
Abstract
Ischemic stroke (IS) causes white matter (WM) injury and disrupts myelin integrity, contributing to long-term cognitive dysfunction. Oligodendrocyte lineage cells, particularly oligodendrocyte precursor cells (OPCs), are essential for post-stroke remyelination, yet their survival, differentiation, and myelin-forming capacity are highly dependent on metabolic state. However, a systematic overview of these metabolic changes post-stroke is still lacking. Here, we review current knowledge on the crosstalk between classical pathways and oligodendrocyte lineage metabolism in IS, integrating evidence from recent studies and complementing them with a re-analysis of three independent single-cell RNA sequencing (scRNA-seq) datasets from mouse stroke models covering hyperacute, acute, and chronic recovery phases. This re-analysis highlighted stage-specific alterations in oxidative phosphorylation, inositol phosphate metabolism, and sphingolipid metabolism within OPCs, alongside activation of cholesterol biosynthesis in mature oligodendrocytes (OLs). These findings are consistent with evidence linking these pathways to oligodendrocyte lineage progression and WM repair. Together, the literature and re-analysis support the notion that oligodendrocyte lineage metabolism is an important regulator of post-stroke remyelination and may provide potential therapeutic targets for promoting cognitive function.
Keywords
Introduction
Ischemic stroke (IS), the second leading cause of long-term disability and mortality worldwide, is caused by a sudden and critical reduction in cerebral blood flow, typically due to arterial occlusion. 1 Beyond acute neuronal injury, IS also triggers long-term cognitive dysfunction. Clinically, the severity of post-stroke cognitive impairment (PSCI) is often underestimated. PSCI affects up to 60% of stroke survivors within the first year, with approximately one-third eventually progressing to dementia.2,3 Effective prevention of PSCI remains a major therapeutic gap in stroke treatment. 4 There is a strong association between white matter (WM) injury and PSCI which is linked to poor outcomes in IS patients.5–8 Brain scans also reveal that WM injury can continue for weeks or months after the stroke. 9 Therefore, new treatments should focus on protecting WM to help patients improve functional recovery.
Within the WM, oligodendrocyte lineage cells including oligodendrocytes (OLs) and oligodendrocyte precursor cells (OPCs) are key components, serving the essential functions of myelin production, maintenance, and regeneration. 10 Mature OLs are the primary myelinating cells in the central nervous system (CNS), responsible for maintaining myelin sheath integrity and providing axonal support. 11 OPCs constitute ~5%–8% of glial cells in the adult CNS, 12 and retain the capacity for self-renewal and differentiation into OLs, thereby contributing to myelinogenesis during development and remyelination following injury.13,14 Under certain conditions, OPCs can also differentiate into astrocytes and Schwann-like cells.15–17 Together, oligodendrocyte lineage cells form a dynamic cellular system that supports WM maintenance and repair after IS.
The high energy demands required to sustain their functions render oligodendrocyte lineage cells highly vulnerable to energy deprivation, leading to severe cellular damage following IS.18,19 Disruption of metabolic state can impair OPCs survival, proliferation, and differentiation, while also compromising OL-mediated myelin maintenance and axonal support. 20 However, due to their relatively low abundance and the technical challenges associated with isolating OPCs and OLs from brain tissue, in vivo investigations of their metabolic changes after IS remain limited. Thus, in addition to summarizing the existing literature, we also re-analyzed three scRNA-seq datasets from mouse models of IS, covering multiple time points from hyperacute, acute, and chronic post-stroke stages. We harmonized transcriptomic profiles across datasets to systematically examine metabolic changes in OLs and OPCs. This integrated transcriptional landscape provides insights for understanding how metabolic reprogramming of oligodendrocyte lineage cells may influence WM injury and repair after IS. In this review, we summarize the current knowledge on the roles of the oligodendrocyte lineage cells in IS, integrate our scRNA-seq re-analysis with a specific focus on their unique metabolic adaptations and their translational potential for targeted therapeutic interventions.
Signaling and metabolic regulation of oligodendrocyte lineage cells following IS
Based on the studies summarized in Table 1, a range of preclinical IS models—including transient and permanent middle cerebral artery occlusion (MCAO) as well as photothrombosis (PT) stroke in mice and rats—have revealed signaling and metabolic pathways that shape the fate of oligodendrocyte lineage cells. Key pathways including LINGO-1, Shh–Gli1, LRP1–PPARγ, Nrf2, Notch signaling pathway, and more, each of which have been associated with oligodendrocyte lineage cells proliferation, migration, differentiation, or remyelination. Injury-related modulators such as BNIP3, Nec-1, CB1, TGF-α, and miR-23a-5p have been implicated in modulating OPCs survival after IS. These IS studies highlight diverse signaling mechanisms that shape OPCs and OLs behavior.
Overview of pathways regulating oligodendrocyte lineage cells following stroke.
