
Abstract
The blood–brain barrier (BBB) has long been regarded as a passive, protective filter for the brain. This review re-evaluates the role of brain microvascular endothelial cells (BMECs)—from viewing them as static structural components to recognizing them as dynamic immunoregulatory sentinels within the neurovascular unit. BMECs actively sense inflammatory and pathogenic signals through pattern recognition receptors, integrate competing pathways such as Wnt/β-catenin and NF-κB, and make context-dependent decisions to balance barrier integrity with immune defense. In response to activation, they orchestrate neuroimmune communication by secreting chemokines, modulating adhesion molecules, and facilitating immune cell trafficking. Dysregulation of these functions contributes to the pathogenesis of various central nervous system disorders, including stroke, Alzheimer’s disease, and multiple sclerosis, where BMECs dysfunction drives neuroinflammation and barrier breakdown. Further progress is required to resolve endothelial heterogeneity, develop precise tools for barrier function assessment, and define targeted interventions that modulate the immunoregulatory functions of BMECs in order to explain the mechanistic heterogeneity observed in neurological disorders. These efforts will deepen our understanding of the BBB’s role in physiology and pathology, offering new strategies for the prevention and treatment of neurological diseases, and highlight BMECs as active participants in brain immunity and potential therapeutic targets.
Keywords
Introduction
The stability of the human brain microenvironment relies on a precisely regulated physiological front—the blood–brain barrier. Traditionally, BMECs have been regarded as a selectively permeable physicochemical barrier: paracellular pathways sealed by tight junctions, nutrient transport mediated by transporters, and toxin clearance facilitated by efflux pumps. This classical paradigm positioned them as a passive interface between the central nervous system and peripheral circulation.
However, growing evidence reveals that BMECs are not just structural components but active immunomodulatory hubs within the neurovascular unit, deeply involved in various pathologies. Beyond forming a physical barrier, they maintain neuroimmune homeostasis via pattern recognition receptor expression, antigen presentation, cytokine secretion, and regulation of immune cell migration 1 . Under pathological conditions, BMECs initiate and amplify immune responses, shifting from passive “barrier victims” to active “immune regulators.”
This review aims to systematically synthesize recent advances in understanding the roles of BMECs in central nervous system diseases. By redefining the identity of BMECs across various pathological conditions from the perspective of their physiological properties and functions, this review reconceptualizes BMECs not as passive targets but as active contributors to disease progression.
Structural and functional characteristics of BMECs
Spatiotemporal heterogeneity of vascular endothelium
Vascular endothelium is not a homogeneous monolayer; instead, endothelial cells exhibit remarkable spatiotemporal heterogeneity in their structure. 1 Spatially, significant differences in structure and function exist not only across different organs and vascular segments but also between neighboring endothelial cells within the same vessel. For example, continuous endothelium (e.g. in large arteries) is rich in tight junctions, fenestrated endothelium (e.g. in renal glomeruli) facilitates filtration, and discontinuous endothelium (e.g. in hepatic sinusoids) allows macromolecular exchange. Functionally, the BBB exhibits the lowest baseline permeability, whereas induced permeability occurs predominantly in postcapillary venules; leukocyte adhesion molecules such as E-selectin are also preferentially expressed on venular endothelium. Temporally, endothelial phenotypes dynamically change during development, inflammation, or within the tumor microenvironment. This heterogeneity arises partly from transient induction by local microenvironmental cues (e.g. blood flow, cytokines) and partly from stable maintenance via epigenetic mechanisms (DNA methylation, histone modifications), and this feature is highly conserved throughout evolution.
Focusing on the specialized barrier structure of the BBB, recent single-cell and spatial transcriptomic studies have revealed the spatiotemporal principles of human BBB development. 2 Temporally, BBB transcriptional signatures (e.g. the transporter MFSD2A, the tight junction molecule CLDN5) begin to emerge at gestational week 8 (GW8); prior to this, brain endothelial cells lack BBB characteristics. Subsequently, distinct endothelial subpopulations gradually mature. Spatially, neural progenitor cells (NPCs) and neurons activate endothelial β-catenin signaling via CDH2 homotypic interactions, inducing the expression of BBB-specific transporters. Concurrently, NPCs secrete PDGFD to promote mural cell proliferation, whereas various signaling pathways between endothelial and mural cells (e.g. PDGFB, TGFB1) become enhanced after GW8 and exhibit regional heterogeneity, with stronger interactions in neuron-rich areas. Furthermore, the histone variant H2A.Z.1 regulates BBB-related genes through interaction with β-catenin; its deficiency leads to BBB leakage. These findings not only validate the spatiotemporal specificity of endothelial cells but also offer new targets for intervention in BBB developmental disorders.
Specialized structure and metabolic features of BMECs
As the innermost lining of capillaries, endothelial cells exhibit remarkable structural and functional specificity. BMECs display distinct characteristics compared to their peripheral counterparts. Reflecting their specialized functions, BMECs express unique types and quantities of ion transporters and receptors. They possess fewer fenestrations and pinocytotic vesicles but harbor more mitochondria than peripheral endothelial cells, indicating sustained high metabolic activity essential for normal brain function (Figure 1). 3 Consequently, disruptions in energy metabolism (e.g. due to hypoxia) impair this activity and can have profound consequences.
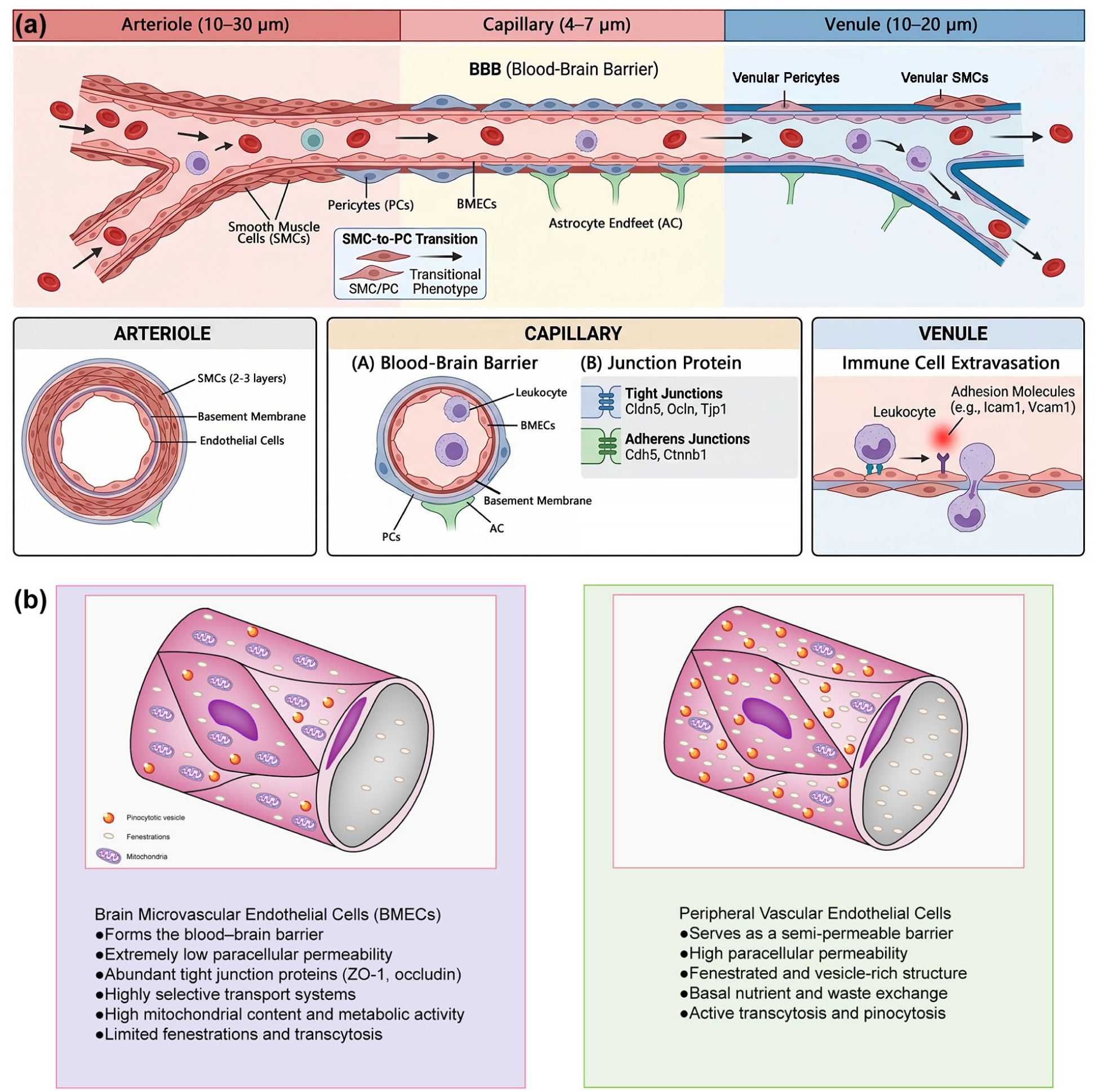
(a) Brain microvascular zonation and (b) comparison of the structural and functional features between BMECs and peripheral vascular endothelial cells. BMECs exhibit continuous, non-fenestrated endothelium with abundant tight junctions, forming the BBB and showing highly selective transport activity. In contrast, peripheral endothelial cells display fenestrated or discontinuous structure, higher permeability, and greater involvement in immune cell trafficking and general nutrient exchange. These distinct characteristics underlie their specialized roles in cerebral versus systemic vascular physiology. Integration of BMECs into the neurogliovascular unit.
A defining feature of BMECs is the formation of a continuous, non-fenestrated endothelium through highly specialized intercellular structures, including tight junction proteins such as ZO-1 and occludin. This structure severely restricts paracellular exchange and facilitates the timely elimination of harmful substances.4,5 Furthermore, BMECs express specific transport proteins that permit the passage of glucose, amino acids, and certain drugs, establishing a highly selective transport system fundamental to the cerebral circulation and constituting their crucial barrier function. Additionally, BMECs contribute to maintaining vascular tone and facilitate paracrine signaling. 6 Damage to BMECs and the consequent dysfunction of these roles can severely impact normal physiological processes.
