aβ from the brain. However, the ApoE4 variant, with a strong genetic risk factor for AD, impairs the removal of Aβ via LRP1. 59 LRP1 is also expressed by smooth muscle cells in the human brain and knock-out of LRP1 in smooth muscle cells in a mouse model of AD (APP/PS1) increased Aβ accumulation within the parenchyma and the vasculature. 60 Further, LRP1-smooth muscle cell knock-out mice on the ApoE4 background, not ApoE3, led to heightened BBB leakage, perivascular glial activation and earlier impairment of spatial memory. 61 These studies point to an important role for the endothelium as well as smooth muscle cells in the transport of Aβ out of the brain. Recent single-cell transcriptomic studies on human brain tissue have shown that ApoE is also highly expressed by astrocytes and meningeal fibroblasts in addition to smooth muscle cells. 62 It is possible other perivascular cells and support tissues like astrocytes and the meninges are involved in the efficient removal of Aβ via the bloodstream and the maintenance of vascular homeostasis.
In addition to passage of Aβ via LRP1–ApoE mediated mechanisms out of the brain via the bloodstream, another proposed mechanism is utilisation of this pathway for degradation of Aβ via enzymatic or lysosomal digestion. 63 Cultured human brain smooth muscle cells were shown to degrade Aβ via LRP1 and lysosomal processes. 60 In pericytes, an Aβ-degrading enzyme called beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) was shown to process Aβ40–Aβ34 suggesting pericytes are capable of directly degrading Aβ. Pericytes from AD patients displayed a reduction in the conversion of Aβ40–Aβ34, indicating a disruption to the degradation of Aβ in AD. BACE1 was also found to be expressed in smooth muscle cells, providing a potential role in the direct degradation of Aβ. 64 Three other major enzymes are involved in Aβ degradation – neprilysin (NEP), insulin-degrading enzyme (IDE) and angiotensin-converting enzyme (ACE). 65 In humans, NEP has been shown to be expressed by smooth muscle cells 66 whereas IDE has been shown to be expressed more broadly in smooth muscle cells, pericytes and endothelial cells. 67 Both NEP and IDE showed decreased enzymatic activity from AD brain tissue. ACE has been shown to localise within the perivascular space among the basement membrane proteins, decorin and fibronectin. While its cellular origin is unknown, the enzymatic activity of ACE from AD brain tissue was heightened. 68 While changes in these particular mural-cell derived Aβ processing enzymes can be noted, changes in overall mural cell populations are confounding. For pericytes, some studies report a decrease in their numbers in AD/CAA pathology 69 while other studies indicate preserved pericyte populations in AD. 70 Loss of smooth muscle cells is a more consistent phenomenon reported by many different studies (Figure 1(c)).9,71,72 However, an interesting finding is the effect Aβ may have on the contractility of pericytes and smooth muscle cells, with pericytes showing contractile responses to Aβ 46 and smooth muscle cells showing impairment in their contractile abilities that may be attributed to phenotypic switching.9,73 How failure of mural cell Aβ transport, processing and control of cerebral blood flow contributes to CAA pathology and overall brain health is unknown, but distinct changes in these populations could contribute to the differing CAA pathologies observed (i.e. type 1 vs type 2 CAA).
Another proposed mechanism for removal of Aβ from the brain relies on movement of CSF in the perivascular space by the oscillatory contraction and dilation of arteries and arterioles where exchange of fluid into the brain occurs through aquaporin-4 (Aqp4) water channels on astrocytic endfeet. Once in the brain parenchyma, CSF mixes with interstitial fluid (ISF) as well as metabolic waste like Aβ, tau and lactate where the pulsation of arteries and arterioles helps move waste-ISF mixtures throughout the brain. Exit of waste from the brain is proposed to occur through three mechanisms – glymphatic clearance, intramural periarterial drainage (IPAD) pathway and the mixing hypothesis. 74 In the glymphatic system, waste exits via the perivascular spaces surrounding venules, potentially specific venules that cross the subarachnoid space and arachnoid barrier into the dura where meningeal lymphatic vessels directly facilitate the removal of waste out of the brain.38,75,76 In the IPAD pathway, waste-ISF drains along the basement membrane of arteries and arterioles in a passive manner out of the brain, against the direction of blood flow. 77 The mixing hypothesis suggests that waste-ISF drains along periarterial channels, via diffusion with waste-solute transport aided by physiological motion (resulting from cardiac pulsatility and vasomotion).78,79
Disruption to the glymphatic and IPAD pathways have been reported in numerous mouse models of AD80,81 however it is unclear whether alterations to different aspects of these mechanisms are either a consequence or contributing factor to vascular Aβ accumulation. Mislocalisation of Aqp4 to astrocytic endfeet surrounding the perivascular space disrupts glymphatic clearance in mice, with loss of perivascular Aqp4 also observed in human AD brain tissue.82,83 Degeneration and/or dysfunction in smooth muscle cell populations is also a likely contributing factor due to poor vasomotor activity. However, whether changes to smooth muscle cells or astrocytic endfeet is a cause or consequence of impaired Aβ clearance is still unknown.
Immune cells like perivascular macrophages can also participate in the removal of Aβ via phagocytic mechanisms. As such, perivascular macrophages are known to take up tracer injected into the brain and the cisterna magna, indicating a role for regulating vascular waste with possible impairment in these mechanisms contributing to CAA. 84 In support of this, depletion of border associated macrophages (BAMs), which include perivascular macrophages (discussed in further detail in section 3), in mice using clodronate-liposomes prior to stroke led to heightened Aβ build-up on blood vessels and reduced glymphatic clearance. 85 This was attributed to a suppression of astrocytic stress due to BAM-derived mesencephalic astrocyte-derived neurotrophic factor. It has also been shown that perivascular macrophages are important for modulating the vascular extracellular matrix which upon BAM depletion, impairs arteriolar pulsatility and CSF flow in the perivascular spaces. Impairment of perivascular macrophage function in this manner as well as their phagocytic activity is proposed to hinder waste clearance in AD. 86 Given their localisation along arteries and arterioles, this may be important for controlling type 2 CAA pathology, however studies have yet to address this.