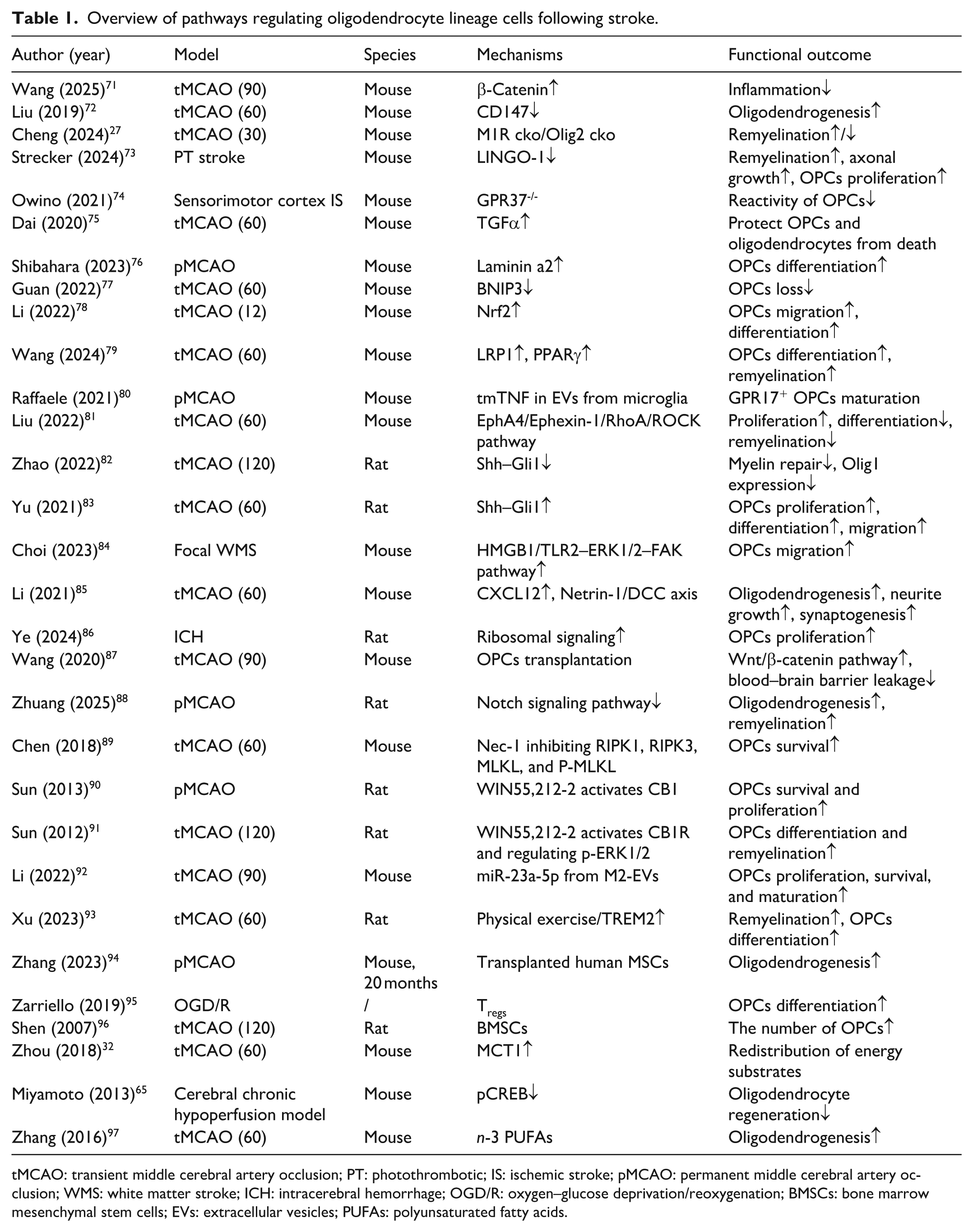
tMCAO: transient middle cerebral artery occlusion; PT: photothrombotic; IS: ischemic stroke; pMCAO: permanent middle cerebral artery occlusion; WMS: white matter stroke; ICH: intracerebral hemorrhage; OGD/R: oxygen–glucose deprivation/reoxygenation; BMSCs: bone marrow mesenchymal stem cells; EVs: extracellular vesicles; PUFAs: polyunsaturated fatty acids.
Accumulating evidence from other neurological diseases suggests that many of these pathways are closely associated with metabolic processes, which are essential for maintaining oligodendrocyte lineage cells function. For instance, Nrf2 is widely recognized for its role in regulating cellular redox homeostasis and mitochondrial function, thereby influencing oxidative metabolism under stress conditions. 21 Similarly, LRP1–PPARγ signaling has been linked to lipid metabolism and fatty acid oxidation, processes that are essential for myelin synthesis.22,23 Notch signaling, beyond its typical role in cell fate determination, has also been implicated in the regulation of glycolysis and mitochondrial activity. 24 In addition, Wnt/β-catenin signaling has been shown to coordinate glucose metabolism and anabolic pathways, thereby supporting biosynthetic demands during differentiation. 25 Collectively, although these pathways are described as regulators of oligodendrocyte lineage cells function after IS, evidence from other diseases indicates that they are frequently coupled to metabolic processes, raising the possibility that metabolic regulation represents a shared downstream mechanism underlying their effects.