Beyond their intrinsic properties, BMECs do not function in isolation; rather, they operate as an integral component of a larger multicellular complex—the neurogliovascular unit (NGVU). The NGVU is a functional assembly in the brain composed of BMECs, pericytes, astrocytes, microglia, and neurons. Its core functions include maintaining BBB integrity, regulating local cerebral blood flow to meet neuronal metabolic demands (neurovascular coupling), clearing metabolic waste, and defending against the invasion of peripheral pathogens and toxins. Different cellular components work synergistically through a sophisticated signaling network: BMECs recruit pericytes via the PDGF‑B/PDGFRβ pathway, promoting vascular stabilization and BBB maturation; astrocytes induce the expression of endothelial tight junctions through Wnt/β‑catenin and Shh signaling, and regulate water transport via polarized AQP4; microglia release factors such as VEGF‑C to assist vascular anastomosis and remodeling, while also maintaining synaptic homeostasis through signals like IL‑33. In hepatic encephalopathy, hyperammonemia and systemic inflammation downregulate PDGFRβ expression and induce TGFβ and MMP‑9, disrupting endothelial‑pericyte signaling, leading to tight junction degradation, BBB leakage, and accumulation of neurotoxins. 7 In the chronic phase of repetitive mild traumatic brain injury (rmTBI), although a compensatory increase in vascular density occurs locally, the lack of pericyte coverage on newly formed vessels and dysregulated PDGF‑B/PDGFRβ signaling result in prolonged arterial transit time and significantly reduced cerebral blood flow and vascular reactivity, indicating that impaired endothelial‑pericyte signaling integration is the structural basis for neurovascular uncoupling. 8 In summary, the homeostasis of the NGVU depends on the integrity of transcellular signaling networks, and targeting the repair of the endothelial‑pericyte signaling axis holds promise as a common strategy to ameliorate neurological deficits in the above‑mentioned diseases.
Although traditionally viewed as passive victims in inflammatory responses, BMECs are far from passive targets. Instead, they act as sentinels capable of active sensing and participation in immune responses. Given their direct contact with circulating blood, BMECs express a comprehensive repertoire of pattern recognition receptors to detect “danger signals” from pathogens or tissue damage. Crucially, upon activation, they initiate a complex, proactive response rather than merely undergoing passive injury. They upregulate specific adhesion molecules (e.g. ICAM-1, VCAM-1) and polarically secrete chemokines, actively guiding and even specializing the transendothelial migration of immune cells.9,10 They can process and present antigens, engaging directly in dialogue with T cells, and are themselves potent producers of inflammatory mediators. 11 In essence, BMECs serve as critical signal integrators and processing centers within the neuroimmune communication network, actively interpreting immune cues and determining whether, and how, the brain responds to peripheral or central inflammatory events.
BMECs as immune sensors
As the first line of defense for the central nervous system, BMECs undertake the critical role of surveilling the circulatory system for potential threats and preventing harmful substances from entering the brain parenchyma. This stringent surveillance system is orchestrated by an array of pattern recognition receptors (PRRs; Table 1). For instance, toll-like receptors (TLRs) detect both pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), thereby initiating immune responses. This capability is vital for the defensive function of the BBB, as it helps control inflammation, maintain barrier integrity, and promote tissue repair. The PRRs on these cells also participate in regulating angiogenesis required for tissue repair, although their dysregulation can contribute to pathologies such as atherosclerosis and neuroinflammation. Specifically, PRRs recognize highly conserved molecular patterns from PAMPs, as well as DAMPs—a class of endogenous ligands released during CNS inflammation, injury, or aging, alongside various disease-specific aberrant pathogenic molecules (e.g. Aβ in Alzheimer’s disease). Ligand-receptor binding subsequently activates downstream signaling pathways, leading to the synthesis and release of inflammatory factors.
Pattern recognition receptors and sensing pathways in BMECs.
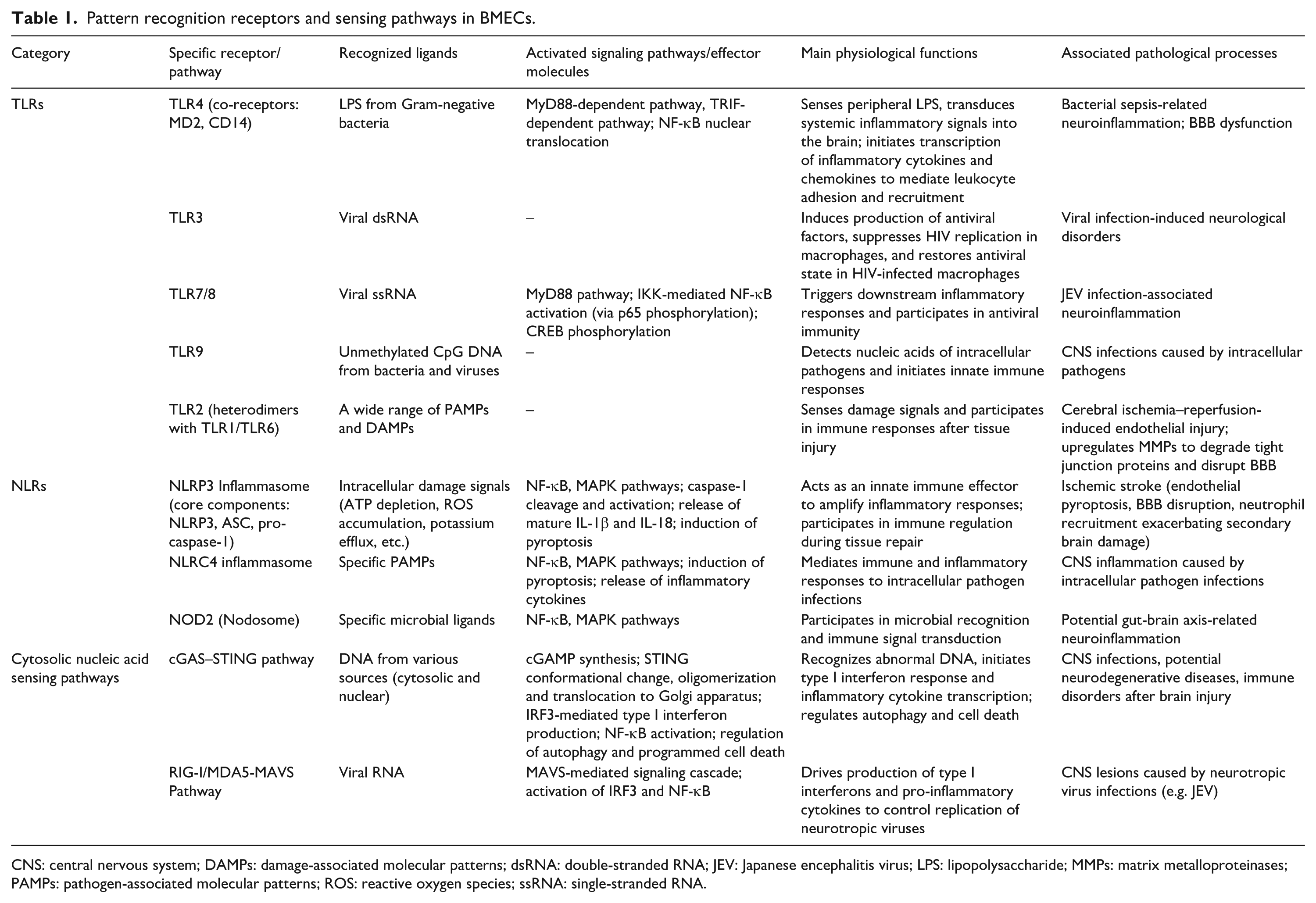
CNS: central nervous system; DAMPs: damage-associated molecular patterns; dsRNA: double-stranded RNA; JEV: Japanese encephalitis virus; LPS: lipopolysaccharide; MMPs: matrix metalloproteinases; PAMPs: pathogen-associated molecular patterns; ROS: reactive oxygen species; ssRNA: single-stranded RNA.
