
Abstract
Ischemic stroke triggers a profound inflammatory response that exacerbates secondary brain injury, with infiltrating macrophages serving as major pathological mediators. Although previous studies have largely focused on immune cell–intrinsic mechanisms, the immunomodulatory role of cerebral endothelial cells in post-stroke inflammation remains poorly understood. In the current study, using single-cell RNA sequencing, we identified extensive inflammatory reprogramming in cerebral endothelial cells after stroke and discovered atypical chemokine receptor 1 (ACKR1) as a prominently upregulated regulator associated with chemokine sequestration and leukocyte recruitment. To determine its functional significance, we selectively overexpressed ACKR1 in cerebral endothelial cells using endothelial-targeted AAV-X1.1 in a mouse model of ischemic stroke. At 1 day after transient middle cerebral artery occlusion, endothelial ACKR1 overexpression preserved blood–brain barrier integrity, evidenced by maintained ZO-1 continuity, enhanced pericyte and astrocytic endfoot coverage, and reduced leakage of both exogenous tracers and endogenous IgG. By day 3, ACKR1 overexpression markedly reduced macrophage infiltration, decreased infarct volume, and improved neurological outcomes. These protective effects persisted through day 28, accompanied by improved sensorimotor recovery and attenuated brain atrophy. Collectively, our findings identify cerebral endothelial cells as active regulators of post-stroke neuroinflammation and reveal endothelial ACKR1 signaling as a potential therapeutic strategy for ischemic stroke.
Keywords
Introduction
Ischemic stroke is a leading cause of neurological disability and mortality worldwide, imposing a heavy burden on public health.1–4 One major contributor to the exacerbated brain tissue damage and resulting long-term neurological deficits is the excessive inflammatory responses after stroke, culminating in an inflammatory storm.5,6 Central to this inflammatory response is the infiltration of peripheral leukocytes into the brain, which triggers and amplifies this inflammatory cascade. 7 Accordingly, strategies aimed at limiting pathological leukocyte infiltration and inhibiting excessive inflammatory responses represent a promising therapeutic strategy for ischemic stroke.
Pathological leukocyte infiltration after ischemic stroke is regulated by a complex interplay of multiple factors. 8 Ischemia-induced activation of resident microglia triggers the massive release of chemokines, establishing a concentration gradient that guides peripheral leukocytes towards the ischemic lesion.9,10 Concurrently, early disruption of the blood–brain barrier (BBB) 11 compromises its ability to prevent abnormal immune cell migration, thereby facilitating infiltration and amplifying pathological inflammatory responses. 12 These processes are highly dependent on the aberrant accumulation of cerebral chemokines, with their concentrations directly influencing the severity of the inflammatory storm cascade. 13
Chemokine receptors play a crucial role in regulating chemokine levels and facilitating immune cell migration. 14 Classical chemokine receptors, such as CCLs and CXCLs, 15 are predominantly found on immune cells and mediate their directional migration by sensing chemokine gradients.16,17 In contrast, atypical chemokine receptors (ACKRs) exhibit distinct functional characteristics.18,19 Rather than participating in classical chemotactic signaling pathways, they regulate chemokine bioavailability by binding and sequestering them, thereby controlling local chemokine concentrations. 20 The function of ACKR is highly context-dependent and may differ substantially across disease models and pathological settings. In cardiovascular diseases such as atherosclerosis, ACKR1 has been shown to participate in macrophage-endothelial crosstalk and neutrophil transendothelial migration.21–23 However, the specific role that ACKRs play in post-stroke immune cell infiltration and BBB integrity remains poorly understood.
In this study, we used single-cell RNA sequencing (scRNA-seq) to examine the rold of ACKR1, which we found to be is specifically upregulated in brain endothelial cells after stroke. Subsequently, we developed a mouse model with targeted overexpression of ACKR1 in cerebral endothelial cells and subjected it to ischemic stroke. Our results show that ACKR1 significantly reduced macrophage infiltration, preserved BBB integrity, and improved post-stroke neurological function, providing a new therapeutic target for ischemic stroke.