To understand how this potential signaling-metabolism crosstalk translates into functional neuroprotection, it is essential to examine the fundamental metabolic demands of WM repair. This repair process can be modulated by the availability of myelin substrates and energy supply.26–28 Therefore, metabolic reprogramming is significant to promote WM recovery after IS.29,30 Due to the high ATP requirements of myelination, oligodendrocyte lineage cells exhibit a specific preference for energy substrates, utilizing lactate for lipid synthesis at a significantly higher rate than neurons and astrocytes. 31 OPCs rely on monocarboxylate transporter 1 (MCT1) for lactate uptake, which is upregulated following stroke, whereas its expression in mature OLs remains unchanged. 32 The distinct MCT1 expression profiles between OPCs and mature OLs dictate their differential vulnerabilities to energy deprivation, indicating that energy substrate redistribution is a key determinant of WM damage.
The metabolic response of OPCs to ischemia also involves active regulatory mechanisms. To adapt to low-glucose environments, OPCs utilize a specific intrinsic metabolic checkpoint. ALDOC, their predominant intracellular aldolase isozyme, undergoes physiological acetylation at the K14 residue, which prevents the activation of the downstream AMPK pathway by low-glucose signals. 33 By bypassing AMPK-mediated energetic dormancy, OPCs maintain the proliferation and differentiation capacities required for initiating repair within the ischemic penumbra. Therefore, metabolic shifts serve as regulatory mechanisms that drive WM recovery after IS.
Nevertheless, a comprehensive and systematic evaluation of global metabolic reprogramming in oligodendrocyte lineage cells following IS is still lacking. To bridge this gap, evidence from other neurological diseases provides important complementary insights into the metabolic regulation of OPCs and OLs. Lipid homeostasis, for example, is essential for oligodendrocyte lineage cells differentiation and myelination. 34 A recent study using a seipin-deficient mouse model, which mimics features of metabolic neurological disorders, demonstrated that seipin loss disrupts lipid homeostasis, leading to abnormal lipid droplet morphology, impaired OPCs differentiation, defective myelination, and subsequent cognitive and motor impairments. 34 In the context of demyelinating diseases such as multiple sclerosis (MS), a metabolomic study revealed that taurine markedly promotes OPCs differentiation by supporting intracellular serine pools required for glycosphingolipid synthesis and myelin formation. 35 Furthermore, a recent review focusing on neurodegenerative diseases including MS, Alzheimer’s disease, and Parkinson’s disease proposed that oxidative and carbonyl stress interfere with mitochondrial function and lipid biosynthesis, thereby impeding the transcriptional and metabolic programs essential for OPCs maturation. 29 Together, findings from related neurological disorders emphasize the critical role of lipid metabolic networks in the oligodendrocyte lineage cells, suggesting that targeting lipid homeostasis may similarly drive WM recovery following IS.
In summary, current literature indicates that various signaling pathways can regulate the survival and function of oligodendrocyte lineage cells after IS. Notably, several of these pathways also exert metabolic regulatory effects. Furthermore, evidence from IS and other CNS disorders suggests that lactate metabolism and lipid homeostasis are critical determinants of oligodendrocyte lineage cells-mediated WM repair. However, direct mechanistic studies investigating the metabolic reprogramming of the oligodendrocyte lineage cells following IS remain unexplored. Therefore, using public scRNA-seq datasets, we systematically analyzed the metabolic reprogramming of the oligodendrocyte lineage cells following IS.
Re-analysis of public scRNA-seq datasets
We examined scRNA-seq datasets from mouse models of IS that were available by March 2025. In total, three datasets comprising 10 samples and ~96,000 cells across several post-stroke time points and two stroke models were included. The characteristics of these three datasets are summarized in Table 2. To preserve biological heterogeneity across different stroke models, we analyzed the datasets independently rather than applying a global integration. While all control samples were batch-corrected and merged, the PT model and datasets from unmatched time points were processed as separate cohorts for dimensionality reduction, clustering, and differential expression. Integration was restricted strictly to the 3-day post-MCAO datasets. The quality control for the extracted data were shown in Figure 1(a). Using known cell markers, we identified 15 main cell populations from the clustering analysis (Figure 1(b) and (c)). The overall cellular composition for each condition is summarized in Figure 1(d). Subsequent analyses focused on OLs and OPCs, which showed distinct patterns of gene expression and metabolic pathway enrichment at different phases after IS. The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment results of eight metabolic pathways for OLs and OPCs were shown in Figure 2.
The scRNA-seq studies included in this integrative analysis.