Toll-like receptors
TLRs represent the most extensively studied and functionally diverse family of PRRs, playing a cornerstone role in the immune surveillance conducted by BMECs. Notably, the TLR4 complex, together with its co-receptors MD2 and CD14, primarily recognizes lipopolysaccharide (LPS) from Gram-negative bacteria.12,13 While the traditional view held that LPS directly disrupts the BBB, emerging evidence positions BMECs as critical intermediaries that sense peripheral LPS and transduce systemic inflammatory signals into the brain. 11 During bacterial sepsis, LPS activates TLR4 on cerebrovascular endothelium. This engagement triggers both the MyD88-dependent and TRIF-dependent signaling pathways, leading to nuclear translocation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and subsequent transcription of various inflammatory cytokines and chemokines, thereby creating a favorable milieu for leukocyte adhesion and recruitment.14,15
TLR3, expressed on BMECs, recognizes double-stranded viral RNA, a function pivotal in the pathogenesis of various viral infection-induced neurological disorders. For instance, TLR3 signaling in human BMECs can induce the production of antiviral factors that suppress HIV replication in macrophages and help restore an antiviral state in HIV-infected macrophages. 16
TLR7/8 are responsible for sensing viral single-stranded RNA, whereas TLR9 detects unmethylated CpG DNA, which is commonly found in bacteria and viruses. These receptors are localized on endosomal membranes, ensuring effective detection of intracellular pathogens. Infection with Japanese encephalitis virus (JEV), for example, has been shown to upregulate the TLR7/MyD88 signaling axis, resulting in the activation of NF-κB via IKK signaling and p65 phosphorylation, as well as the phosphorylation-mediated activation of the cAMP response element-binding protein (CREB), collectively driving subsequent inflammatory responses.17,18
Notably, in most cell types (e.g. macrophages, neurons, hepatocytes), CREB is generally considered to exert anti‑inflammatory and pro‑survival functions. It induces the expression of the anti‑inflammatory cytokine IL‑10, suppresses NF‑κB transcriptional activity by competing for the shared co‑activator CBP/p300, and upregulates anti‑apoptotic proteins such as Bcl‑2, thereby limiting tissue damage and promoting cell survival. 19 However, in the unique context of cerebrovascular pericytes infected with JEV, CREB exhibits a pro‑inflammatory role. This striking functional duality can be attributed not only to differences in downstream pathways triggered by distinct stimuli (JEV activates the signaling axis via TLR7) but also to cell‑specific factors in pericytes, the involvement of TLR7‑downstream ERK signaling, and the amplifying effect of a positive feedback autocrine loop mediated by PGE2. These mechanisms collectively enable CREB to cooperate with NF‑κB in driving the expression of pro‑inflammatory factors such as IL‑6 and RANTES. 17
TLR2, capable of recognizing a broad spectrum of damage signals, typically forms heterodimers with either TLR1 or TLR6 on the cell surface to detect a wide array of PAMPs and DAMPs. It is currently implicated in endothelial injury following cerebral ischemia–reperfusion events, where its activation can upregulate the expression of matrix metalloproteinases (MMPs) in the cerebrovascular endothelium, consequently leading to the degradation of tight junction proteins.18,20,21
Nucleotide-binding and oligomerization domain like receptors (NLRs)
The NLR family represents one of the largest classes of pattern recognition receptors in the body. Upon recognition of their specific molecular patterns, NLRs are believed to undergo oligomerization, assembling into large signaling complexes. Prominent examples include the NLRP3 and NLRC4 inflammasomes, as well as the NOD2-associated “Nodosome.” These complexes activate key signaling pathways such as NF-κB and MAPK, induce pyroptosis, and promote the release of inflammatory cytokines including TNF-α, IL-1β, and IL-18, thereby mediating a cascade of downstream immune and inflammatory responses. 22
The most extensively studied NLR is NLRP3. The NLRP3 inflammasome, a crucial component of the innate immune system, is a multi-protein complex comprising three core components: the NLRP3 protein molecule, the apoptosis-associated speck-like protein containing a CARD (ASC), and pro-caspase-1. If TLRs function as alarm systems that signal potential damage and activate inflammatory pathways, the activation of NLRs represents a more direct effector mechanism. TLR-mediated activation of the NF-κB pathway enhances the transcriptional upregulation of NLRP3 and pro-IL-1β, which in turn facilitates the subsequent activation of the NLRP3 inflammasome. Inflammasome “activation” is primarily characterized by the cleavage and activation of caspase-1. The activated caspase-1 then processes pro-IL-1β and pro-IL-18 into their mature, highly inflammatory forms, concurrently triggering a form of inflammatory cell death known as pyroptosis.23–25
Numerous pathological conditions can activate the NLRP3 inflammasome in BMECs, inducing pyroptosis, disrupting the BBB, and amplifying the initial injury. A classic example is ischemic stroke. Cerebral ischemia and hypoxia directly lead to endothelial ATP depletion, reactive oxygen species accumulation, and potassium efflux, creating a perfect “storm” for NLRP3 inflammasome activation in these cells. Endothelial-derived IL-1β further recruits neutrophils, exacerbating secondary brain damage. 25
Cytosolic nucleic acid sensing pathways
Beyond the receptors on the plasma membrane and endosomes, the cytosol contains specialized defense systems capable of directly sensing nucleic acids. The cGAS-STING pathway has emerged as a major research focus in neuroscience. Upon DNA recognition, cGAS catalyzes the synthesis of the second messenger cGAMP, which then binds to and activates STING. Activated STING subsequently initiates the production of type I interferons via IRF3. cGAS can detect DNA from various sources, is distributed in both the cytosol and nucleus, and its activity is regulated by multiple factors. Following cGAMP binding, STING undergoes conformational changes, oligomerizes, and translocates to the Golgi apparatus. In addition to inducing type I interferon responses, it can also activate NF-κB, promote the transcription of inflammatory cytokines, and participate in processes such as autophagy and programmed cell death.26,27
RIG-I and MDA5 are key sensors for viral RNA in the cytosol. Upon binding to viral RNA, they initiate a signaling cascade through the mitochondrial antiviral signaling protein (MAVS), ultimately leading to the activation of IRF3 and NF-κB. This drives the production of type I interferons and pro-inflammatory cytokines, a response critical for controlling the replication of neurotropic viruses such as Japanese encephalitis virus. 28
Building upon this capacity for active immune sensing, a core function of BMECs lies in the precise integration and decision-making regarding diverse signals, which constitutes another critical dimension of their role as commanders of the neuroimmune interface.
Cooperation and crosstalk among immune sensors
The above-mentioned PRR pathways do not operate in isolation; rather, they form a tightly integrated network across spatial and functional dimensions. Taking TLR4, the most extensively studied receptor, as an example: upon activation, TLR4 at the plasma membrane initiates the MyD88 pathway, whereas following internalization into early endosomes it switches to the TRIF pathway to induce type I interferon synthesis and secretion. 12 This “internalization switch” critically determines the intensity and duration of the inflammatory response. Notably, the expression of CD14 itself is regulated by age-related circulatory factors: aged plasma upregulates Cd14 and multiple TLR‑related genes in BMECs, while young plasma partially reverses this aging transcriptional signature, 29 suggesting that systemic environmental factors influence BBB immune sensing by modulating the expression and distribution of PRRs.
At the inflammasome level, non‑canonical (caspase‑11‑GSDMD) and canonical TLR4 signaling are functionally coupled. Wei et al. demonstrated that circulating LPS, after internalization into BMECs via the LBP‑CD14 axis, directly activates cytosolic caspase‑11, which cleaves GSDMD to form membrane pores, leading to endothelial pyroptosis and a dramatic increase in BBB permeability. 30 This process is independent of TLR4‑dependent cytokines but requires CD14‑mediated LPS internalization, illustrating hierarchical synergy along the “recognition‑internalization‑effector” axis. Similarly, full activation of the NLRP3 inflammasome depends on a TLR‑provided “priming signal” (upregulation of NLRP3 and pro‑IL‑1β) and a second signal (e.g. ATP, ROS), forming a two‑signal checkpoint. 10
Furthermore, cooperation among immune sensors extends beyond the same cell type to different cell types. Leow‑Dyke et al. found that activation of neuronal TLR4 by LPS triggers the release of chemokines (e.g. KC/CXCL1), which act on BMECs to upregulate ICAM‑1/VCAM‑1 and promote neutrophil transmigration across the endothelium. 31 This indicates that even when BMECs themselves are not directly stimulated, TLR4 signaling in adjacent neurons can indirectly activate the endothelium via paracrine mechanisms, forming a “neuron‑endothelium” immune crosstalk. In summary, immune sensors in BMECs engage in multilayered crosstalk: TLRs determine signal direction through internalization/trafficking; CD14 serves as a key auxiliary molecule bridging extracellular recognition and intracellular effects; NLRP3 and GSDMD act as downstream executors initiating pyroptotic BBB disruption; and TLR4 signals from other cell types (e.g. neurons) can also indirectly regulate endothelial function. Analyzing these integrated mechanisms will help design precise intervention strategies targeting specific nodes (e.g. CD14, GSDMD) to mitigate BBB damage while preserving essential immune defense.
Integration of diverse signals by BMECs
To distinguish from the traditional view of BMECs as a passive barrier, we reconceptualize them as active homeostatic interveners and decision‑makers in response to stress. Previous descriptions have either portrayed BMECs as a structural barrier or merely listed individual immune molecules without a unifying logic. In contrast, we define BMECs as genuine decision‑makers that weigh opposing signals—such as the pro‑inflammatory NF‑κB pathway versus the barrier‑protective Wnt/β‑catenin and NRF2 pathways—and make calibrated choices that determine whether the brain mounts an immune response at the cost of transient barrier leakage or prioritizes barrier integrity by suppressing inflammation. This integrative perspective shifts the core question from “which molecules do BMECs express?” to “how do BMECs resolve conflicting signals to produce a functional outcome?” The framework takes four categories of continuous inputs: pathogen‑/damage‑associated molecular patterns (e.g. LPS, viral nucleic acids), inflammatory cytokines (e.g. TNF‑α, IL‑1β), homeostatic and metabolic cues (e.g. Wnt ligands, Ang‑1), and cellular crosstalk signals from pericytes, neurons and glia. Integrating these inputs yields three major output modes: a barrier maintenance mode (upregulation of tight junctions, suppressed transcytosis); an immune defense mode (chemokine secretion, adhesion molecule upregulation, and in extreme cases pyroptosis or EndoMT); and an intermediate reparative mode (TGF‑β‑mediated repair and vascular stabilization). By making this framework explicit, we provide a conceptual map that organizes complex and often contradictory observations of BMEC behavior across diseases, and highlights key signaling nodes as potential therapeutic control points.
Negative regulation of barrier integrity by pro-inflammatory signaling
The activation of pro-inflammatory signaling pathways often antagonizes those responsible for maintaining barrier function, thereby compromising BBB integrity. While this process may facilitate immune cell entry into the central nervous system, it concurrently exacerbates neuroinflammation. The NF-κB pathway is a canonical pro-inflammatory cascade, whereas the Wnt/β-catenin pathway sustains BBB integrity by regulating BMECs differentiation and tight junction protein expression. 32 Studies indicate that LPS or pro-inflammatory cytokines activating the NF-κB pathway can suppress Wnt/β-catenin signaling in BMECs.33,34 Furthermore, LPS activates the caspase-4/11-GSDMD pathway in BMECs, increasing plasma membrane permeability and facilitating the exchange of intra- and extracellular substances. 30 Excessive GSDMD activation also triggers BMECs pyroptosis, further disrupting the BBB (Figure 2).