Another important aspect of the vasculature that can contribute to the tendency of vascular Aβ accumulation and loss of blood vessel integrity is thickening and alterations of the vascular basement membrane (Figure 2). One of the major structural components found on all vascular zones is collagen IV which has shown widely varying changes across mouse and human studies, some reporting no changes, while others show increases or decreases in collagen IV presence on the brain vasculature. 87 Endothelial cells, mural cells and fibroblasts all express collagen IV and therefore the discrepancies in these changes could lie in cell types that are affected by Aβ presence and/or the inconsistency in vascular zones being studied. Fibronectin is a glycoprotein that also makes up the vascular basement membrane, with increases associated with heightened Aβ deposition on blood vessels. 88 It is postulated other basement membrane proteins such as the heparin sulfate proteoglycans, perlecan and agrin, are also reported to be increased along the brain vasculature. 89 Perlecan may accelerate Aβ accumulation and promote fibril formation. 90 Intriguingly, agrin appears to favour vascular build up and senile plaques in Dutch type CAA. 91 The importance of studying specific changes in these proteins along the different vascular zones is becoming more apparent and could provide further insight into the different CAA pathologies and how vascular function and integrity is altered. 92 It is possible changes in the vascular basement membranes could alter the profiles, activity or presence of the immune cells like perivascular macrophages and microglia along the vascular wall, resulting in worsening Aβ accumulation and vascular stability.
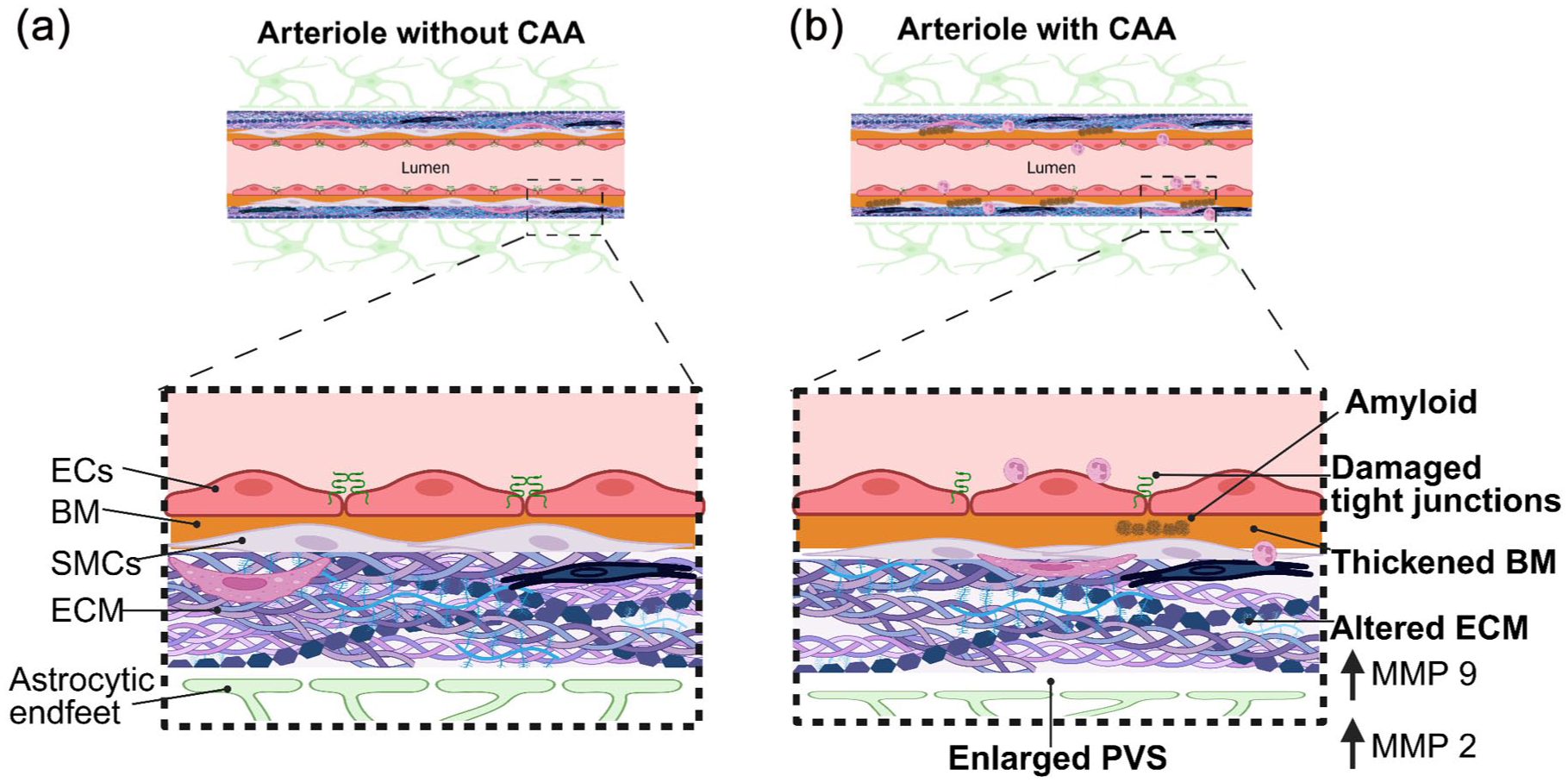
An illustrative schematic, focussed on perivascular changes in vessels without (a) and with CAA (b). In CAA Aβ is deposited within the basement membrane of arterioles. Aβ deposition damages the structure of vessels including damaging tight junctions between ECs, resulting in ‘leaky’ vessels and impaired blood brain barrier. In CAA the BM also thickens, resulting in stiffer vessels. Accompanying the above changes are changes to the ECM, whereby certain MMPs become upregulated, including MMP2 and MMP9.