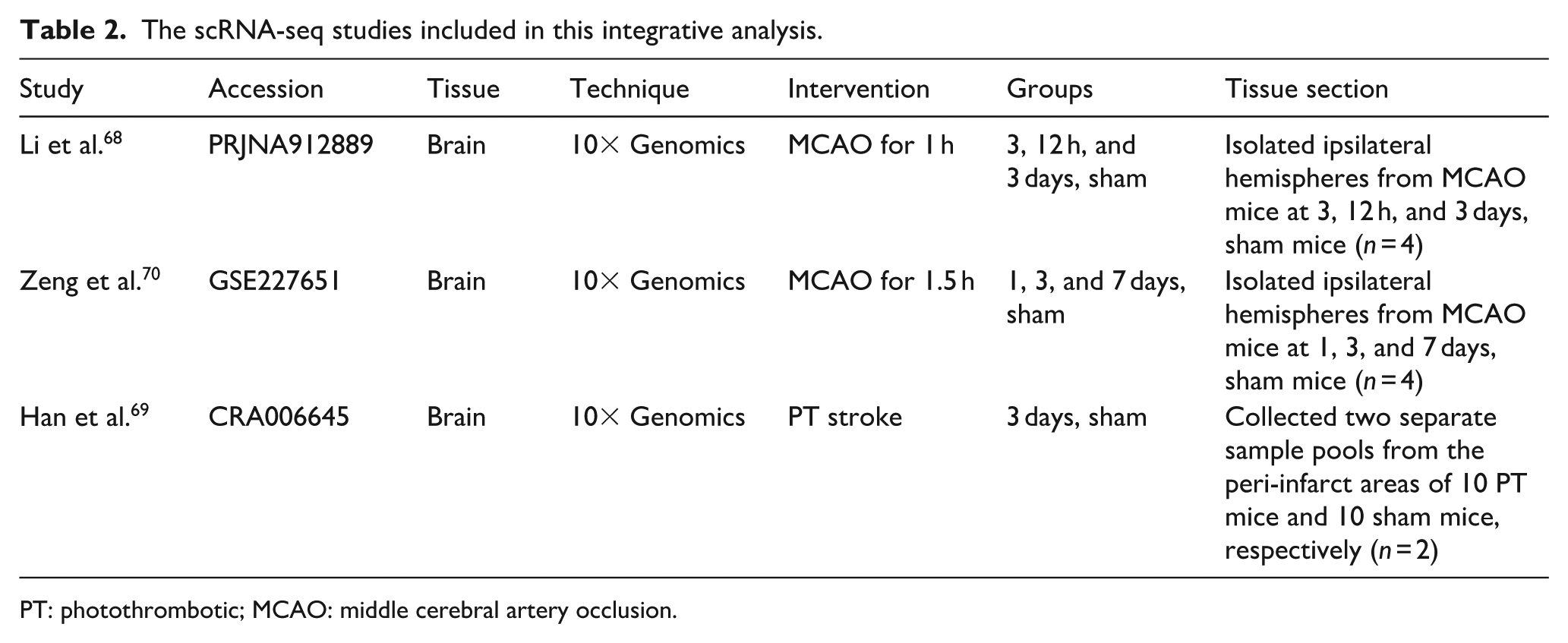
PT: photothrombotic; MCAO: middle cerebral artery occlusion.
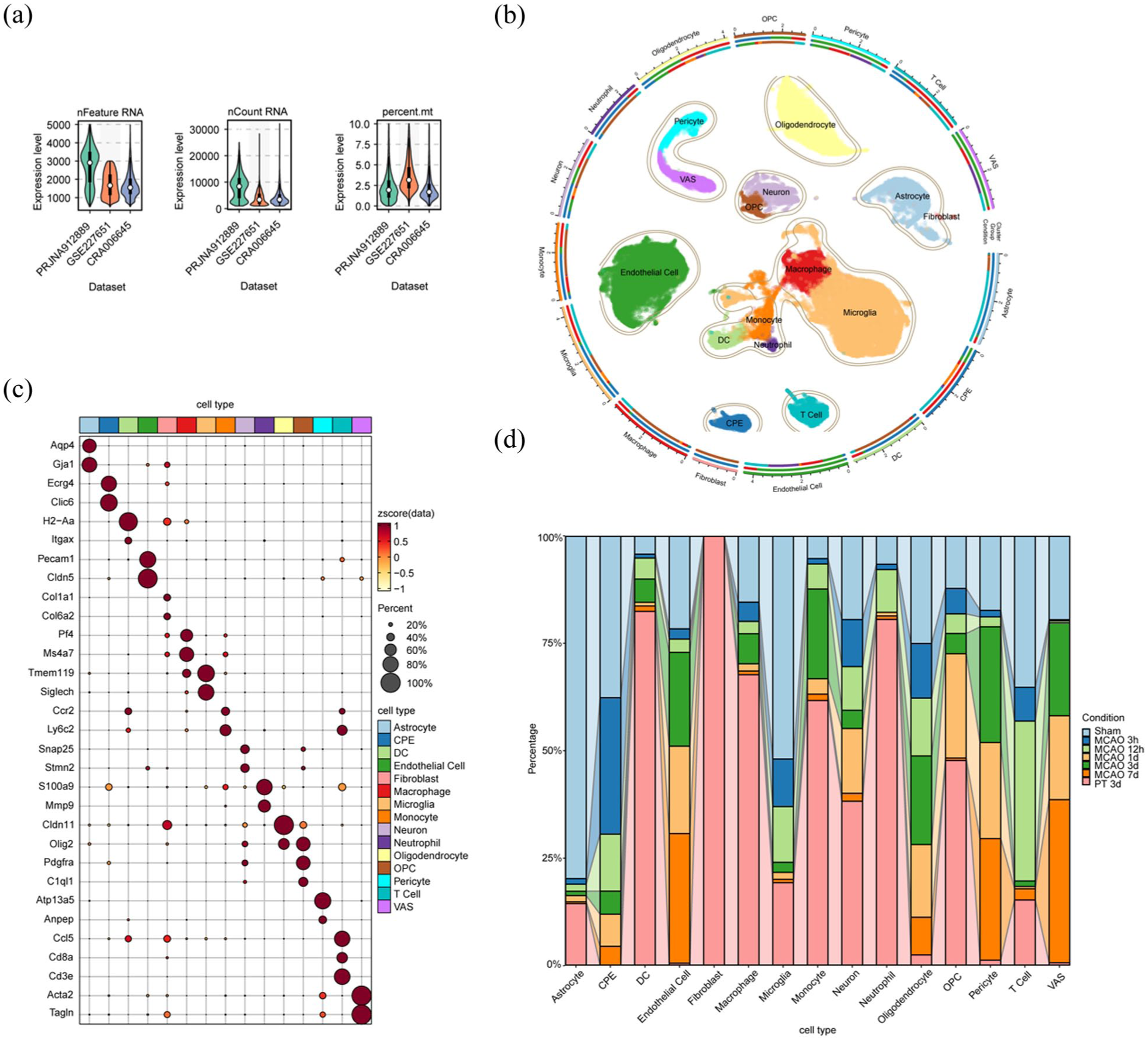
Quality control and cell annotation for the extracted data: (a) quality control after filtering, (b) the UMAP representation of the cells after correction and doublet removal, circularized, (c) a dot plot of the cell-type marker merge RNA data, and (d) cell component per cell type percent group by condition after annotation.
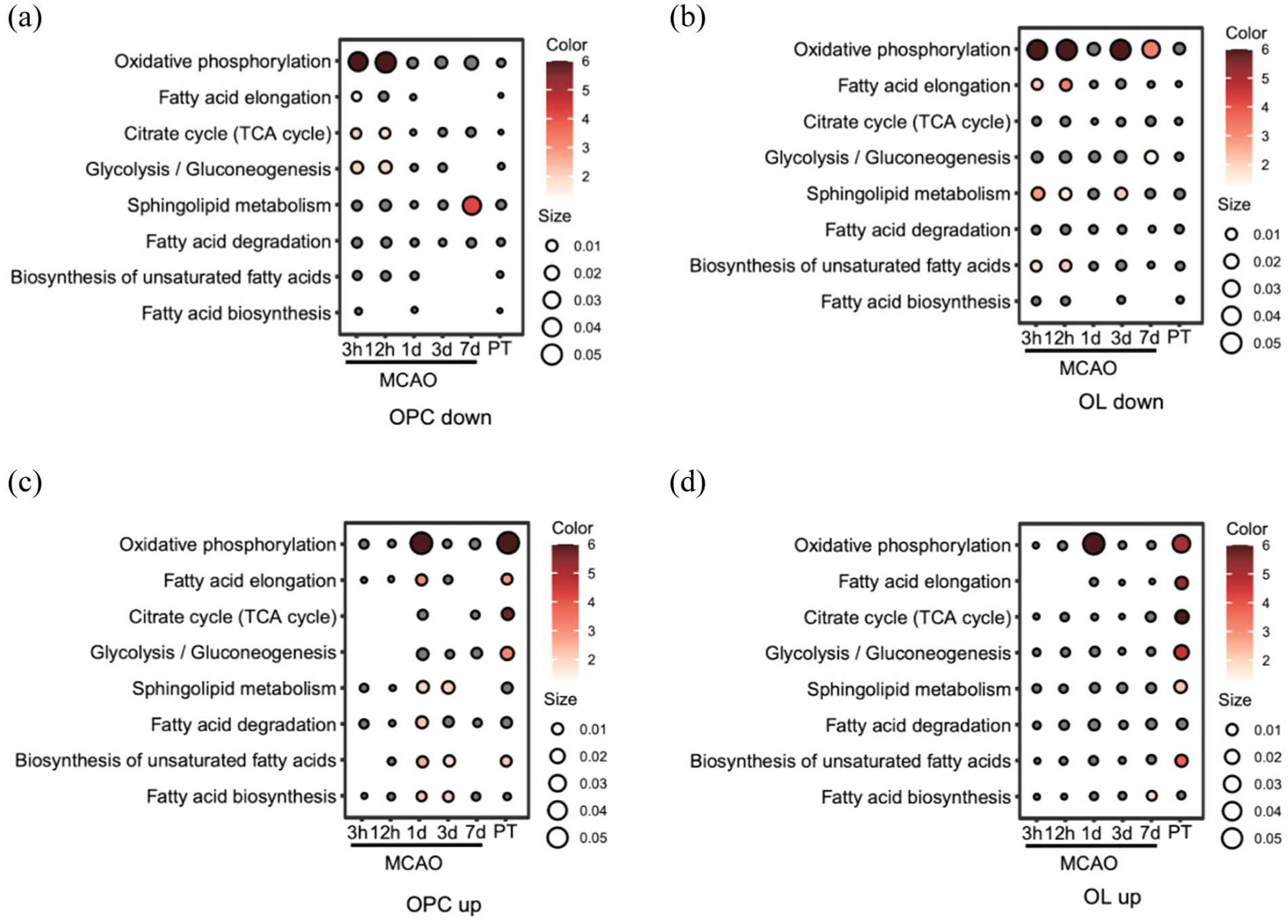
KEGG enrichment results of eight metabolic pathways for OPCs and OLs. Red bubbles indicate upregulated or downregulated pathways, with dot size representing gene count and color intensity reflecting the adjusted p value: (a) downregulated pathways in different time-points within eight metabolic pathways for OPCs, (b) downregulated pathways in different time-points within eight metabolic pathways for OLs, (c) upregulated pathways in different time-points within eight metabolic pathways for OPCs, and (d) upregulated pathways in different time-points within eight metabolic pathways for OLs.