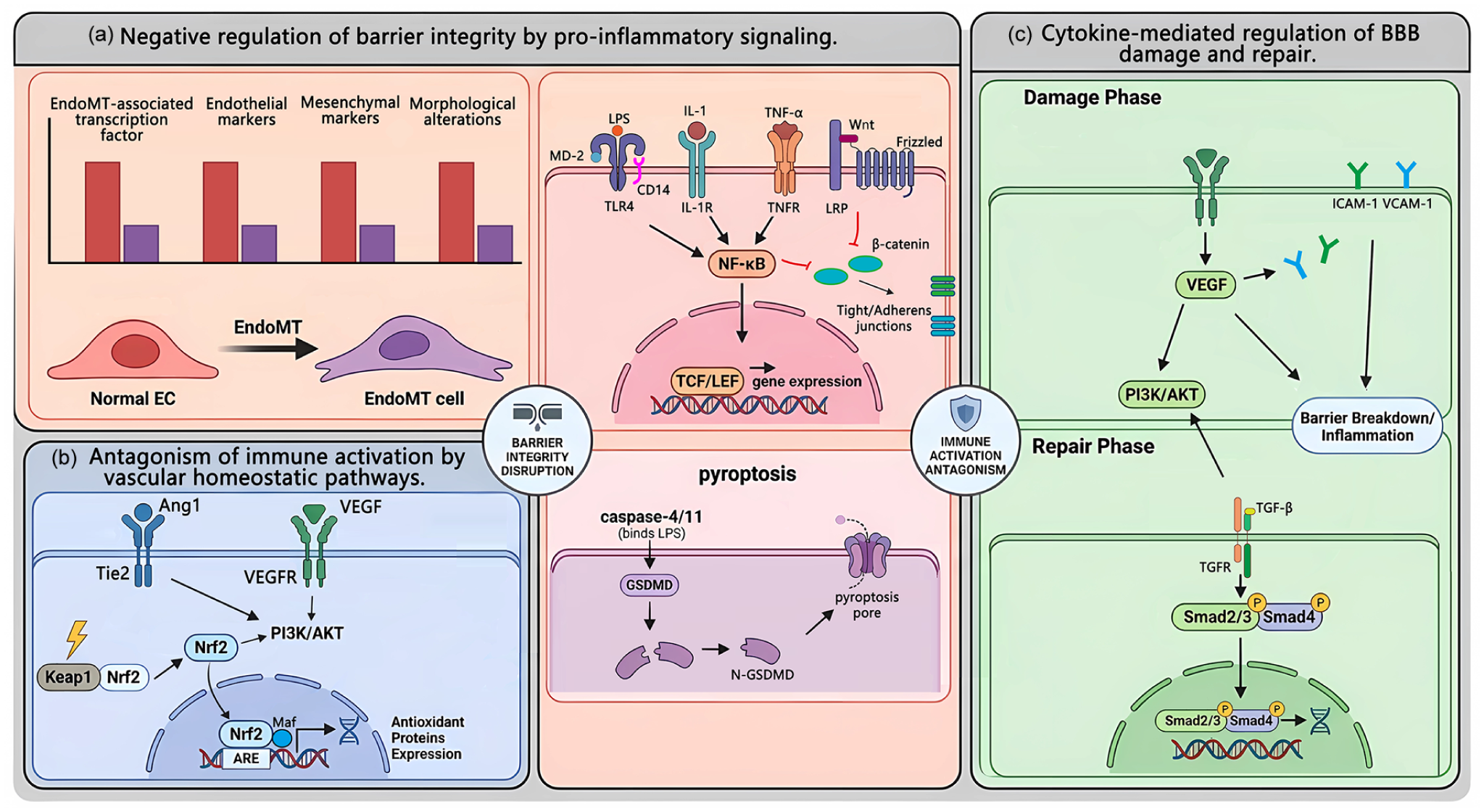
(a) Negative regulation of barrier integrity by pro-inflammatory signaling. Stimulation of various membrane receptors—including the LPS-sensing complex (TLR4/MD-2/CD14) by LPS, IL-1R by IL-1, and TNFR by TNF-α—activates the NF-κB pathway, initiating inflammatory responses and contributing to endothelial injury and barrier impairment. NF-κB signaling further suppresses the Wnt/β-catenin pathway in BMECs, disrupting intercellular junctions and directly compromising barrier function. LPS can also trigger endothelial pyroptosis via the caspase-4/11–GSDMD pathway. Additionally, inflammatory cytokines promote EndoMT, (b) antagonism of immune activation by vascular homeostatic pathways. Under hypoxic stress, activation of the Nrf2 pathway in BMECs induces downstream protective responses, including the SLC7A11/GPX4 axis to inhibit ferroptosis and the PI3K/AKT pathway to suppress inflammation. Ang‑1, secreted by pericytes and astrocytes, binds to the Tie2 receptor on BMECs, promoting vascular stability and barrier function. Activated Tie2 signaling counteracts vascular leakage induced by VEGF and inflammatory cytokines, thereby protecting the BBB, and (c) cytokine-mediated regulation of BBB damage and repair. During the inflammatory phase, activated endothelial and glial cells produce VEGF, which directly increases BBB permeability by disrupting intercellular junctions, upregulates ICAM‑1 and VCAM‑1 expression, and stimulates chemokine production, collectively promoting immune cell adhesion and migration. In the repair phase, TGF‑β binds to its receptor TβR and activates the Smad pathway, modulating inflammatory responses and facilitating tissue repair.
In addition to pathways that suppress protective mechanisms and induce apoptosis, inflammatory factors can also promote endothelial-to-mesenchymal transition (EndoMT). EndoMT is a pathological process in which BMECs lose their specialized functions and dedifferentiate into mesenchymal-like cells. This transition is characterized by an upregulation of EndoMT-associated transcription factors (TFs), downregulation of brain endothelial markers, upregulation of mesenchymal markers, and accompanied by morphological alterations such as cytoskeletal reorganization. Studies have demonstrated that stimulation of BMECs with TGF-β1 and IL-1β accelerates the EndoMT process (Figure 2). 35
Collectively, these findings demonstrate that BMECs actively participate in the negative regulation of the BBB under the influence of inflammatory signals.
Antagonism of immune activation by vascular homeostatic pathways
To counteract the detrimental effects of inflammatory responses on vascular function, intrinsic homeostatic signaling pathways in BMECs serve to oppose inflammation and maintain stability. Key protective homeostatic signals include the NRF2 pathway, a primary cellular defense mechanism against oxidative stress. Under basal conditions, Nrf2 is predominantly retained in the cytoplasm by its binding partner Keap1, maintaining low expression levels. 36 During ischemic stroke, excessive oxidative stress triggers the dissociation of Nrf2 from Keap1, allowing its translocation to the nucleus where it binds to antioxidant response elements to activate downstream pathways. 37 These include the SLC7A11/GPX4 pathway, which inhibits ferroptosis, 38 and the PI3K/AKT pathway, which suppresses inflammatory responses (Figure 2). 39
Angiopoietin-1 (Ang-1), secreted by pericytes and astrocytes, binds to the Tie2 receptor on BMECs, promoting vascular stability and barrier function. Activated Tie2 signaling counteracts vascular leakage induced by vascular endothelial growth factor (VEGF) and inflammatory cytokines, thereby conferring protection to the BBB.40,41
Cytokine-mediated regulation of BBB damage and repair
VEGF is a potent pro-angiogenic factor and a well-established mediator of increased vascular permeability. 42 In neuroinflammation, activated BMECs and glial cells can produce VEGF. VEGF not only directly increases BBB permeability by disrupting intercellular junctions but also upregulates ICAM-1 and VCAM-1 expression and stimulates chemokine production, thereby actively promoting immune cell adhesion and migration (Figure 2). 43
During the later stages of inflammation, distinct immune cell populations secrete pro-repair factors. For example, alternatively activated macrophages and regulatory T cells produce transforming growth factor-beta (TGF-β), which not only suppresses the expression of pro-inflammatory genes but also facilitates pericyte recruitment and extracellular matrix deposition, contributing to BBB repair and scar formation (Figure 2).44,45
Key regulatory nodes integrating inflammatory and homeostatic pathways
To help the reader navigate the complex signaling network, we highlight three key regulatory nodes that represent the balance between immune defense and barrier preservation. The first node is the NF‑κB and Wnt/β‑catenin antagonism, where activation of NF‑κB by inflammatory stimuli suppresses Wnt/β‑catenin signaling, leading to downregulation of tight junctions and increased permeability; this node determines whether the barrier remains intact or becomes leaky. The second node is the NRF2‑controlled oxidative stress checkpoint, which counteracts NF‑κB‑driven inflammation and pyroptosis. When NRF2 is activated, it induces antioxidant programs (SLC7A11/GPX4) and inhibits ferroptosis, whereas failure of NRF2 permits ROS accumulation and NLRP3 inflammasome activation, tipping the balance toward endothelial damage. The third node is the Tie2/Ang‑1 and VEGF axis, where pericyte‑derived Ang‑1 stabilizes the endothelium and opposes the pro‑permeability and pro‑inflammatory effects of VEGF. Collectively, these three nodes serve as hubs where competing signals converge, and their relative activity dictates whether BMECs adopt a barrier‑protective, immune‑defensive, or reparative phenotype. Recognizing these nodes provides a roadmap for designing interventions that selectively modulate BMEC function without abolishing essential immune surveillance.
Therefore, BMECs are far from passive signal recipients. By integrating antagonistic pathways such as NF‑κB versus Wnt/β‑catenin, the NRF2 checkpoint, and the Tie2/Ang‑1‑VEGF axis, they actively weigh “barrier maintenance” against “defense initiation.” The outcome of this integration directly determines the course of the neuroimmune response. This process extends beyond signaling and translates into concrete proactive defense behaviors, transforming BMECs from a “decision hub” into “defense executors.”