Materials and methods
Animals
Specific pathogen-free (SPF) male C57BL/6J mice (2-month-old, 20–25 g; 4-week-old for AAV injection) were obtained from SPF (Beijing) Biotechnology Co., Ltd. and housed under SPF conditions. Four-week-old mice were used for AAV-mediated ACKR1 overexpression to allow a 3–4-week latency period, ensuring that mice reached an optimal body weight for tMCAO surgery at the time of overexpression. All protocols were approved by the Animal Welfare and Ethics Committee of Capital Medical University (permit no. AEEI-2021-058). All procedures were performed in compliance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011) and the ARRIVE guidelines. 24
Transient middle cerebral artery occlusion (tMCAO) model
Following established experimental protocols, 25 tMCAO was induced in 2‑month‑old male C57BL/6J mice. Surgical anesthesia was initiated with isoflurane (3%–4% for induction, 1.5%–2% for maintenance). The right middle cerebral artery (MCA) was occluded by a 6–0 silicone‑coated monofilament (0.20 ± 0.02 mm in diameter, coated segment 5–6 mm long) inserted through the external carotid artery. After 90 min of occlusion, the filament was withdrawn to allow reperfusion. Regional cerebral blood flow (CBF) was continuously monitored using laser speckle contrast imaging (LSCI); successful occlusion was defined as a reduction in CBF to ⩽30% of baseline, and successful reperfusion was confirmed by a return to ⩾70% of baseline. Animals that died intraoperatively or failed to meet the CBF criteria were excluded. Sham‑operated mice underwent identical surgical procedures except for filament insertion. Both the surgical team and the outcome assessors were masked to group assignment. Mice were randomly allocated into tMCAO or sham groups before surgery.
Single‑cell RNA sequencing and analysis
Single‑cell suspensions were prepared from the ischemic ipsilateral hemisphere of mice 3 days post‑tMCAO and from the corresponding brain tissue of sham‑operated controls. Libraries were generated using the 10× Genomics Chromium platform, and sequencing data were processed with the Cell Ranger pipeline. Quality control, normalization, integration, clustering, and cell‑type annotation were performed using the Seurat package (v4.4.0) in R. Low‑quality cells were filtered (transcripts <1500 or >30,000, mitochondrial fraction >20%), data were normalized, and batch effects were corrected with Harmony. Graph‑based clustering and UMAP were applied for dimensionality reduction, and cell types were identified by canonical marker genes. Differential expression, Gene Ontology enrichment, and endothelial subclustering analyses were conducted as previously described. 26 Briefly, differential expression between tMCAO and sham groups was tested using the Wilcoxon rank‑sum test (FindMarkers function). Genes expressed in <10% of cells in either group were discarded (significance required |log2FC| > 0.25 and Bonferroni‑adjusted p < 0.05). Gene Ontology enrichment (Biological Process) was performed with clusterProfiler, using Benjamini‑Hochberg adjusted p < 0.05 and enrichment factor >1.5 as thresholds. For endothelial subpopulation analysis, endothelial cells were subsetted by marker genes and re‑clustered (resolution 0.5). Distinct subclusters were visualized by UMAP. Expression of Ackr1 and other atypical chemokine receptors was profiled across cell types and conditions using violin and feature plots.
Assessment of brain injury
Brain injury assessment was performed as described previously.25,27 Magnetic resonance imaging (MRI) was conducted on a 7 T Bruker BioSpec 70/30 system. Three days after reperfusion, T2‑weighted images were acquired with 0.5 mm slice thickness. Hyperintense regions on T2‑weighted scans denoted infarcted tissue. Infarct volume was calculated by summing the manually or semi‑automatically delineated infarct areas across all slices and multiplying by slice thickness (0.5 mm). Edema percentage was determined as ((ipsilateral hemisphere volume − contralateral hemisphere volume)/contralateral hemisphere volume) × 100. All T2‑based quantifications were performed by an investigator blinded to group assignment using ITK‑SNAP and ImageJ. For TTC staining, after final MRI scans, mice were euthanized, and brains were sectioned into 1‑mm coronal slices. Sections were incubated in 2% 2,3,5‑triphenyltetrazolium chloride at 37 °C for 20 min, then fixed in 4% paraformaldehyde. Infarct areas appear white.