Hyperacute phase (3 and 12 h)
In the hyperacute phase (3 and 12 h), the OXPHOS pathway was strongly suppressed in OPCs (Figure 2(a)) and OLs (Figure 2(b)). Unlike mature OLs that rely on glycolysis, OPCs depend on OXPHOS to meet the high energy demands of proliferation and differentiation.36,37 Therefore, suppression of OXPHOS may critically impair OPCs function. Our compound–reaction–enzyme–gene network analysis further confirmed that key mitochondrial enzymes, including Complex I, II, and V, were influenced in OPCs (Figure 3(a)). Although the glycolytic pathway appeared stable in OPCs, it may not be sufficient to support cell survival. This observation aligns with a previous finding that OPCs upregulate MCT1 expression following IS, 32 suggesting that lactate may serve as a crucial alternative energy substrate for OPCs survival during the hyperacute phase.
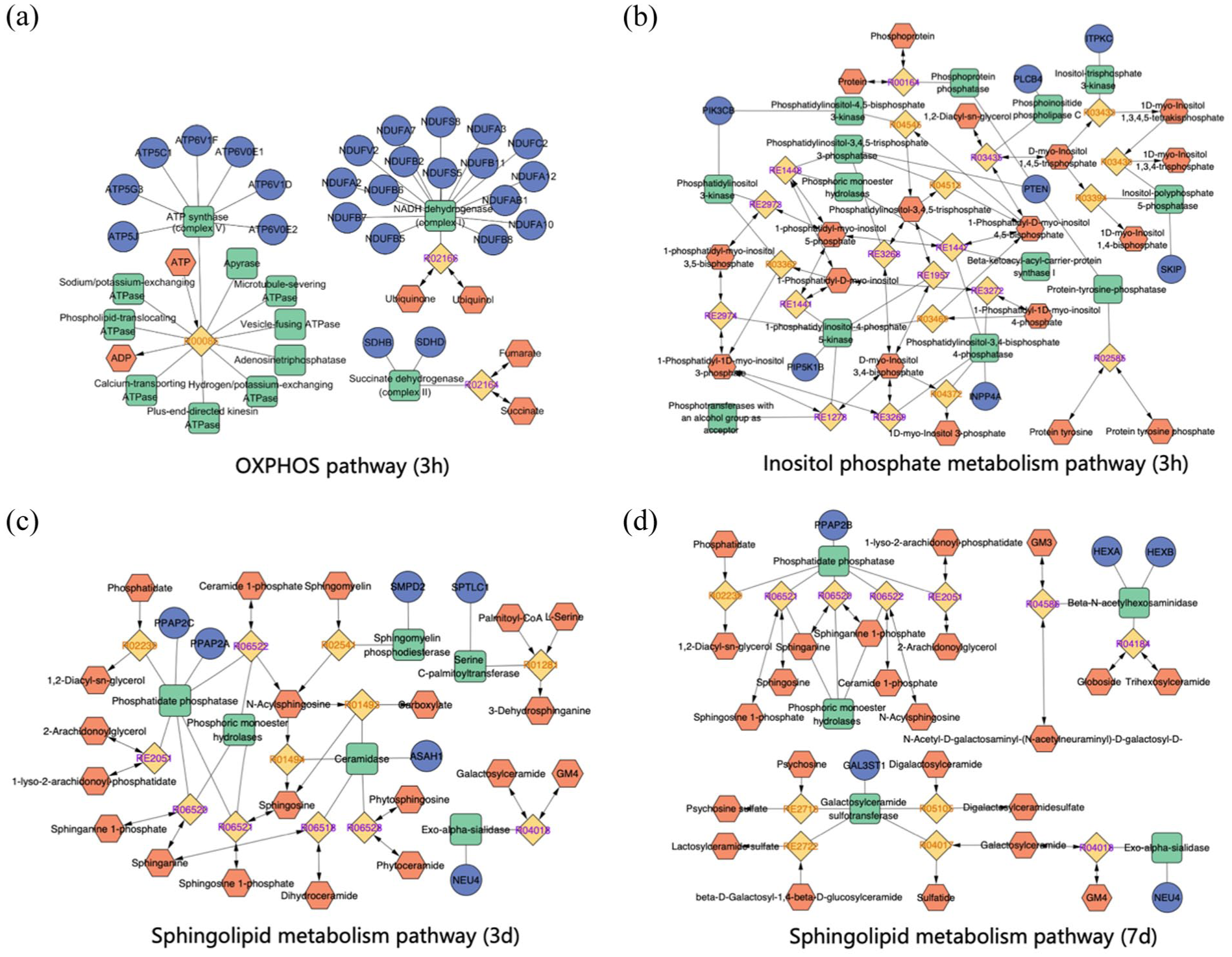
Compound-reaction-enzyme-gene networks for OPCs. The dark blue circle represents the key target genes, the orange hexagon represents the key metabolite, the green square represents the key enzymes, and the yellow square represents the reactions: (a) OXPHOS pathway at 3-h post-stroke, (b) inositol phosphate metabolism pathway at 3-h post-stroke, (c) sphingolipid metabolism pathway at 3-day post-stroke, and (d) sphingolipid metabolism pathway at 7-day post-stroke.