The proactive defense of BMECs
Recruitment of immune cells and trans-endothelial migration by BMECs
Upon stimulation, BMECs synthesize and secrete chemokines, forming the foundation of their active defense capabilities. Studies have demonstrated that bEnd.3 cells significantly increase production of CCL2 and CXCL1 following pro-inflammatory cytokine stimulation. 46 Furthermore, CCL2, CCL5, CCL20, and IL17 facilitate Th17 cell migration across bEnd.3 monolayers, indicating that BMECs actively recruit specific immune cell subsets, ultimately exacerbating BBB disruption through enhanced immune infiltration. Additionally, BMECs promote CD8+ T cell infiltration into the brain via CXCL12 secretion, participating in immune regulation. Targeted inhibition of the CXCL axis represents a potential therapeutic strategy for suppressing immune infiltration in the central nervous system.46,47
Guided by chemokine gradients and upregulated endothelial E-selectin and P-selectin, circulating leukocytes initiate the initial rolling phase. 48 Subsequently, high-affinity interactions between endothelial ICAM-1/VCAM-1 and leukocyte integrins mediate firm adhesion. 49 BMECs dynamically regulate leukocyte transmigration by modulating the phosphorylation status and subcellular localization of junctional proteins. For instance, with participation of junctional adhesion molecules such as JAM-A, BMECs can guide leukocytes through the paracellular pathway. When intercellular junctions are particularly tight, they may alternatively initiate the transcellular pathway, permitting leukocytes to traverse directly through the BMECs body. 50
Antigen presentation
BMECs not only function as sentinels of innate immunity but also serve as potential bridges connecting innate and adaptive immunity. They constitutively express MHC class I molecules, enabling them to present endogenous antigens to CD8+ T cells. When infected by intracellular pathogens, BMECs can directly present pathogen-derived antigens to cytotoxic T cells, thereby becoming targets for recognition and elimination by CD8+ T cells (Figure 3). Research indicates that MHC I-mediated antigen presentation at the BBB provides a “stop signal” to CD8+ T cells, inhibiting their migration and promoting their retention on the luminal side. T cells that recognize cognate antigens become arrested and persistently scan the endothelial surface, ultimately leading to endothelial apoptosis and disruption of the BBB monolayer. 51 Furthermore, under stimulation by inflammatory cytokines such as IFN-γ, BMECs can be induced to express MHC class II molecules and co-stimulatory molecules. This equips them with the potential to present exogenous antigens to CD4+ T cells (Figure 3). 52
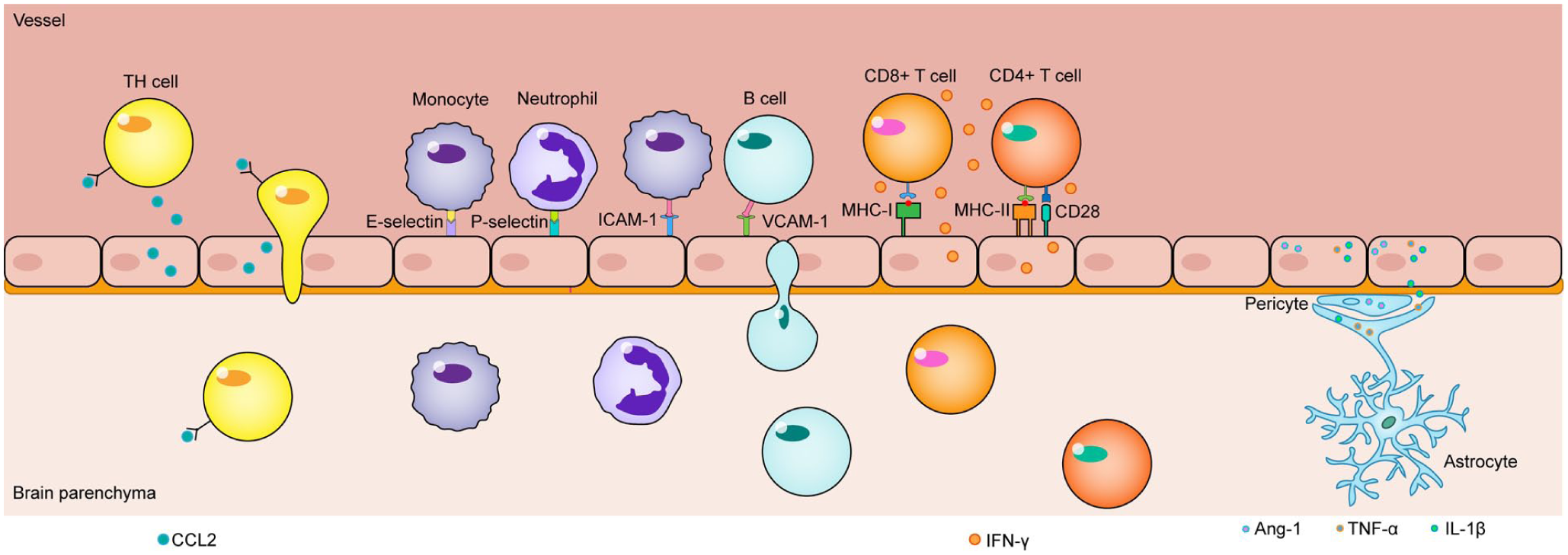
Upon stimulation by pro-inflammatory cytokines, BMECs secrete chemokines (e.g. CCL2) into the vascular lumen, which directs the migration of Th17 cells towards the tissue. Guided by this chemokine gradient and the upregulated expression of E-selectin and P-selectin on BMECs, leukocytes initiate tethering and rolling along the vessel wall. Firm adhesion and subsequent transmigration are mediated by high-affinity interactions between endothelial adhesion molecules (ICAM-1 and VCAM-1) and their integrin counterparts on leukocytes. BMECs constitutively express MHC class I molecules, enabling the presentation of endogenous antigens to CD8+ T cells. Under inflammatory conditions (e.g. upon IFN-γ stimulation), they can also upregulate MHC class II and co-stimulatory molecules, allowing for the presentation of exogenous antigens to CD4+ T cells. This local immune response is amplified by pro-inflammatory mediators (such as TNF-α and IL-1β) released from activated microglia and astrocytes. In contrast, pericytes attempt to restore homeostasis and exert negative regulation, in part through the secretion of stabilizing factors like Ang-1.
Synthesis and regulation of complement components
BMECs synthesize various complement components, including C3, C5, and Factor B. Activated complement fragments serve as potent chemoattractants, while the membrane attack complex can directly perforate the BMECs membrane, leading to cytolysis and complete loss of barrier function. 53 Research indicates that C3/C3aR activation may trigger intracellular Ca2+ release, subsequently activating calcium/calmodulin-dependent kinase. This kinase then phosphorylates the myosin light chain motor protein at Ser19, promoting stress fiber formation and ultimately disrupting VE-cadherin integrity and BBB function. Consequently, inhibition of C3 and modulation of the complement C3/C3aR pathway have emerged as promising therapeutic targets for ischemic stroke. 54
Bidirectional immune communication within the neurovascular unit
Beyond antigen presentation, BMECs engage in multifaceted immune communication with other cells in the neurovascular unit. Endothelial-derived IL-1β and CCL2 serve as potent activation signals for microglia (Figure 3). During systemic inflammation, endothelial-derived IL-1β has been identified as the initial trigger driving microglial morphological changes and pro-inflammatory cytokine production. 55 Activated BMECs also release VEGF and MMPs, remodeling the basement membrane covered by astrocytic endfeet and altering astrocyte gene expression toward a pro-inflammatory phenotype.56,57 Activated microglia and astrocytes in turn produce substantial amounts of TNF-α and IL-1β, further amplifying endothelial immune responses and establishing a positive feedback loop that sustains inflammation.58,59 In contrast, pericytes attempt to stabilize the endothelium through secretion of factors such as Ang-1 (Figure 3), exerting negative regulatory influence. 60
Transcytosis and clearance mechanisms of BMECs
The transcytosis and clearance mechanisms of BMECs primarily consist of receptor‑mediated transcytosis (RMT), adsorptive‑mediated transcytosis (AMT), and the ATP‑binding cassette (ABC) efflux system. 4 RMT represents a key pathway for BMECs to clear toxic proteins such as Aβ: LRP1 on the basolateral side binds Aβ, followed by clathrin‑ and PICALM‑dependent endocytosis to form vesicles, which are then sorted via the Rab5/Rab11 pathway and transcytosed to the luminal side for release, thereby achieving brain‑to‑blood efflux. 61 Conversely, RAGE on the luminal side mediates the reverse influx of circulating Aβ, exacerbating pathological deposition. 4 In addition, the transferrin receptor (TfR) is a classic RMT target; anti‑TfR antibodies can deliver therapeutic molecules into the brain via clathrin‑mediated transcytosis. 62 AMT does not rely on specific receptors but rather triggers endocytosis through charge‑ or glycosylation‑dependent interactions. For example, cationized albumin is transported across the BBB via the caveolin‑1 (Cav1) pathway. Under physiological conditions, Mfsd2a actively suppresses caveolae formation by altering membrane lipid composition, thereby limiting AMT and non‑specific transcytosis. The ABC efflux system, represented by P‑glycoprotein (P‑gp/ABCB1), is localized on the luminal membrane and uses ATP to pump drugs, toxins, and Aβ back into the bloodstream. In Alzheimer’s disease, downregulation of P‑gp function leads to impaired Aβ clearance. The clathrin and caveolin pathways exhibit distinct preferences in RMT and AMT: TfR and LRP1 primarily engage the clathrin pathway, whereas certain forms of AMT and transcytosis under pathological conditions depend on Cav1.
In pathological conditions, these transcytosis pathways are frequently hijacked or dysregulated. Recent studies have revealed diverse strategies by which pathogens exploit BBB transport systems. Neonatal meningitis Escherichia coli, group B Streptococcus, and Streptococcus pneumoniae co-opt TfR transcytosis by fusing their bacteria‑containing vesicles with TfR vesicles via a TRAF3–RalA–exocyst complex; this fusion depends on SEC6–SNAP23 interaction, while basolateral release requires VAMP3 and syntaxin 4.63,64 Zika virus and S. pneumoniae can also enter BMECs via caveolae‑mediated AMT, with heterogeneous expression of virulence factors regulating their intracellular fate and transcytosis efficiency.65,66 In type 2 diabetes, monocytes release non‑enzymatic pro‑cathepsin D, which binds endothelial LRP1, upregulates Cav1, and abnormally enhances caveolae‑mediated transcytosis, leading to BBB leakage and cognitive impairment. 67 These examples illustrate how BMECs can be subverted from their protective roles under pathological stimuli.
In summary, during immune responses, BMECs not only control gateway access for immune cells but also actively transmit immune signals through multiple mechanisms. Furthermore, they modulate the intensity and scope of inflammatory reactions through diverse pathways. This functional evolution redefines their role from traditional gatekeepers to strategic commanders of inflammatory responses at the BBB.
However, when these physiological immune surveillance and defense functions become dysregulated or are subjected to excessive, sustained pathological stimuli, BMECs can transform from protectors into drivers of injury, deeply participating in the pathological processes of various central nervous system disorders (Table 2).
Core pathological mechanisms and therapeutic directions of BMEC dysfunction in CNS diseases.