Flow cytometry
Three days after tMCAO or sham surgery, flow cytometry was conducted as previously described.28,29 Briefly, for flow cytometric analysis, deeply anesthetized mice were transcardially perfused with normal saline. The ipsilateral cerebral hemisphere was collected and processed using the Neural Tissue Dissociation Kit (Cat. 130‑092‑628; Miltenyi Biotec). Tissue dissociation was performed on an automated RWD DSC-400 Single-Cell Suspension Dissociator (RWD Life Science), following the manufacturer’s instructions. The resulting homogenate was filtered through a 70 µm cell strainer and resuspended to a density of 1 × 106 cells/mL. Cell suspensions were incubated with a Zombie Aqua Fixable Viability Kit (Cat. 423102; BioLegend) to discriminate live/dead cells, followed by staining with fluorescence-conjugated antibodies against: CD45 (PerCP/Cyanine5.5, Cat. 103132; BioLegend), CD11b (APC, Cat. 101212; BioLegend), Ly6G (Brilliant Violet 421, Cat. 127628; BioLegend), and CX3CR1 (PE, Cat. 149006; BioLegend). Stained samples were acquired on an LSRFortessa flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (FlowJo, LLC).
Immunofluorescence, 3D reconstruction, and histology
For immunofluorescence staining, mice were transcardially perfused with 4% paraformaldehyde (PFA) under deep anesthesia. The extracted brains were immersed in the same fixative at 4 °C for 24 h, followed by sequential dehydration in 20% and 30% sucrose solutions (each step until the tissue sank). The dehydrated samples were embedded in optimal cutting temperature (OCT) compound and stored at −20 °C. Coronal sections (40 µm thick) were cut on a cryostat. After washing with phosphate buffered saline (PBS), sections were blocked for 1 h at room temperature in PBS containing 0.2% Triton X‑100 and 3% bovine serum albumin, then incubated with primary antibodies overnight at 4 °C. The following primary antibodies were used: Lectin 649 (Vector Labs, DL-1178-1, 1:300), CD11b (Servicebio, GB115689, 1:500), CCR2 (Proteintech, 16153-1-AP, 1:200), ACKR1 (MCE, HY-P81909, 1:50), Flag (Invitrogen, MA1-91878, 1:200), NeuN (Proteintech, 66836-1, 1:100), GFAP (Abcam, ab7260, 1:1000), ZO-1 (Abcam, ab221547, 1:200), AQP4 (CST, 59678s, 1:500), PDGFRβ (Huabio, ET1605-20, 1:200), and MAP2 (Proteintech, 17490-1-AP, 1:500). After washing, sections were incubated with species‑matched fluorophore‑conjugated secondary antibodies for 1–2 h at room temperature. Secondary antibodies included: Alexa Fluor 488 Goat Anti-Mouse IgG (Invitrogen, A11029, 1:500), Alexa Fluor 488 Goat Anti-Rabbit IgG (Invitrogen, A11034, 1:500), and Alexa Fluor 594 Goat Anti-Rabbit IgG (Invitrogen, A11037, 1:500). Nuclei were counterstained with DAPI. For three‑dimensional morphological analysis, Z‑stack images were acquired using a confocal microscope with a step size of 1 µm under consistent acquisition parameters. The Z‑stack sequences were imported into Imaris software (v10.1.0; Oxford Instruments). Regions of interest (ROIs) were defined from the DAPI channel, and the target fluorescence channels were segmented and 3D‑reconstructed within the surface module using uniform intensity thresholds across all samples. For splenic histology, spleens were collected, fixed in 4% PFA, and paraffin‑embedded. Sections (5 µm) were stained with hematoxylin and eosin (H&E). Digital images were captured from multiple randomly selected fields of view. Using ImageJ software, the areas of red pulp and white pulp were measured per field of view. All quantifications were performed by an investigator blinded to group assignment.