Furthermore, we observed that apart from OXPHOS, several lipid metabolic pathways, including sphingolipid metabolism, fatty acid elongation, and the biosynthesis of unsaturated fatty acids, were significantly downregulated in mature OLs in the hyperacute phase (Figure 2(b)). These findings suggest that, in response to IS induced energy depletion, mature OLs may undergo marked metabolic reprogramming by broadly suppressing energy-demanding lipid biosynthetic pathways.
In contrast, inositol phosphate metabolism was significantly activated in OPCs and OLs in the 3-h post stroke group (Supplement Figure 1). Key genes such as Plcb4, Itpkc, Pik3cb, and Pten were enriched in OPCs (Figure 3(b)). This pathway is critical for intracellular calcium homeostasis and signal transduction.38–40 Although direct evidence is limited, the upregulation of these genes suggests a potential protective mechanism. For instance, Itpkc regulates IP3/IP4 levels, which may modulate calcium channels, thereby affecting cell migration.41,42 Interestingly, we observed the simultaneous upregulation of Pten (which inhibits PI3K/Akt signaling) and Pik3cb (which activates it). This suggests a complex, dynamic regulation of the PI3K/Akt pathway, which plays a bidirectional role in IS. 43 Furthermore, since neuron-OPC synapses rely on Ca2+-dependent signaling, 44 the activation of inositol phosphate metabolism may enhance the ability of OPCs to receive neuronal inputs. This could be a crucial early step for activity-dependent remyelination.
Acute phase (1 and 3 days)
At 1-day post-stroke, we observed a significant upregulation of OXPHOS pathway both in OPCs (Figure 2(c)) and OLs (Figure 2(d)). This indicates that oligodendrocyte lineage cells might shift from an energy-deficient state to a recovery phase. This rapid restoration of mitochondrial function is likely essential for them to support WM repair.
At 1-day post-stroke, mature OLs responded to IS by rapidly activating cholesterol biosynthesis pathways which including terpenoid backbone and steroid biosynthesis (Supplement Figure 2). Given that cholesterol constitutes 25%–30% of myelin, this upregulation likely represents an endogenous repair mechanism to support remyelination and generate neuroprotective metabolites like coenzyme Q10.45–47
By 1- and 3-day post-stroke, OPCs shifted toward metabolic recovery, marked by the activation of sphingolipid metabolism (Figure 2(c)). As a central intermediate of sphingolipid metabolism, physiological levels of ceramide are essential for myelination. Genes like Smpd2 and Sptlc1 were upregulated in the acute phase to generate ceramide (Figure 3(c)). Furthermore, its downstream metabolite, sphingosine-1-phosphate (S1P), actively promotes OPCs survival and differentiation.48–50
Chronic phase (7 days)
Conversely, excessive ceramide accumulation, a pathological feature in IS patients, exerts potential neurotoxicity by triggering mitochondrial dysfunction and oxidative stress.51–53 Given these adverse effects, the precise control of sphingolipid metabolism is vital. Interestingly, we observed a biphasic regulation of OPCs sphingolipid metabolism: an initial upregulation at 1- and 3-day post-stroke, followed by a significant downregulation at 7 days with genes like Smpd2 and Sptlc1 declined (Figure 3(d)). This late-stage downregulation indicates a protective negative feedback mechanism. This adaptation enables OPCs to avert ceramide-induced neurotoxicity during the chronic phase.