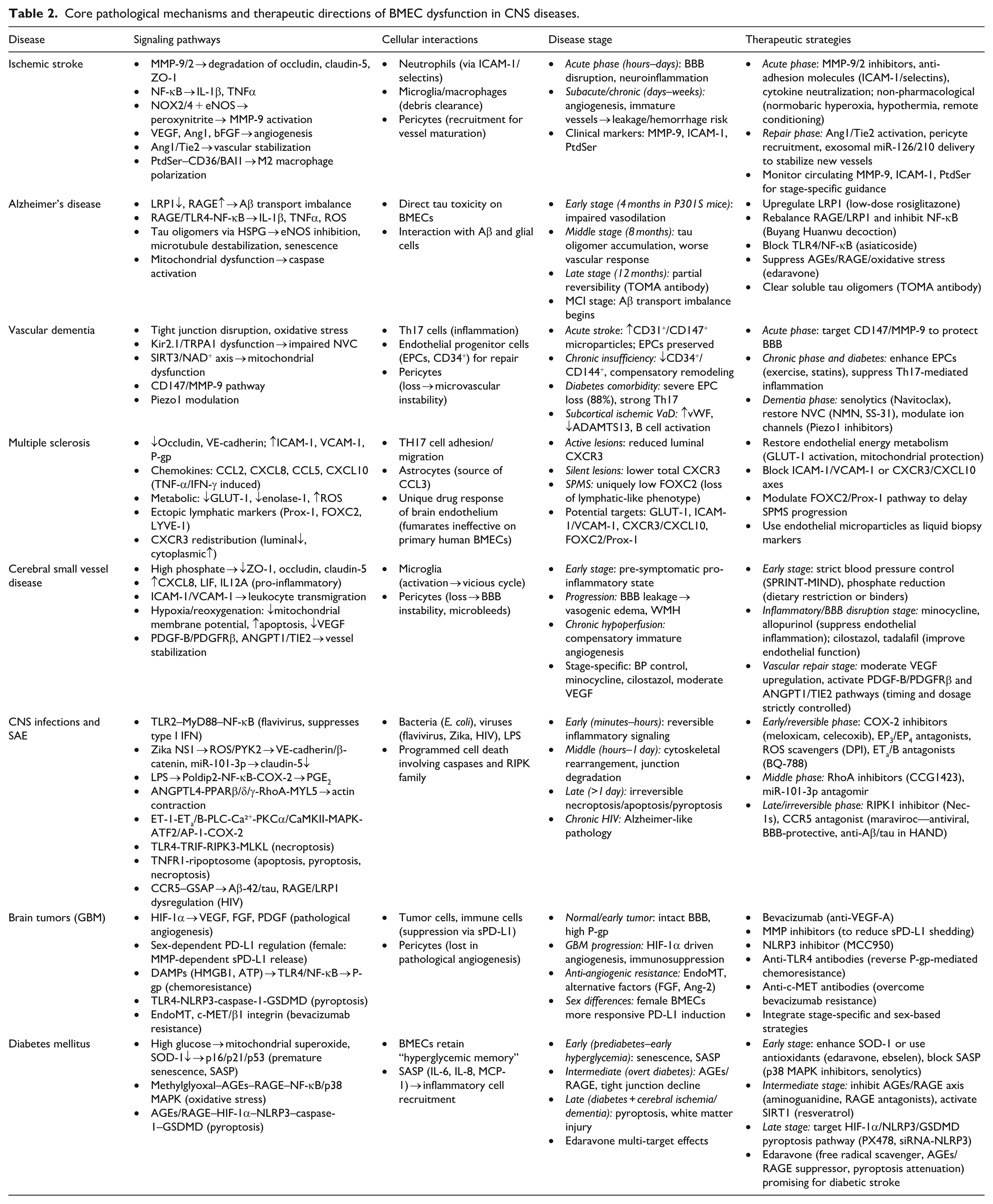
Pathological mechanisms and therapeutic targeting of BMECs in central nervous system disorders
Ischemic stroke
Distinct roles of BMECs in ischemic stroke at different stages call for stage‑specific therapeutic targets. In the acute phase (hours–days after ischemia–reperfusion), BMECs act as key executors of BBB disruption and neuroinflammation. 68 Yang et al., in studies of acute phase MCAO models and plasma from acute ischemic stroke patients, found that BMECs produce MMP‑9/2, which degrade tight junction proteins (occludin, claudin‑5, ZO‑1). 69 Adhesion molecules such as ICAM‑1 and selectins mediate massive neutrophil infiltration. Superoxide generated by NOX2/NOX4 reacts with excessive nitric oxide derived from eNOS to form peroxynitrite, further activating MMP‑9 and directly damaging the endothelium. Pro‑inflammatory cytokines IL‑1β and TNFα exacerbate these injuries via the NF‑κB pathway. Therapeutic targets in this stage focus on inhibiting MMP‑9/2, blocking adhesion molecules or neutralizing inflammatory cytokines, as well as multimodal non‑pharmacological interventions such as normobaric hyperoxia, hypothermia, and remote ischemic conditioning. 69 In the subacute/chronic phase (days–weeks after injury), BMECs switch to a reparative role: driven by VEGF, Ang1, and bFGF, they proliferate, sprout, and form new blood vessels to restore perfusion to the penumbra. However, this process is a double‑edged sword. Immature nascent vessels lack adequate pericyte coverage and intact tight junctions, which may instead increase BBB leakage and exacerbate vasogenic edema or even hemorrhagic transformation. 70 This phenomenon was demonstrated by Fang et al. and Xu et al. in rodent models of VEGF‑induced angiogenesis (subacute phase, days 3–7) and discussed in clinical observations of the peri‑infarct area in stroke patients (days 5–14), where enhanced but immature vessels are observed. Therefore, repair‑phase therapies should not only promote angiogenesis but also emphasize vascular stabilization: strategies such as the Ang1/Tie2 axis, pericyte recruitment, or exosome‑mediated delivery of miR‑126/210 can facilitate maturation of new vessels. Meanwhile, endothelial‑exposed phosphatidylserine (PtdSer) guides microglia/macrophages toward an M2 reparative phenotype via CD36/BAI1 receptors, helping to clear leakage‑associated debris. This mechanism was identified by Liu et al. and Xu et al. in rodent MCAO models (subacute phase, days 3–7) and in vitro studies using OGD‑treated endothelial cells; PtdSer has been proposed as a reversible “molecular window” biomarker in the subacute stage of ischemic stroke. Notably, MMP‑9 and MMP‑13 participate in vascular remodeling and neuronal plasticity at this stage, and excessive inhibition may worsen outcomes. In clinical practice, circulating levels of MMP‑9, ICAM‑1, and PtdSer can be used to dynamically assess the balance between BBB injury and repair, guiding stage‑specific treatment. For preclinical research, models incorporating aging, comorbidities, and reperfusion, combined with endothelial‑specific knockout and spatial transcriptomics, are needed to dissect the transition of BMECs from barrier disruptors to repair promoters, and to explore combination strategies that promote vascular maturation while preventing early leakage.68,70
Alzheimer’s disease
In AD, BMECs are not merely passive victims of Aβ deposition but actively participate in neurovascular unit dysfunction. Using in vitro models of primary human or rat BMECs with acute Aβ exposure, Moon et al. and Liu et al. demonstrated that impaired Aβ transport across the BBB (decreased LRP1‑mediated efflux and increased RAGE‑mediated influx, leading to Aβ accumulation).71,72 Further in vitro studies by Liu et al., Song et al., and Li et al. revealed that Aβ or advanced glycation end‑products activate the RAGE/TLR4‑NF‑κB pathway, promoting the release of inflammatory cytokines (IL‑1β, TNF‑α), adhesion molecules (ICAM‑1, VCAM‑1), and reactive oxygen species, thereby inducing endothelial injury.72–74 Li et al. specifically used a combination of methylglyoxal‑induced chronic injury and acute oxygen‑glucose deprivation in human BMECs to model diabetic cerebrovascular complications. In addition, using the P301S tauopathy mouse model at distinct ages (4, 8, and 12 months, representing early, middle, and late stages, respectively) together with human BMEC cultures, Hussong et al. discovered that soluble tau oligomers enter BMECs via heparan sulfate proteoglycans, trigger endogenous tau phosphorylation, microtubule destabilization, inhibition of eNOS and NO production, and potently induce endothelial cell senescence. 75 Song et al. also showed in in vitro human BMECs that Aβ leads to mitochondrial membrane potential reduction, caspase activation, and apoptosis, disrupting BBB integrity.
Based on these mechanisms, several therapeutic targets have been proposed, including upregulating LRP1 (low‑dose rosiglitazone); rebalancing RAGE/LRP1 and inhibiting NF‑κB (Buyang Huanwu decoction); blocking TLR4/NF‑κB (asiaticoside); suppressing AGEs/RAGE/oxidative stress (edaravone); and clearing soluble tau oligomers (TOMA antibody).
BMEC dysfunction evolves across AD stages. Using P301S tauopathy mice, Hussong et al. showed that endothelium‑dependent vasodilation is impaired at 4 months (early stage); tau oligomers accumulate and vascular responses worsen by 8 months (middle stage); and at 12 months (late stage), TOMA treatment still partially restores function, indicating late‑stage reversibility. Aβ‑related transport imbalance and inflammation are presumed to begin at the mild cognitive impairment stage.
Vascular dementia
During the progression of VaD, dysfunction of BMECs persists but exhibits marked heterogeneity across different stages. Mechanistically, BMECs are core components of the BBB. Studies using models of chronic cerebral hypoperfusion have found that disruption of tight junctions, oxidative stress, and impaired neurovascular coupling directly lead to cerebral hypoperfusion. In brain microvascular models, dysfunction of the endothelial Kir2.1 and TRPA1 channels has been shown to impair neurovascular coupling, as demonstrated by Harraz et al. and Thakore et al.76,77 Investigations in aging models have revealed mitochondrial dysfunction involving the SIRT3/NAD+ axis, a finding also observed in models of chronic cerebral hypoperfusion by Chen et al. 78 Regarding dysregulated metal ion homeostasis, abnormal iron deposition has been found to activate the AMPK/autophagy pathway in VaD models, while copper has been shown to enhance zinc‑induced neurotoxicity in neuronal models of vascular dementia.79,80
Clinical insights into stage‑specific characteristics come from an original cross‑sectional study by Goncharov et al., 81 which enrolled 120 elderly subjects, including patients with acute ischemic stroke, patients with chronic cerebral circulation insufficiency, patients with newly diagnosed or untreated type 2 diabetes mellitus, patients with subcortical ischemic vascular dementia, and age‑matched healthy controls. In patients with acute ischemic stroke, representing the acute phase, acute endothelial injury predominates, characterized by elevated CD31+ and CD147+ microparticles, whereas endothelial progenitor cell levels remain relatively preserved and the immune response shows only mild Th17 activation. In patients with chronic cerebral circulation insufficiency, defined as sustained hypoperfusion for at least 2 months, endothelial repair capacity begins to decline (as reflected by reduced CD34+ and CD144+ expression), and the lack of specific biomarkers suggests a state of compensatory remodeling. In patients with type 2 diabetes mellitus, the most severe impairment of endothelial repair capacity is observed (CD34+ decreased by ~88%, CD144+ by ~78%), accompanied by the strongest Th17 response, presenting moderate microvascular damage but a profoundly pro‑inflammatory environment. Finally, in patients with established subcortical ischemic vascular dementia, microvascular injury is most prominent (von Willebrand factor increased by ~86%, ADAMTS13 activity decreased by about 18%, and reductions in albumin and total protein). Notably, endothelial progenitor cell levels show no marked decline—possibly due to an elevated baseline caused by age‑related diseases or a decompensated state—along with aberrant activation of plasmablasts and B cells, indicating chronic antigenic stimulation.