Endothelial-specific ACKR1 overexpression
Four‑week‑old male C57BL/6J mice underwent bilateral intracerebroventricular stereotaxic injection. Under isoflurane anesthesia and with a stereotaxic frame, a 10 μL Hamilton syringe was used to deliver the viral solution at coordinates (from bregma): AP −0.5 mm, ML ±1.0 mm, DV −2.0 mm. Each hemisphere received 5 μL of AAV‑X1.1 30 encoding Ackr1 carrying the ACKR1 overexpression cassette (CAG‑ACKR1‑3×FLAG‑T2A‑EGFP‑SV40 PolyA) or an empty control vector (CAG‑3×FLAG‑T2A‑EGFP‑SV40 PolyA). The titer was 1 × 1013 vg/mL, corresponding to a total viral load of 5–8 × 1010 vg per animal (2.5–4 × 1010 vg per hemisphere). The injection speed was 0.5 μL/min, and the needle was kept in place for 2 min after injection to prevent reflux. Postoperatively, animals received analgesic and antibiotic care and were allowed 3–4 weeks for stable transgene expression before tMCAO surgery.
Blood–brain barrier (BBB) permeability assessment
For evaluation of BBB leakage, two fluorescent tracers were administered via tail vein injection 15 min before euthanasia: AF647‑conjugated cadaverine (1 kDa, 100 μg per mouse, 100 μL; Invitrogen, A30679) and FITC‑conjugated dextran (20 kDa, 2 mg per mouse, 100 μL; Sigma, FD20S). At the indicated time after tracer injection, mice were deeply anesthetized with isoflurane and transcardially perfused with ice‑cold physiological saline. The ipsilateral ischemic hemisphere was rapidly dissected, frozen on dry ice, and stored at −80 °C for later sectioning. Coronal sections (60 µm thick) were prepared using a cryostat. For detection of endogenous IgG (150 kDa) extravasation, sections were blocked in PBS containing 3% BSA for 1 h at room temperature, then incubated overnight at 4 °C with Alexa Fluor 594‑conjugated goat anti‑mouse IgG (1:200; Invitrogen, A11032). After washing, sections were mounted and imaged. The volume of tracer leakage or IgG extravasation was quantified as previously described. 31
Neurological function assessments
Behavioral tests were performed as described previously. 25 All assessments were conducted 3 days after tMCAO by an investigator blinded to group assignment. The modified Garcia score (0–15, higher indicates better function) evaluated overall neurological status based on body proprioception, posture symmetry, forelimb extension, corner turning, and gripping ability. Sensorimotor function was assessed using the adhesive removal test, recording the time to first contact and to completely remove a piece of tape from the contralateral forepaw (cutoff 60 s). Motor coordination was measured using the grid walking test, calculating the percentage of contralateral forelimb foot faults relative to the total number of steps. Spontaneous locomotor activity was evaluated in an open-field arena, where the total distance traveled over 5 min was recorded using SMART 3.0 video tracking software (Panlab SMART Video Tracking System; Panlab, Barcelona, Spain).
Statistical analysis
All data are shown as mean ± SD. Normality was evaluated using the Shapiro–Wilk test. Two-group comparisons were made using unpaired two‑tailed Student’s t‑test or Mann–Whitney U test, as appropriate. For comparisons involving three or more groups, one‑way or two‑way ANOVA was applied, followed by Tukey’s post hoc test. Correlations were assessed with Pearson’s (normal data) or Spearman’s (non‑normal or clustered data) test. A significance threshold of p < 0.05 was set. GraphPad Prism 10 was used for all analyses.
Results
Macrophage infiltration dominates cellular remodeling at day 3 in tMCAO mice
We first established the tMCAO model of focal cerebral ischemia–reperfusion injury and conducted key quality-control experiments to confirm its validity. Ipsilateral CBF was monitored in real time using LSCI (Figure 1(a) and (b)). Compared with baseline (Pre), CBF decreased sharply during middle cerebral artery occlusion (Occ) and was restored after reperfusion (Rep), confirming successful induction of ischemia and reperfusion. At 3 days post-surgery, both MRI and TTC staining revealed well-defined infarcts in the ipsilateral hemisphere of tMCAO mice (Figure 1(c)). Quantitative MRI analysis revealed a substantial infarct volume (Figure 1(d)), confirming robust damage in the ischemic model.