As discussed here, sufficient clearance of waste from the brain relies on many vascular-related mechanisms, many of which rely on healthy perivascular cells and environment with more recent studies pointing toward some immune involvement. Alterations to any of these mechanisms/cells are potential points for initiating CAA or potentially crossing the threshold to CAA-ri. Thus, understanding these mechanisms is important for many facets of CAA and AD.
Inflammatory cells in the perivascular environment in CAA
Numerous cells involved in immune responses can influence the perivascular environment, including border associated macrophages and microglia, as well as fibroblasts, and in some cases, peripheral immune cells. This section will discuss how each of these cell types can impact the pathophysiology of CAA.
Border associated macrophages
Macrophages are resident immune cells which play important roles in both tissue injury and repair. 93 Several types of macrophages exist within the central nervous system (CNS). Within the CNS, parenchymal macrophages are classified as microglia. Macrophages found within the borders of the parenchyma at the interface between the BBB and CSF, for example, within the meninges, perivascular spaces and choroid plexus are classified as border associated macrophages or BAMs. BAMS are distinct from microglia, evidenced by the expression of different scavenger receptors, including CD163 94 and the mannose receptor (CD206). 95 BAMs themselves can also be divided into even more specific cell types, 96 including subdural/leptomeningeal macrophages, observed within the pia mater, dural macrophages found within the dural meninges, stromal choroid plexus macrophages located within the stroma of the choroid plexus and epiplexus macrophages, which are located on the apical epithelial surface.97,98 Perivascular macrophages are also another subdivision of BAMs, which are located alongside cerebral blood vessels, within the perivascular space, specifically they localise to arteries, arterioles, venules and veins.34,99 Due to the location of perivascular macrophages, these cells are regularly exposed to Aβ deposited within arteriolar walls in CAA. Studies have begun to investigate the impact CAA may have upon BAMs or the effect these cells may have on CAA and vascular function.
In mice where perivascular macrophages were selectively depleted with clodronate-liposomes there was an observed increase in CAA, evidenced by a greater number of blood vessels positively stained with Thioflavin-S, a common stain to identify Aβ. This group also pharmacologically increased the turnover of perivascular macrophages and in turn observed a reduction in CAA, with perivascular macrophages, not astrocytes or microglia colocalised with Aβ. 100 Part of this could be attributed to their role in removal of Aβ via phagocytosis as described above, however other studies have implicated perivascular macrophages in the pathophysiology of CAA, demonstrating their role in CAA may be more complex.
Numerous studies suggest that vascular dysfunction could be due to oxidative stress.101,102 As perivascular macrophages are a known producer of reactive oxygen species (ROS), 103 Park et al. aimed to investigate whether perivascular macrophages may mediate the impaired vascular responses which have previously been associated with the deposition of Aβ. They showed that following the reduction of perivascular macrophages, the vascular impairment and oxidative stress often reported in response to Aβ (either directly applied to the neocortex or in the Tg2576 model) was not observed. 104 This suggests that perivascular macrophages may play an important role in the vascular dysfunction observed in response to Aβ. However, the study only included young mice (3–4 months of age), Aβ levels within the brain are increased at this age, but Aβ plaque pathology is not observed. 105 Therefore, this study was unable to provide an insight into how perivascular macrophages may be involved in the pathophysiology of CAA, which occurs later in disease. To address this gap, a recent study used mice aged 3, 18 and 22 months old. CD36, an Aβ-binding scavenger receptor (associated with vascular dysfunction and increased oxidative stress observed in response to Aβ) 106 was removed from perivascular macrophages. Following the removal of CD36 they reported no impairment of neurovascular function, reduced ROS production and a reduction in Aβ40, the species most commonly associated with vascular amyloid deposition. 107 Interestingly, a reduction in CAA was also observed in parallel with less smooth muscle cell damage in aged mice. Alongside these vascular improvements, cognitive function was also improved in these mice. However, the deletion of CD36 had no impact on Aβ42 or overall amyloid plaque deposition, highlighting a specific impact on the vascular species of Aβ. Collectively, these findings suggest that perivascular macrophages may play an important role in the pathophysiology of CAA and most importantly these immune cells may be an important therapeutic avenue for CAA treatments, which currently are incredibly limited. It is important to note that while most studies have focussed on the relationship between CAA and perivascular macrophages, given the vascular pathologies, many tools to manipulate their populations also disrupt other BAMs, including those in the meninges and choroid plexus and therefore we cannot rule out their role in vascular Aβ pathology and immune responses. However, until more specific tools become available to separate out perivascular, meningeal and choroid plexus-residing macrophages, their distinct roles in the progression of CAA or immune responses are not known.
Fibroblasts
Fibroblasts are the main cellular make-up of the meninges outlining the outside of the brain where they are in close proximity to many immune cells like BAMs and the occasional T cell. 108 A subpopulation of fibroblasts, known as perivascular fibroblasts (PVFs) create a sheet around the leptomeningeal arteries and veins and their penetrating vessels where they are found on all arterioles and larger diameter venules in the cortex. 109 They are immediately outside the smooth muscle cell layer on arterioles and sometimes found near perivascular macrophages. 35 PVFs are thought to maintain the vascular wall due to the high expression of extracellular matrix proteins like Collagen-type 1, lumican and decorin. 110 In a single-nuclei transcriptomic study on isolated vessels from patients with AD, PVFs were shown to be reduced in number and have heightened TGFβ and IFN signalling. 111 Additionally, depletion of perivascular macrophages led to an increase in extracellular matrix gene expression in PVFs suggesting a potential homeostatic interplay between perivascular macrophages and fibroblasts. 86 So far, studies are limited on PVFs in AD/CAA however these studies point to a potential influence of PVFs on the inflammatory state in the arteriolar perivascular space as well as the compliance of the arteriolar wall with implications in the pathophysiology of CAA.