Based on these stage‑specific differences, appropriate therapeutic strategies should be tailored accordingly. For the acute phase, targeting the CD147/MMP‑9 pathway may protect the BBB, as supported by clinical evidence from stroke patients and further demonstrated in stroke models where CD147 knockout reduces BBB disruption. 82 For the chronic phase and in diabetic patients, efforts should focus on enhancing endothelial progenitor cell levels (e.g. through exercise and statins) and suppressing Th17‑mediated inflammation. 83 For the dementia phase, integrated approaches are required, including senolytic therapy (e.g. Navitoclax) to improve functional hyperemia, NMN and SS‑31 to restore neurovascular coupling responses, and Piezo1 inhibitors to prevent cognitive impairment. 84 In conclusion, the imbalance between endothelial injury and repair is central to VaD, and evaluating the endothelial phenotype may guide stage‑specific interventions.
Multiple sclerosis
BMECs play an active and complex pathological role in MS. Beyond physical barrier disruption, BMECs contribute to immune cell recruitment through metabolic reprogramming, chemokine dysregulation, and aberrant neurolymphatic expression. Specifically, using serum from untreated relapsing-remitting MS (RRMS) patients applied to primary human BMECs, Sheikh et al. demonstrated that pro-inflammatory factors in MS serum downregulate tight junction proteins (occludin, VE-cadherin) and upregulate ICAM-1, VCAM-1, and P-glycoprotein, thereby promoting TH17 cell adhesion and migration. 85 BMECs are also a major source of chemokines. Subileau et al., based on analyses of post-mortem MS brain tissues (including active, chronic active, and silent lesions) as well as primary and immortalized human BMECs, found that BMECs constitutively secrete CCL2 and CXCL8; that TNF-α induces CCL2, CCL5, and CXCL8; that IFN-γ specifically upregulates CXCL10; and that CCL3 is mainly derived from astrocytes. 86 At the metabolic level, using serum from treatment-naïve RRMS patients on human BMECs, Sheikh et al. further showed that RRMS serum reduces glycolytic activity, downregulates GLUT-1 and enolase-1, impairs mitochondrial respiration, and increases reactive oxygen species, indicating endothelial energy stress. Moreover, Yun et al., in a cohort study of 150 RRMS patients, 26 secondary progressive MS (SPMS) patients, and 60 healthy controls combined with in vitro cytokine stimulation experiments on human BMECs, demonstrated that BMECs ectopically express lymphatic markers (Prox-1, FOXC2, LYVE-1). 87 Serum levels of these proteins are reduced in MS patients, and FOXC2 is uniquely lower in SPMS than in RRMS, suggesting progressive loss of the lymphatic-like endothelial phenotype. Regarding lesion activity, Subileau et al., using immunohistochemical and immunogold electron microscopy analyses of MS brain tissues at different lesion activity stages, observed that CXCR3 on BMECs shows reduced luminal expression and increased cytoplasmic accumulation in active lesions, and that total CXCR3 expression is lower in silent lesions than in normal white matter, indicating that subcellular redistribution of chemokine receptors correlates with inflammatory activity. Notably, Haarmann et al., in a comparative in vitro study, found that while fumaric acid esters inhibit NF-κB and ICAM-1 in human umbilical vein endothelial cells, they lack direct effects on primary human BMECs (non-immortalized, low-passage) under both basal and IL-1β-stimulated inflammatory conditions, highlighting a unique drug-response phenotype of brain endothelium. 88 Based on the above mechanisms, potential therapeutic targets include restoring endothelial energy metabolism (GLUT-1/mitochondrial protection), blocking the ICAM-1/VCAM-1 or CXCR3/CXCL10 axes, modulating the FOXC2/Prox-1 pathway to delay SPMS progression, and using endothelial microparticles as liquid biopsy markers. Given stage-specific pathological alterations in BMECs, stage-targeted interventions may provide new MS treatment strategies.
Cerebral small vessel disease
BMECs are central participants in the pathological process of CSVD, with their dysfunction evolving across disease stages.89,90 In a community‑based study of patients without stroke or dementia, Chung et al. found that elevated serum phosphate (>3.925 mg/dL) is closely associated with white matter hyperintensities; their subsequent in vitro experiments on cultured human BMECs demonstrated that high phosphate directly downregulates tight junction proteins (ZO-1, Occludin, and Claudin-5), leading to BBB leakage. 91 Using plasma from asymptomatic patients with early‑stage CSVD, Cifù et al. treated human BMECs in vitro and observed upregulation of pro‑inflammatory genes (CXCL8, LIF, IL12A) and cytokine‑mediated signaling pathways, indicating that endothelial cells enter a pro‑inflammatory state even before clinical symptoms emerge. 92
With disease progression, sustained endothelial activation disrupts tight junctions and increases BBB permeability, causing vasogenic edema and white matter hyperintensities. In an in vitro hypoxia/reoxygenation model that mimics acute ischemic injury, Li et al. found that endothelial cells upregulate adhesion molecules ICAM‑1 and VCAM‑1, which promote leukocyte transmigration and microglial activation, thereby forming a vicious cycle of inflammation and white matter injury. 93 The same study also reported that hypoxia/reoxygenation reduces mitochondrial membrane potential, increases apoptosis, and decreases VEGF expression, all of which impair vascular repair capacity. In a chronic cerebral hypoperfusion model, Uemura et al. observed that compensatory angiogenesis yields structurally immature vessels, and that pericyte loss further destabilizes the BBB, increasing the risk of microbleeds. 90
Stage-specific interventions may be considered. In early stages, strict blood pressure control (SPRINT‑MIND) and phosphate reduction (dietary restriction or binders) mitigate initial injury. At the inflammatory/BBB disruption stage, minocycline and allopurinol suppress endothelial inflammation, while cilostazol and tadalafil improve endothelial function. During vascular repair, moderate VEGF upregulation or activation of PDGF‑B/PDGFRβ and ANGPT1/TIE2 pathways may promote vessel stabilization, though timing and dosage require strict control. Thus, BMECs undergo a dynamic evolution from silent activation to inflammatory injury and repair decompensation, supporting multi‑target, stage‑specific therapeutic strategies integrating anti‑inflammation, BBB protection, and vascular repair.
Central nervous system infections and sepsis-associated encephalopathy
BMECs are central to BBB disruption in bacterial meningitis, viral encephalitis, and sepsis‑associated encephalopathy (SAE). The injury mechanisms evolve with disease stage.
In the early stage (minutes–hours), reversible inflammatory signaling dominates. Using an acute in vitro model of human BMECs, Bhide et al. demonstrated that flavivirus envelope protein DIII activates the TLR2–MyD88–NF‑κB pathway, inducing cytokines and TAM receptors while suppressing type I interferon responses via JAK–STAT/SOCS1/3. 20 In acutely exposed human BMECs (24 h), Rastogi et al. found that Zika virus NS1 triggers ROS/PYK2‑dependent phosphorylation of VE‑cadherin and β‑catenin, and Bhardwaj et al. further showed that NS1 upregulates miR‑101‑3p, both leading to reduced claudin‑5 expression.94,95 In an acute mouse model of SAE (intraperitoneal LPS injection, 6–18 h), Kikuchi et al. reported that LPS induces Poldip2 expression, which drives NF‑κB‑mediated COX‑2 and PGE2 production, rapidly increasing endothelial permeability. 96 Moreover, using acutely stimulated human BMECs, Dalvi et al. demonstrated that exogenous arachidonic acid is metabolized by COX‑2 to PGE2, which acts on EP3 and EP4 receptors to elevate intracellular Ca²+ and remodel the cytoskeleton within 2–30 min. 97
In the middle stage (hours–1 day), cytoskeletal rearrangement and junctional degradation occur. In both human BMECs in vitro and an acute mouse model (tail vein injection, 24 h), Liu et al. showed that meningitic Escherichia coli upregulates ANGPTL4 via PPARβ/δ/γ, which inhibits ARHGAP5 and activates the RhoA–MYL5 axis, causing actin contraction and intercellular gap formation without net loss of tight junction proteins. 98 In acutely stimulated bEnd.3, Lin et al. found that endothelin‑1 (ET‑1), which is elevated during cerebral ischemia or trauma, activates ET3 receptors and the PLC–Ca²+–PKCα/CaMKII–MAPK pathway, promoting ATF2/AP‑1 and p300 binding to the COX‑2 promoter and sustaining PGE2 release. 99
In the late stage (>1 day), irreversible programmed cell death predominates. Using human BMECs in vitro and a neonatal mouse intraperitoneal infection model, Wang et al. demonstrated that during neonatal meningitic E. coli infection, the TLR4–TRIF pathway activates RIPK3–MLKL necroptosis as early as 1–2 h, followed by TNFR1‑dependent assembly of the ripoptosome complex (RIPK1/RIPK3/caspase‑8), which simultaneously triggers apoptosis, pyroptosis and necroptosis. 100 Although caspase‑8 cleaves RIPK1 as a negative feedback mechanism, prolonged infection breaks this balance, resulting in massive BMEC death and marked loss of claudin‑5, ZO‑1, and ZO‑2. In a chronic HIV infection model (hu‑PBL‑NSG mice, 3 weeks post‑infection), Bhargavan et al. showed that CCR5 signaling upregulates GSAP, increases Aβ‑42 generation and tau hyperphosphorylation, and dysregulates RAGE and LRP1, leading to Alzheimer‑like pathology. 101
Stage‑dependent therapeutic targets—Early/reversible phase: COX‑2 inhibitors (meloxicam, celecoxib), EP3/EP4 antagonists, ROS scavengers (DPI), or ET_B antagonists (BQ‑788). Middle phase: RhoA inhibitors (CCG1423) or miR‑101‑3p antagomir. Late/irreversible phase: RIPK1 inhibitor (Nec‑1s) or CCR5 antagonist (maraviroc), the latter offering antiviral, BBB‑protective and anti‑Aβ/tau actions in HIV‑associated neurocognitive disorders.