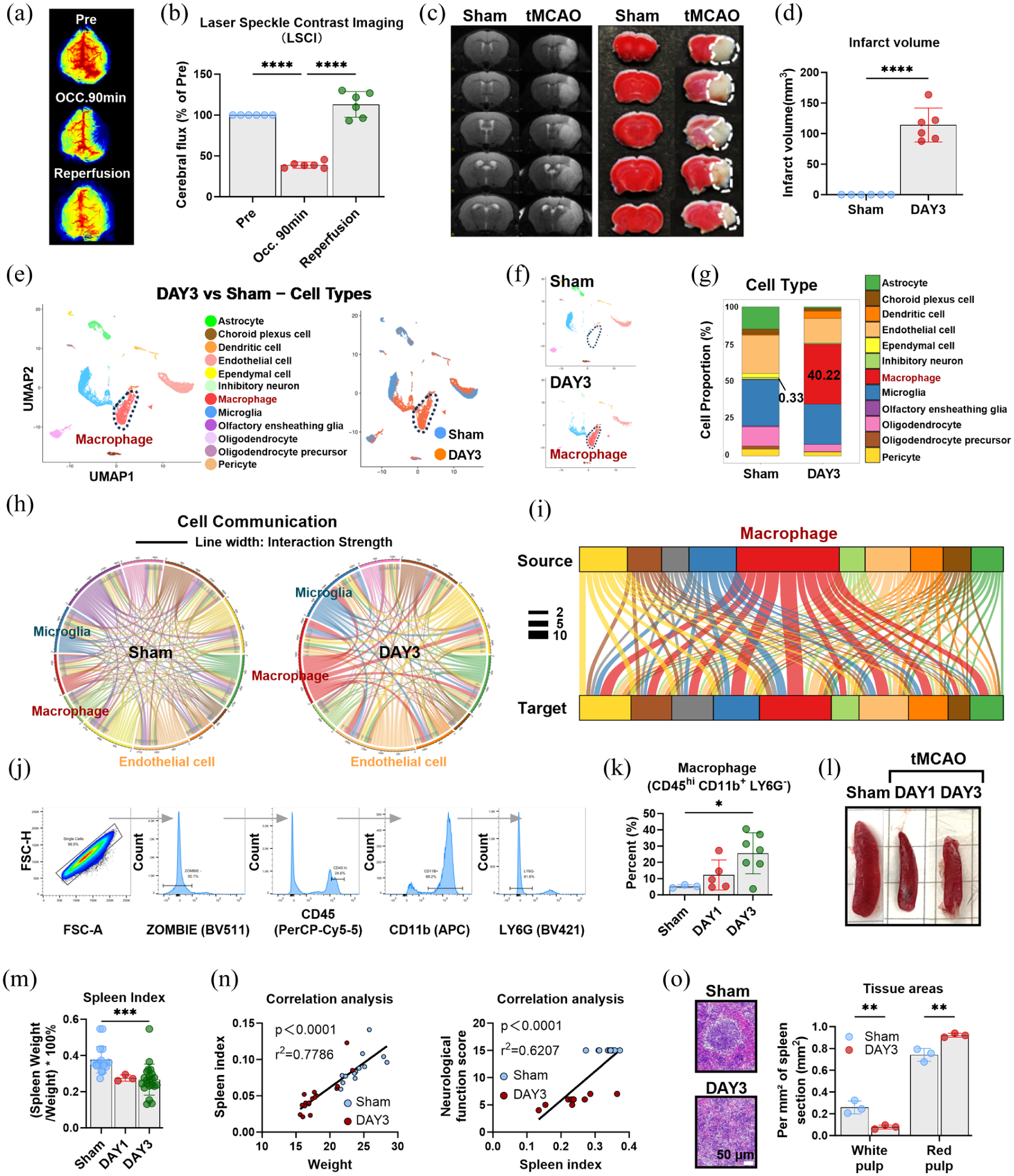
Macrophage infiltration drives acute-phase cellular remodeling after tMCAO: (a) representative LSCI of CBF at baseline (Pre), during middle cerebral artery occlusion (Occ, 90 min), and after reperfusion (Rep), (b) quantification of ipsilateral CBF normalized to baseline, (c) T2-weighted MRI and TTC staining at 3 days post-tMCAO showing infarct lesions, (d) quantification of infarct volume based on MRI, (e) UMAP visualization of major cell types from scRNA-seq of sham and tMCAO (3 days) brain tissues, (f) enlarged UMAP highlighting the expansion of the macrophage cluster after tMCAO, (g) quantification of cell-type proportions, showing the relative abundance of each cell type, (h) chord diagram illustrating strengthened interactions among macrophages, microglia, and endothelial cells after tMCAO, (I) sankey diagram showing macrophages as a central communication hub in the ischemic brain, (j) flow cytometry gating strategy for macrophages (CD45+CD11b+Ly6G−), (k) quantification of macrophages at 1 and 3 days post-tMCAO, (l) gross appearance of spleens from sham and tMCAO mice (1 and 3 days), (m) spleen index (spleen weight/body weight ratio) in sham and tMCAO mice, (n) correlation analysis between spleen index and body weight (left) or neurological score (right) in tMCAO mice, and (o) H&E staining of spleen sections (left) and quantification of splenic white pulp area (right). Data are presented as mean ± SD (n = 3–22); statistical significance was determined by one-way ANOVA followed by Tukey’s post hoc test. Bar = 50 μm in (o).