Microglia
Microglia are the most abundant immune cell type residing within the brain. In human tissue, it is estimated that these immune cells account for between 0.5% and 16.6% of cells within the parenchyma 112 and in mice, microglia are suggested to make up between 5% and 12% 113 of brain cells. These cells have a complex role with regards to Aβ. 114 It has been reported that microglia play an early role in the clearance of Aβ, with studies evidencing that microglia phagocytose amyloid fibrils 115 and plaques 116 and other studies revealing that microglia use digestive exophagy to digest amyloid plaques extracellularly. 117 However, homeostatic microglia may initially seed Aβ plaques, but later in disease, activated microglia can make plaques denser (Figure 1(c)). 118
To assess the role of microglia on amyloid pathology many studies have used genetic methods to deplete microglia in mouse models of AD. In one study, depletion of microglia resulted in a reduction in parenchymal plaque deposition but an increase in vascular amyloid deposition/CAA. This points to an important role microglia may play in limiting CAA. The increase in CAA was also accompanied by an increase in brain haemorrhages (a common consequence of CAA), an increase in brain calcification and early mortality. However, when microglia were repopulated, this pathology was prevented. 119 Another study showed that sustained depletion of microglia using the colony stimulation factor-1 receptor inhibitor, PLX5622, in 5xFAD mice also reduced parenchymal Aβ plaques and increased accumulation of Aβ on the brain vasculature. 120 These studies indicate the importance of this immune cell in the protection against CAA and vascular instability.
Recent studies are also highlighting the unique cellular interactions microglia make with the brain vasculature. Ultrastructurally, microglia make contact mainly with capillary vessels rather than arterioles and venules through gaps in astrocytic endfeet creating direct contact points called microglia plugs. 36 While the importance of microglia plugs is unknown, it is becoming increasingly evident that microglia can influence capillary diameter and therefore blood flow through capillary networks. This seems to be due to interactions with pericytes given microglia associated with pericytes can influence capillary width. 121 This is further supported by studies showing that microglia can influence cerebral blood flow through the PANX1-CD39-P2YR12 purinergic signalling cascade.122,123 In AD, the association of pericytes with microglia decreases specifically in the superior frontal gyrus, a brain region heavily burdened by AD pathology. 121 Expression of P2YR12 in microglia also decreases in human AD tissue and mouse models of AD, where microglia instead take on disease associated transcriptional profiles. 124 This is supported by recent work showing activated CD68+ microglia around vessels with higher grades of CAA pathology in human tissue. 12 It is likely that microglia switch from supporting vascular function to contributing to some perivascular inflammation, however it is unclear if their role for protecting against CAA pathology diminishes over time in ageing populations or individuals at risk for developing AD. Additionally, it is possible that the perturbations in microglia function in controlling Aβ pathology could be more problematic for capillary pathology and function, however studies have yet to address this.
In recent single-cell transcriptomic studies on human and mouse AD tissue, the disease-associated microglia (DAM) commonly found in other diseases 125 are present and appear to be TREM2- (trigger receptor expressed on myeloid cells 2) dependent in the 5xFAD mouse model. 126 In human AD tissue, while they did find expression of genes indicative of DAM profiles (TREM2, MHCII, CD68 and APOE) they also reported expression of other genes, SORL1, A2M and CHI3L1, which are not typical of the DAM phenotype. 126 Interestingly, microglia with more homeostatic signatures (TMEM119, P2YR12, CX3CR1) were more abundant but also highly expressed interferon regulatory factor 8 (IRF8) which is known to be expressed by reactive microglia in peripheral nerve injury. 127 However, whether the typical DAM profile appears in CAA pathology is unknown, more studies are shifting in studying the major driver of DAM phenotypes like TREM2.
A therapeutic target of interest and known genetic risk factor for AD that may modulate microglia is TREM2. 128 Conflicting observations have been reported with regards to studying TREM2 deficiency in AD mouse models. Studies reveal that TREM2 deficiency impacts Aβ deposition, however the changes in Aβ pathology occur at different times and in some cases different directions,129,130 although it appears that later on in disease stage TREM2 deletion results in an increase in Aβ pathology. 131 The above studies did not specifically focus on how TREM2 deficiency may impact CAA pathology, as these studies had focussed on mouse models which favoured parenchymal plaque deposition of Aβ. In a CAA-related study, a TREM2 knockout mouse line was bred with the TgSwDI CAA mouse model. In TREM2 deficient CAA-prone mice (TgSwDI), Aβ deposition was increased compared to WT and TREM2 heterozygous mice. Alongside total Aβ load being increased, it appeared that TREM2 deletion resulted in a reduction in CAA. 132 Suggesting that TREM2 may affect vascular and parenchymal amyloid deposition in distinct ways. These findings were investigated further using single nucleus RNA sequencing analysis. Although microglia were activated, it was observed that TREM2 deletion trapped them in a transition, not fully reactive state. Additionally, a subset of perivascular macrophages in the TREM2 knockout mice were also observed, with upregulation observed in pathways associated with immune function, protein synthesis and cell junction pathways, suggesting possible changes to BBB integrity. TREM2 deletion also impacted other cells within the perivascular environment, including smooth muscle cells, pericytes and astrocytes. 132 Together, these findings highlight that alterations to TREM2 can impact many aspects of the perivascular environment. TREM2 targeting antibodies, as well as Aβ targeting antibodies are both therapeutic focusses, yet this work suggests that targeting TREM2 may impact many aspects of the perivascular environment. Some trials have already been halted due to moderate haematologic effects133,134 and the INVOKE-2 clinical trial failed to slow AD progression. 135 It will be interesting to see the results of other clinical trials targeting TREM2, 136 however it is still unknown if targeting TREM2 will be effective in patients with CAA.
Influence of peripheral immune cells (T cells, neutrophils and monocyte-derived macrophages) on the perivascular environment in CAA
Peripheral immune cells, including lymphocytes (such as T and B cells), myeloid cells (such as monocytes, macrophages, neutrophils) and other immune cells like natural killer (NK) cells all play an important role in regulating homeostasis and protecting the body from invading pathogens, ultimately playing roles in both innate and adaptive immune responses. Roles for these cells in the pathophysiology of CAA, especially for passing the threshold for developing CAA-ri is becoming increasingly important to understand as possible contributors and therapeutic targets.