Brain tumors
BMECs exhibit stage‑dependent pathological mechanisms in glioblastoma (GBM), offering multiple therapeutic targets. 102 Navratil et al., in their immunohistochemical analysis of normal human cerebral cortical and subcortical tissues obtained from surgical or autopsy specimens, demonstrated that BMECs maintain BBB integrity through tight junctions, low expression of leukocyte adhesion molecules, and high activity of P‑glycoprotein, thereby restricting tumor and immune cell extravasation in normal brain and early‑stage tumors. 103 During GBM progression, hypoxia‑induced HIF‑1α upregulates VEGF, FGF, and PDGF, triggering pathological angiogenesis.102,104 Under inflammatory cytokines and VEGF, Baggio et al., in their in vitro study using primary human umbilical vein endothelial cells isolated from male and female donors (non‑tumor tissue), discovered a sex‑dependent PD‑L1 regulation: female‑derived BMECs increased total PD‑L1 and released soluble PD‑L1 via matrix metalloproteinase‑dependent shedding, creating an immunosuppressive microenvironment, whereas male‑derived cells exhibited higher basal PD‑L1 expression but weaker responses to stimulation. 105 Furthermore, Acioglu and Elkabes summarized that acute‑phase data from animal models of ischemic stroke, traumatic brain injury, and infection together with human post‑mortem findings, reported that damage‑associated molecular patterns such as high mobility group box 1 and ATP activate the TLR4/NF‑κB pathway in BMECs, upregulating P‑glycoprotein and causing chemoresistance, while simultaneously activating the NLRP3/caspase‑1/GSDMD pathway, which induces endothelial pyroptosis and severely disrupts BBB integrity. 10 In the anti‑angiogenic therapy‑resistant stage, Hovis et al., synthesizing patient‑derived and animal model studies including chronic resistance models, as well as Ahir et al. incorporating data from preclinical resistance models and clinical trials, demonstrated that BMECs undergo endothelial‑to‑mesenchymal transition, upregulate alternative pro‑angiogenic factors such as fibroblast growth factor and angiopoietin‑2, and activate the c‑MET/β1 integrin axis, leading to resistance to anti‑VEGF agents like bevacizumab. Potential therapeutic targets include bevacizumab (VEGF‑A), MMP inhibitors (sPD‑L1), MCC950 (NLRP3), anti‑TLR4 antibodies (P‑gp‑mediated resistance), and anti‑c‑MET antibodies (EndoMT). Integrating stage‑specific and sex‑based strategies may improve GBM therapy.
Diabetes mellitus
In diabetes-related cerebrovascular injury, BMECs progressively transition from premature senescence and inflammation to pyroptosis. In the early stage (prediabetes to early hyperglycemia), high glucose induces stress‑induced premature senescence. Using a mouse model of short‑term sustained hyperglycemia (7 days) and cultured human endothelial cells, Prattichizzo et al. found that hyperglycemia leads to mitochondrial superoxide overproduction and insufficient SOD‑1 compensation, resulting in upregulation of p16/p21/p53 and the establishment of a senescence‑associated secretory phenotype (SASP) including IL‑6, IL‑8, and MCP‑1. Notably, the same study demonstrated that BMECs retain a “hyperglycemic memory” even after glucose normalization, indicating long‑lasting imprinting. 106 Working with cultured human BMECs exposed to methylglyoxal for 24 h followed by oxygen‑glucose deprivation to mimic acute ischemia, Li et al. showed activation of the RAGE/NF‑κB/p38 MAPK pathway, amplifying oxidative stress and inflammation. 74 BBB tight junctions begin to decline, and cerebral blood flow is impaired.107,108 In the late stage (diabetes with cerebral ischemia/dementia), under hypoxic conditions, endothelial cells undergo pyroptosis. Using mouse BMECs subjected to OGD and a mouse model of acute ischemic stroke induced by middle cerebral artery occlusion, Han et al. demonstrated that the AGEs/RAGE axis upregulates NLRP3 via HIF‑1α, activating caspase‑1 and cleaving GSDMD to form membrane pores, which release IL‑1β and IL‑18, thereby disrupting the BBB and damaging the neurovascular unit, ultimately accelerating white matter injury and cognitive decline. 23
Therapeutic strategies should be stage‑specific. Early stage: enhance SOD‑1 or use antioxidants (edaravone, ebselen), and block SASP (p38 MAPK inhibitors, senolytics). Intermediate stage: inhibit the AGEs/RAGE axis (aminoguanidine, RAGE antagonists) or activate SIRT1 (resveratrol). Late stage: target the HIF‑1α/NLRP3/GSDMD pyroptosis pathway (PX478, siRNA‑NLRP3). Edaravone, with its multiple actions (free radical scavenging, AGEs/RAGE suppression, and pyroptosis attenuation), holds translational promise for diabetic stroke patients.
Future research directions and translational prospects
By systematically examining the characteristics of BMECs and their roles in specific diseases, we have re-evaluated brain endothelial function from an immunological perspective. This paradigm shift reconceptualizes BMECs not merely as passive structural components for defense, but as dynamic sentinels capable of actively sensing, integrating, and executing immune instructions. This evolving perspective not only deepens our understanding of the pathogenesis of various neurological disorders but, more importantly, unveils novel therapeutic targets for future interventions.
Clinical and preclinical implications of the revised perspective
Viewing BMECs merely as passive structural components of the BBB is no longer sufficient to explain their complex roles in neurological disorders. From an immunological perspective, the BMECs acts as a sentinel that actively senses, integrates, and executes immune instructions. For example, in acute ischemic stroke and other neurological diseases, BMECs regulate immune cell recruitment, adhesion, and transendothelial migration through pattern recognition receptors (e.g. TLR4), chemokines (e.g. CCL2), and autophagic signals (e.g. phosphatidylserine), thereby determining whether inflammation exacerbates injury or promotes repair. This paradigm shift has important implications for clinical practice and preclinical research. At the clinical level, endothelium‑derived soluble molecules (e.g. circulating MMP‑9, soluble CD36, HMGB1) can serve as dynamic biomarkers to predict BBB disruption, hemorrhagic transformation risk, and post‑reperfusion immune status, aiding patient stratification and individualized therapy. Endothelial‑targeted interventions (e.g. normobaric hyperoxia, remote ischemic conditioning, selective hypothermia) have shown promise in protecting the barrier and fostering a reparative immune shift. At the preclinical level, spatial transcriptomics, intravital imaging, and endothelial‑specific knockout models should be used to dissect the immune heterogeneity of BMECs across disease stages and brain regions, and to validate interventions targeting endothelial immune molecules such as PtdSer receptors (CD36/BAI1) and tight junction regulatory pathways.46,69,109
Emerging directions driven by technological innovation
Breakthroughs in research are often propelled by technological advances. Immune activities involve extensive collaboration among multiple cells and systems. The application of spatial transcriptomics and single-cell multi-omics technologies is crucial for mapping cellular expression profiles across different disease stages, brain regions, and even distinct vascular segments. This enables a deeper understanding of BMEC roles in various neurological diseases and their complex interactions with neighboring cells. For instance, Pfau et al., utilizing single-cell and spatial transcriptomics, revealed that endothelial molecular specialization and their unique interactions with pericytes constitute the foundation for BBB functional heterogeneity, supporting the development of region-specific therapies for the central nervous system. 110 Similarly, Hansen et al., through analysis of stroke mice, delineated the spatial molecular signatures underlying BBB disruption and loss of vascular tone following ischemia–reperfusion. 111
Intravital two-photon microscopy allows direct observation of interactions between endothelial and immune cells in live animals. Complementing this, human pluripotent stem cell-derived BBB organoids and organ-on-a-chip models enable genetic and pharmacological manipulations within a highly controlled human-relevant system, facilitating precise mechanistic dissection. 112 For example, Ben et al. coupled three organ-chips to simulate trans-BBB input, brain parenchyma simulation, and trans-BBB output steps, modeling the physiological effects of intravascular administration of the psychostimulant methamphetamine and, for the first time, uncovered previously unknown metabolic coupling mechanisms between the BBB and neurons. 108
Transitioning from broad-spectrum inhibition to precision medicine
Current pharmacological interventions for CNS disorders largely rely on broad suppression of inflammatory cascades. Future drug development targeting BBB injury should focus on endothelial-specific immunomodulatory molecules, such as ALCAM and MCAM, aiming to inhibit pathogenic cell infiltration while preserving beneficial immune surveillance functions. Furthermore, as inflammatory responses are essential host defenses against pathogens, not all NF-κB or NLRP3 activation is detrimental. Future therapeutic strategies should aim for precise inhibition of aberrant signaling within specific BMEC subpopulations or at specific timepoints in disease contexts, while preserving their fundamental host defense capabilities. In treating CNS disorders, the scope should extend beyond targeting the lesion site alone to include promoting BMEC repair and enhancing their resilience against inflammatory insults, representing a highly promising neuroprotective strategy.
Summary
The BBB represents a dynamic neuroimmune interface whose core functions extend beyond maintaining cerebral homeostasis to encompass its role as an active immunological sentinel. As the central component of the BBB, BMECs precisely regulate immune cell migration and inflammatory responses through their expression of pattern recognition receptors and integration of multiple signaling pathways. Current research has progressively unveiled the molecular mechanisms by which BMECs sense systemic inflammation and mediate protective neural responses under physiological conditions.
However, with intensifying or sustained inflammatory challenges, the BBB exhibits supra-physiological reactions: BMECs undergo tight junction remodeling, disrupted polarized secretion, and facilitate abnormal immune cell infiltration, ultimately leading to functional decompensation. These pathological alterations closely correlate with clinical manifestations across various neurological disorders. Multiple intrinsic and extrinsic factors—including genetic background, age, sex, and comorbid conditions—may modulate the specificity and intensity of BBB responses by influencing BMEC behavior.
Future investigations should focus on deciphering endothelial heterogeneity via high-resolution technologies, developing precise tools for barrier function assessment, and designing targeted interventions that selectively modulate BMEC immune regulatory functions. Such advances will undoubtedly open new avenues for the prevention and treatment of neurological diseases.
Footnotes
Author contributions
JO, KH, and YF wrote the main manuscript text and prepared all figures. YL, QY, MS, SJ, JZ, YW, KL, ZL, and MF reviewed and revised the manuscript. JO, KH, YF, YW, and KL participated in data collection. ZL and MF contributed to the conceptualization of the study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Science Foundation of China (nos. 82402013, 82271747, and 82571979), the Wu Jieping Medical Foundation (no. 320.6750.2025-23-16), Science and Technological Innovation Project for College Students in Zhejiang Province (Xinmiao Talent Plan 2025R413A028), National Innovation and Entrepreneurship Training Program for Undergraduate (202510343029), The “Thousand-Hundred-Ten” Talent Cultivation Project (WMLEFH2025003).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical considerations
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Data availability statement
Not applicable.