Conclusions
Specifically, our findings synthesize how stage-dependent metabolic shifts directly regulate distinct phases of oligodendrocyte lineage cells. In the early phase, the acute restoration of OXPHOS acts as an energetic checkpoint, providing the necessary ATP to overcome ischemia-induced cell-cycle arrest thereby enabling OPCs proliferation and supporting OLs recovery. Subsequently, the dynamic activation of inositol phosphate metabolism supports calcium-dependent signaling, which is essential for OPCs to sense neuronal signals and migrate toward demyelinated areas. 54 In the later recovery stages, the robust shift toward sphingolipid and cholesterol metabolism serves as a biosynthetic checkpoint. This late-stage reprogramming provides not only the massive structural lipids required for membrane wrapping 55 but also specific signaling molecules (such as S1P) that drive terminal differentiation and remyelination. 56
In summary, our analyses indicate that OPCs and mature OLs adopt distinct but coordinated metabolic programs during recovery from IS, reflecting their different functional roles in WM repair. Importantly, these findings suggest that metabolic reprogramming in OPCs is not merely a passive response to ischemic stress, but an active determinant of their functional state and fate.
Future directions
The oligodendrocyte lineage cells undergo distinct metabolic reprogramming following IS, suggesting that metabolic plasticity is a core driver of OPCs proliferation, migration, and remyelination. Regulators such as ATP synthase subunits, Itpkc, Pten, and S1P present viable intervention points to support cell survival and repair. Given that WM injury and failed remyelination largely contribute to long-term functional deficits, targeting these specific pathways may improve neurological recovery.
Translating these findings into clinical practice, however, poses significant challenges regarding cell-specific targeting and concentration control. While systemic AMPK activation offers acute neuroprotection,57,58 it risks disrupting subsequent WM repair. Specifically, OPCs rely on ALDOC-mediated AMPK suppression to preserve their capacity for remyelination. 33 Metabolic substrates like palmitic acid and ceramides exhibit biphasic or toxic effects at high doses.59,60 Therefore, localized delivery via CNS-targeted nanocarriers will be necessary to minimize off-target toxicity. 61
Clinical heterogeneity further complicates metabolic interventions. OPCs regulatory networks and metabolic capacities decline with aging and exhibit sex-specific differences, which inevitably alter therapeutic response thresholds.62–64 Spatial differences between the ischemic core and penumbra also dictate local metabolic demands. Future therapeutic designs will need to incorporate stratified models that account for age, sex, and specific stroke subtypes, such as isolated WM infarction.
Any metabolic therapy must also align with standard reperfusion timelines. While reperfusion restores oxygen and glucose, the concurrent surge in reactive oxygen species (ROS) damages OPCs mitochondria.29,30 Early-stage interventions might therefore require mitochondrial protection via antioxidants or NRF2/PPAR-γ activation,23,65 whereas later recovery phases would benefit from lipid metabolism modulation and substrate supplementation, such as taurine, to support remyelination.35,66,67
Ultimately, generalized nutritional support or simple energy supplementation is insufficient to drive effective WM repair. Advancing these metabolic concepts into practical therapies will require integrating targeted delivery, patient stratification, and stage-specific timing. Future in vivo studies validating these dynamic shifts will be critical for developing targeted rehabilitation strategies.
Our study has several limitations. The included datasets varied in ischemia duration and had limited sample sizes, which may not capture the full spectrum of oligodendrocyte metabolic remodeling. Furthermore, scRNA-seq captures transcriptional activity, which does not always strictly correlate with direct metabolic flux. The lack of spatial transcriptomic data also restricts our ability to distinguish metabolic responses between the ischemic core, penumbra, and remote regions. Lastly, despite batch correction and baseline merging, cross-study technical variations remain a potential source of bias.
Supplemental Material
sj-docx-1-jcb-10.1177_0271678X261458133 – Supplemental material for Metabolic reprogramming of oligodendrocyte lineage cells in ischemic stroke: Insights from literature review and a single-cell transcriptomic re-analysis
Supplemental material, sj-docx-1-jcb-10.1177_0271678X261458133 for Metabolic reprogramming of oligodendrocyte lineage cells in ischemic stroke: Insights from literature review and a single-cell transcriptomic re-analysis by Huijia Zhuang, Xisha Tang, Yu Zhang, Yang Han, Zhiyu Huang, Juan Xin and Hai Yu in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Acknowledgements
Author contributions
Hai Yu conceived and designed the study. Huijia Zhuang and Xisha Tang drafted the manuscript. Huijia Zhuang, Xisha Tang, and Yu Zhang performed the data analyze. Yang Han and Zhiyu Huang performed literature review and figure editing. Huijia Zhuang, Juan Xin, and Hai Yu revised and finalized the manuscript. All authors read and approved the final version of the paper.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (82372208, 82072132, and 82201341) and partly supported by the “Qimingxing” Research Fund for Young Talents (HXQMX0083).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical considerations
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Data availability statement
The transcriptomic and single-cell datasets analyzed in this study were obtained from public databases, including PRJNA912889 (NCBI SRA), CRA006645 (CNSA), and GSE227651 (GEO). The original data were generated and published by the respective authors cited in the reference list.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.