Neutrophils
Emerging evidence suggests a link between peripheral immunity and the development of brain disorders including AD. In a prospective study using data from the UK Biobank a relationship was observed between an increase in levels of innate immune markers (neutrophils, neutrophil–lymphocyte ratio (NLR), platelets, monocytes and systemic-immune-inflammation index (SII)) and an elevated risk of developing a brain disorder. Of these markers, neutrophils had the greatest significant correlation with the risk of developing dementia and stroke. 137 Thus, highlighting a potential link between peripheral immunity and the development of certain brain disorders. However, this study was unable to assess the link between these innate immune markers and CAA. But, a more recent study investigated immune cell infiltration, using peripheral blood in CAA and insomnia. When comparing CAA to age and sex matched controls, they observed an increase in monocyte levels as well as a decrease in the amount of activated NK cells, 138 suggesting CAA may be associated with an inflammatory response. However, this analysis was only conducted on 11 CAA patients and 11 controls and only two of the immune cell types (of the 14 included in the statistical analysis) were significantly different from the controls. Thus, caution should be taken, as studies with larger samples will need to be conducted to establish the strength of these findings. To further establish how CAA may be linked to peripheral immune changes, mechanistic experimental studies will need to be conducted. Yet, these findings do add to the previous work suggesting inflammatory responses may be an important factor to consider in the pathophysiology of CAA.
Neutrophils, in particular, have been linked to the progression of CAA (Figure 1(c)). In a recent study, neutrophils were found in human AD brains as well as the APP/PS1 mouse model of AD with their localisation most prominently along the vasculature with some having infiltrated into the brain parenchyma. This was associated with BBB leakage as well as neutrophil extracellular traps (NETs) which are web-like structures created by neutrophils that are important to trap pathogens and protect against infection. 139 However, NETs have also been attributed to the development of autoimmune disorders as well as the occlusion of the vasculature. 140 Future studies could focus on the recruitment of neutrophils and the formation of NETs as a possible contributor to vascular damage in CAA as well as the sudden inflammatory responses observed in CAA-ri and/or ARIA.
Neutrophils have also been shown to impact cerebral blood flow in both APP/PS1 and 5xFAD models of AD. Two-photon imaging revealed an increased number of ‘stalled’ capillaries in these models compared to WT mice, with the majority of these stalled capillaries showing positive labelling of leukocytes. Treatment with an antibody against Ly6G (a neutrophil surface marker) led to a reduction in neutrophils, the number of stalled capillaries, and an increase in blood flow coupled with improved short-term memory. Most interestingly, administering anti-Ly6G for a month resulted in the reduction of Aβ40 but not Aβ42. 141 Therefore, the study poses the question, could reducing neutrophils result in an increase in Aβ clearance, and could this potentially impact vascular amyloid deposition? Although, it is important to note that the neutrophils targeted within this study were in the vasculature, not within the brain parenchyma. This study highlights the potential role of targeting neutrophils in diseases of amyloidosis in order to improve cerebral blood flow and vascular Aβ accumulation.
In a separate study using 5xFAD and the 3xTg-AD models, neutrophils were also implicated in amyloid pathology. Authors observed a higher expression of vascular adhesion molecules at 4 and at 6 months in 5xFAD and 3xTg-AD models, respectively. Accordingly, an increase in CD45+ immune cells and CD18+ cells, an integrin beta-2 subunit important for cell adhesion were mainly stuck in the meningeal and cortical vessels, at early ages and this was associated with cognitive impairment. The authors suspected the majority of the vascular associated cells migrating into the parenchyma were neutrophils given expression of the neutrophil marker, Gr1. This was confirmed using immunohistochemistry and in vivo two photon imaging demonstrating neutrophils sticking to the endothelium and migrating into parenchymal tissue. In parallel, they also observed the presence of neutrophils adhering to blood vessels and within the parenchyma in human AD postmortem tissue. 142 Although these studies suggest an important role for neutrophils in amyloid related diseases it would be helpful for studies like these to be completed in more CAA-prone models, to assess the role of neutrophils in vascular Aβ accumulation and the inflammatory progression in CAA-ri and or ARIA. Further, recruitment of neutrophils in neurological diseases is widely reported around capillaries and venules, however involvement of arterioles is not clearly demonstrated or ruled out. There are still many outstanding questions regarding how neutrophils could be targeted in CAA pathology. However, studies investigating the role of neutrophils in experimental models of ICH (which CAA can result in) have shown that reducing the number of neutrophils (prior to ICH) can reduce BBB damage, axonal injury and reduce astrocytic and macrophage responses in the surrounding area of the haematoma. 143 Thus, highlighting that reducing neutrophil activity in CAA may improve the health of the perivascular environment.
T cells
As for other peripheral immune cells like T cells, very little is known about their presence in the perivascular space and influence on CAA progression. Recently, CD8 effector T cells were found in the CSF of patients with AD. 144 In a CAA-ri case study, immunoreactive T cells were observed around Aβ burdened vessels and the subarachnoid space. 24 Their role within the perivascular space is not known, however it is speculated that T cells may trigger adaptive immune responses due to the exposure to foreign proteins like, Aβ and other CNS specific molecules. Therefore, they may play a substantial role in the sudden inflammatory profile observed in CAA-ri or ARIA. To our knowledge the presence and/or role of B cells or NK cells in the perivascular space in CAA, CAA-ri or ARIA has not been solidified. Thus, whether they play a role in CAA progression and related inflammatory responses is unclear.
Monocytes and monocyte-derived macrophages
Monocytes, were found to be elevated in a study using data from the UK Biobank, investigating associations between peripheral immunity and the development of brain disorders. 137 Monocytes are circulating undifferentiated cells that localise to tissues and either differentiate into macrophages or dendritic cells depending on the inflammatory cues. 145 Heightened numbers of monocytes and macrophages are observed in CAA brain tissue, however the specific role of monocyte-derived macrophages in perivascular inflammation and CAA pathology is poorly understood. 146 Studies by Hu et al., showed that monocyte-derived macrophages, along with BAMs and microglia, secrete migrasomes upon exposure to Aβ40. Migrasomes are extracellular vesicles that are produced during cell migration and contain complement associated proteins like CD5L. They find these CD5L-containing migrasomes attach to endothelial cells and elicit BBB leakage in the TgSwDI/B CAA mouse model. 147 It is unclear if monocyte-derived macrophages play a unique role in CAA pathology, however it appears there is an interplay between them and perivascular macrophages in potential ARIA events. For example, the anti-Aβ immunotherapy (3D6) leads to the formation of an antibody immune complex with vascular Aβ deposits. This activates CD169+ perivascular macrophages resulting in a robust immune response and infiltration of inflammatory monocytes around vascular Aβ which is also associated with haemosiderin deposits indicative of microbleeds. 20 These studies suggest that monocyte-derived macrophages likely heighten CAA pathology and contribute to perivascular inflammation, that is, indicative of CAA-ri and/or ARIA.
Blood–brain barrier impairment in CAA
Studies on human post-mortem brain tissue have been essential in assisting with our understanding of the potential pathological stages of CAA. BBB impairment could provide unregulated entry of circulating immune cells into the perivascular space and brain parenchyma in CAA and studies on human post-mortem brain tissue have been essential in assisting in our understanding of this. Samples can also be taken in life, during surgical biopsy, however, this is only completed to get a definitive diagnosis 8 and is often completed for suspected CAA-ri cases. 148 Although studies have shown that in vivo imaging may be just as effective at diagnosing CAA-ri. 149 Through these approaches, the pathological progression of CAA alongside BBB leakage and immune responses have come further into light.
Numerous studies have suggested that BBB leakage may be associated with CAA.150,151 The BBB is mainly established by the endothelial cells creating the lumen of the vessel where adherens and tight junctions are crucial to preventing leakage of blood components into the brain parenchyma. This is further supported by the presence of mural cells and astrocytes surrounding the endothelium. 152 One study revealed that the presence of Aβ40 on isolated rat microvessels resulted in changes to the BBB, including a reduction in the tight junction proteins, claudin 1 and 5 as well as an increase of some matrix metalloproteinases (MMPs) including MMP 2 and 9 (Figure 2). Similar findings were also observed in microvessels of a transgenic mouse model (hAPP), with increased vessel permeability alongside reduced expression of claudin 1 and 5 and increased MMP 2 and 9 expression, 151 evidencing that Aβ40 can result in BBB damage. Assessment of post-mortem tissue by Freeze et al., showed IgG and fibrin deposition on blood vessels, indicative of BBB leakage, in CAA cases. 153 Kozberg et al., took this one step further and related BBB leakage and perivascular inflammation in individual vessels using the Vonsattel vessel grade system, ranking vessels from grade 0 to 4, with grade 4 vessels being the most diseased vessels. 12 Additionally, they completed two investigations, one in targeted cases, whereby CAA disease was severe with samples having a higher density of cerebral microbleeds, and the second in non-targeted consecutive cases with differing degrees of CAA disease severity. In both targeted and non-targeted cases, they observed a greater number of fibrin positive vessels in higher graded vessels (grade 2 and 3) however, they also observed BBB leakage within early graded vessels (grade 0 and 1), indicating that BBB leakage may occur early on in disease pathophysiology. In the targeted cases, perivascular inflammation was seen to increase with vessel stage, as observed by a higher density of GFAP positive staining (reactive astrocytes) and CD68 (activated microglia) positive cells around advanced vessels, with this observation being most striking closer to the vessel. Interestingly, in non-targeted, consecutive cases with differing degrees of CAA disease severity, perivascular inflammation was still observed (as assessed with positive staining of reactive astrocytes) however there was no significant effect of activated microglia (CD68 staining) in these cases, with more variability in the staining in this cohort compared to the targeted cohort. These studies point to a possible progression of CAA pathology beginning with BBB leakage and ending with perivascular inflammation and vessel rupture.
Even though post-mortem tissue has been instrumental in understanding CAA pathophysiology, identifying early changes to the vasculature and factors that lead to dramatic inflammatory responses as seen in CAA-ri and ARIA will likely be limited due to higher probability of tissue collection at late-stages of disease. Thus, reverse translational studies including experimental studies using models of CAA will be crucial in elucidating these mechanisms.
Insights on perivascular inflammation in other neurological disorders
Perivascular inflammation occurs in many other neurological disorders and insight into these could provide a unique perspective to pathologies that are uniquely caused by Aβ and the resulting inflammatory responses. For example, in multiple sclerosis, immune infiltration of T cells, B cells and macrophages is heightened along post-capillary venule zones and venules in a phenomenon called perivascular cuffing. 154 Ischaemic stroke and reperfusion injury generally impacts perivascular environments around arterioles and capillaries with mainly endothelial activation and BBB disruption driving transient neutrophil, monocyte/macrophage infiltration that eventually subsides. 155 Traumatic brain injury results in more diffuse perivascular involvement of arteries, arterioles, capillaries and venules possibly due to heavy involvement of neutrophils, microglia and monocyte-derived macrophages. These types of injuries generally damage the meninges which could lead to more widespread and unique immune responses not typically observed in other disorders. 156 It will likely be important to contrast and compare the different vascular pathologies and associated immune responses among various neurological disorders with AD/CAA to open more opportunities in therapeutic development as well as understanding the unique pathophysiology Aβ has on the brain vasculature.
Synthesis of arteriolar perivascular pathologies and the immune landscape
Collectively, the multitude of studies so far demonstrate that CAA progression is intertwined with immunological changes within the perivascular space. Early endothelial and mural cell dysfunction along with basement membrane alterations may prime this niche for barrier dysfunction and altered homeostatic signalling. This may facilitate the activation of resident macrophages and microglia. As vascular pathology advances, recruitment of peripheral immune populations further modifies the local immune landscape while potentially amplifying vascular damage and dysfunction (Figure 3(a) and (b)). Dysregulation in these pathological events could pass the threshold for development of the inflammatory profiles observed in CAA-ri and ARIA. Thus, perivascular pathology and immunological changes are highly dynamic and interwoven processes. Efforts should be focussed on detecting and targeting these changes within the clinic to preserve vascular health.
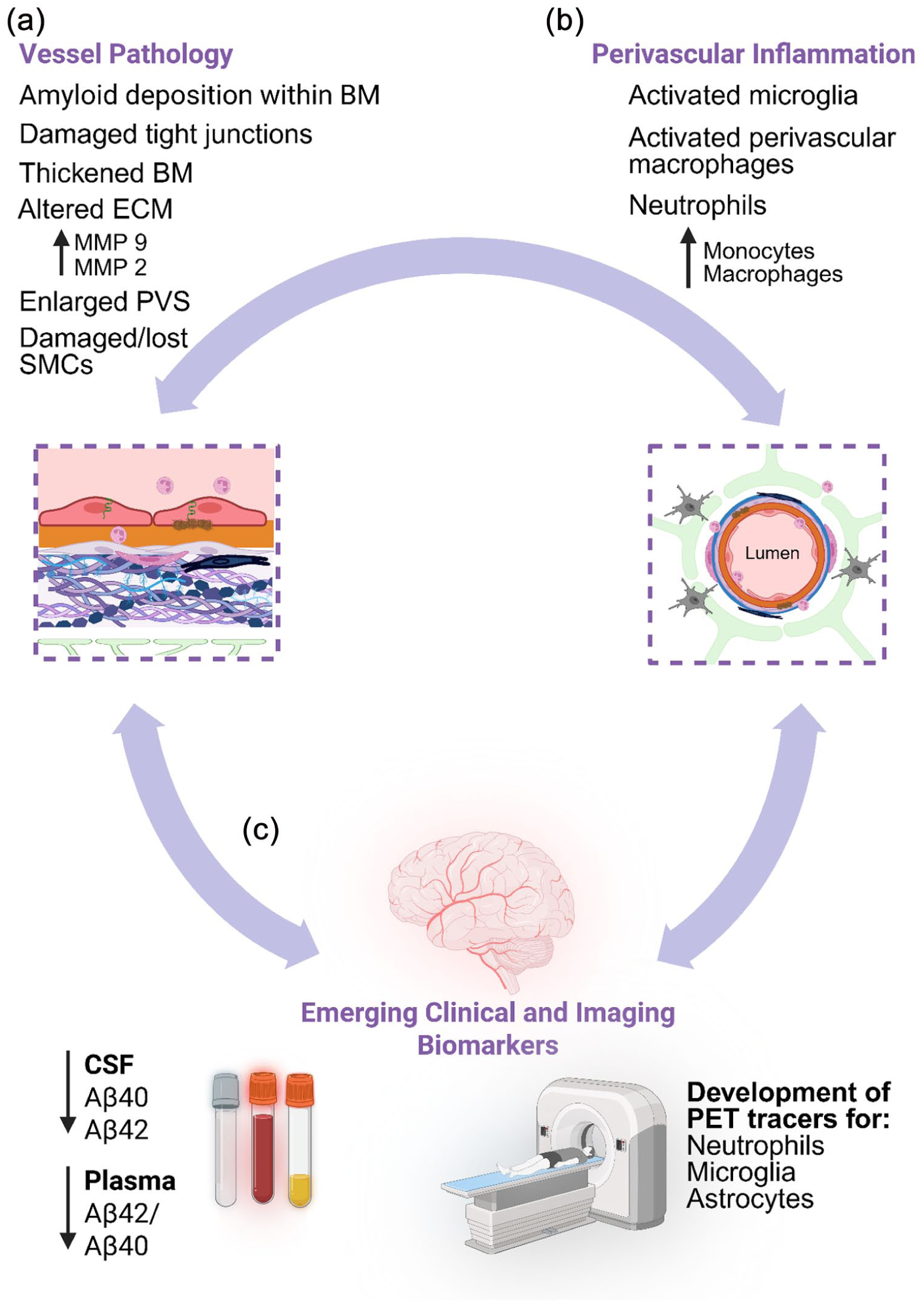
An illustrative schematic showcasing the vessel pathology, perivascular inflammation and emerging clinical and imaging biomarkers: (a) CAA heavily impacts the structure and function of blood vessels, with alterations to the BBB, ECM and a thickening of the BM. We do not fully understand if CAA results in perivascular inflammation or if it results in CAA but evidence highlights increased levels of perivascular inflammation in CAA, including activated microglia, activated astrocytes and increase in neutrophils along blood vessels and within the brain parenchyma, with some reporting an increase in monocytes and monocyte derived macrophages in CAA brain tissue (b). There is still a vast amount of discovery to be made relating to clinical and imaging biomarkers (c) however, CSF samples suggest that individuals with CAA have reduced amount of both Aβ40 and 42 and within the plasma a reduction in the ratio between Aβ42 and 40 is observed. There are still very few ways to ‘image’ inflammation in the brain in humans, however promising avenues could be the development of PET tracers, specifically focussed on neutrophils, microglia and astrocytes.
Clinical and imaging biomarkers for CAA and perivascular pathology
If treatments for CAA are to be aimed at reducing inflammation within the perivascular space, then it is integral that effective and valid clinical and imaging biomarkers are used, which will allow us to diagnose both CAA and inflammatory responses associated with CAA and allow for the monitoring of disease progression. Measuring proteins within the CSF has previously been utilised to measure levels of Aβ species and autoantibodies (Figure 3(c)). However, there is conflicting findings related to the use of autoantibodies to diagnose CAA-ri19,26 – suggesting this biomarker may not be appropriate to measure inflammatory responses relating to CAA-ri yet. However, studies have consistently highlighted that CAA is associated with a reduction in Aβ40 and Aβ42 within the CSF.157–159 Aβ40 is a more soluble isoform of Aβ that tends to accumulate on the vasculature, while Aβ42 primarily forms dense fibrils in the brain parenchyma. It is speculated that certain biochemical properties of Aβ40 favours interactions with vascular basement membrane proteins, such as perlecan, and the solubility of Aβ40 also allows for it to move within fluid-filled spaces more readily. It is likely these properties explain the species-specific accumulation along the brain vasculature and the reduction of Aβ40 in CSF is an indication of heightened vascular accumulation. 160 Additionally, other species (Aβ38, sAβPPα and sAβPPβ) have also been found to be lower in CAA patients than in control and AD patients in addition to the increase in neurofilament light chain (NfL), an indicator of neuronal degeneration. 159 Therefore, theoretically CSF could be an important clinical biomarker researchers and physicians could use in CAA therapeutic trials. However, CSF extraction is not without risk and is still an invasive way of collecting biological information from individuals.
Recently there has been increasing interest in the use of plasma biomarkers in both research and clinical trials, due to the ease and scalability of this method (Figure 3(c)). One study in individuals with probable CAA (diagnosed using the Boston 2.0 criteria) observed a reduced Aβ42/40 ratio, increased p-tau 181 and NfL, and similar GFAP levels as compared to controls. 161 The study highlighted that plasma biomarkers may be able to distinguish between individuals with CAA and healthy controls, which would assist with potential early detection, and monitoring of disease severity. Although, this study did not include an AD comparison group and as a small sample size was used additional studies are warranted in order for plasma biomarkers to be truly incorporated into CAA research practise.
The use of positron emission tomography (PET) imaging is a potentially useful way to track inflammatory cells like neutrophils and has been successfully used in a mouse model of AD where they observed increased neutrophil accumulation in the brain (Figure 3(c)). 162 However, technologies to image neutrophils in humans are limited and have yet to be applied in patients with AD, CAA or CAA-ri. 163 Additionally, imaging of astrocytes and microglia have received increasing interest in recent years. Studies have shown that tracers targeted at astrocytes may have promise, however more research is needed regarding microglia specific tracers. 164 PET imaging seems like a promising avenue for potentially tracking vascular Aβ, however it is generally in a more diffuse, soluble form and turns over quickly, 165 thus the development of an efficient PET tracer has been difficult. It will likely require multiple assessments, like pairing PET and CSF/plasma biomarker analyses to confidently diagnose individuals with CAA.
While tracking of inflammation and Aβ pathology using PET imaging is in its infancy, there are currently many MRI studies aimed at tracking CAA-associated pathologies to improve diagnosis and prognosis. The HIFI-CAA trial is employing highly frequent-monthly MRIs of CAA patients to capture cortical superficial siderosis and subarachnoid haemorrhage events to provide prognostic information. 166 Further, a meta-analysis of PET imaging studies using amyloid-binding tracers determined that amyloid-PET imaging could be a relatively suitable approach for differentiating patients with probable CAA from cognitively normal subjects and patients with deep ICH. 167 Recently, 18F-flutemetamol, which is used to detect fibrillar amyloid in PET imaging, assisted in reclassifying patients with either ICH or SAH into those with amyloid-driven CAA versus arteriolosclerosis where the CAA group had a higher incidence of recurrent haemorrhage events. 168
Opportunities for therapeutic targeting of the perivascular space in CAA
When considering treatments that target the perivascular environment, there are many challenges and questions to consider. One of our biggest challenges is accessing this closed environment due to the highly protective BBB. A major field of study in drug discovery is aimed at circumventing the BBB, 169 for example, by using nanoparticles, ultrasound to open the BBB and even by administering drugs into the CSF intrathecally. Some of these techniques are newer than others and thus are only in their early stages of development, whereas other techniques are more invasive. Thus, therapeutic avenues aimed at targeting cells within the perivascular environment also need to consider these administrative methods.
Additionally, the studies in this review have shown that the perivascular environment is a highly connected and linked environment, with impacts on one cell type likely to influence other cells within this environment. Thus, it’s important to consider how targeting one cell type may impact the environment as a whole. However, it is likely that any successful treatments for CAA and AD may need to be multifaceted in targeting multiple cells of this important environment. Spatial multi-omics approaches would be incredibly useful tools to aid in the understanding of how specific treatments may impact different cells within the perivascular environment. These tools may also give us detailed insights related to specific disease pathways to target.
Overall, it is evident that immune responses are integral in the pathophysiology of CAA, and it is likely that immune targets within the perivascular environment may be important therapeutic avenues. Diagnosing CAA early in individuals as well as stage of disease, especially regarding inflammation will be key. Identification of biomarkers for CAA and disease stages should likely be step one. This will allow for appropriate targeting of relevant immune cells, limiting of vascular instability and vascular Aβ accumulation. Both CSF and plasma biomarkers for CAA are being investigated, however, both are still in early stages of development, and large-scale studies are still warranted. The BATMAN clinical trial170,171 which is investigating the use of minocycline to limit inflammation in individuals with sporadic and hereditary CAA, will hopefully be informative for how targeting the immune system could alter CAA progression. Small studies in patients with probable CAA have indicated that this treatment is mostly tolerable and even showed a reduction in the recurrence of ICH (although findings from larger studies will be needed to clarify if this was due to the drug). 172 It would be especially interesting for reverse translational studies; to be conducted, whereby longitudinal imaging of vessels is conducted in CAA models treated with minocycline. Evidence suggests that this treatment can reduce haemorrhages in CAA mouse models, 173 however, it would be informative to establish how this treatment impacts differently graded CAA vessels. Studies like the BATMAN trial, reverse translation studies and the development of both CSF and plasma biomarkers will be crucial to tackle these important questions and challenges.
Footnotes
Acknowledgements
The authors thank members of the Brain clearance in CAA Leducq Foundation Network for their thoughtful discussions on these topics. Figures created in BioRender.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was funded by the Leducq Foundation (23CVD03) (BE) as well as R00AG080034, AARG-25-1488773 and VCID-UMD-26-1529753 to (SKB).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.