To characterize the global cellular landscape of the ischemic brain during the acute stage, scRNA-seq was performed on brain tissues from sham and tMCAO mice at 3 days post-injury. UMAP visualization showed a striking shift in cellular composition following tMCAO, marked by a pronounced expansion of the macrophage cluster (Figure 1(e) and (f)). Quantitative analysis confirmed that macrophages dramatically increased from 0.33% in sham mice to 40.22% in tMCAO mice (Figure 1(g)), indicating that this expansion represented the most significant cellular change in the ischemic brain and predominantly contributed to acute-phase cellular remodeling.
To validate the scRNA-seq observations, flow cytometry was performed on brain tissues at 1 and 3 days post-tMCAO. Using a gating strategy to identify infiltrating myeloid cells (CD45hiCD11b+LY6G−), we detected a progressive increase in their percentage from days 1 to 3 (Figure 1(j) and (k)), consistent with the scRNA-seq data.
To further clarify how ischemia reshapes intercellular signaling networks within the cerebral microenvironment, cell-cell communication analysis was performed. In sham mice, intercellular communication primarily involved interactions among resident brain cells, including neurons, glial cells, and vascular-associated cells. In contrast, tMCAO induced a profound shift in the communication landscape, significantly enhancing interactions among macrophages, microglia, and endothelial cells (Figure 1(h)). Notably, macrophages emerged as a central communication hub in the ischemic brain, displaying extensive signaling crosstalk with nearly all major cell types (Figure 1(i)).
Given the spleen’s function as a major reservoir for myeloid cells during systemic inflammation, splenic responses were assessed in tMCAO mice. Gross anatomical examination showed marked splenic atrophy post-tMCAO (Figure 1(l)) and a significantly reduced spleen index (spleen weight/body weight ratio) compared with sham mice (Figure 1(m)). Correlation analysis revealed a positive correlation between spleen index and both body weight and neurological function scores in tMCAO mice (Figure 1(n)), indicating that splenic atrophy is closely linked to the severity of ischemic injury. Histological analysis revealed profound structural disruption and marked shrinkage of the splenic white pulp, with a relative expansion of the myeloid-rich red pulp compartment in tMCAO mice (Figure 1(o)).
Collectively, these findings indicate that macrophage infiltration serves as the primary driver of acute-phase cellular remodeling in the tMCAO model. This highlights the importance of elucidating mechanisms that regulate macrophage recruitment into the ischemic brain.
Endothelial cells display augmented chemotactic activity and immune activation, along with impaired BBB homeostasis in tMCAO mice
Building on our finding that macrophage infiltration primarily drives acute-phase cellular remodeling in tMCAO mice, we next sought to identify the key regulators of this inflammatory cascade. Microglia are known as the primary innate immune responders in the ischemic brain, acting as early initiators of neuroinflammation. Consistent with this, our transcriptomic analysis of microglia revealed robust upregulation of genes associated with type II interferon signaling, innate immune activation, leukocyte migration, and cell chemotaxis post-tMCAO (Figure 2(a) and (b)), confirming their early pro-inflammatory activation during the acute ischemic phase.
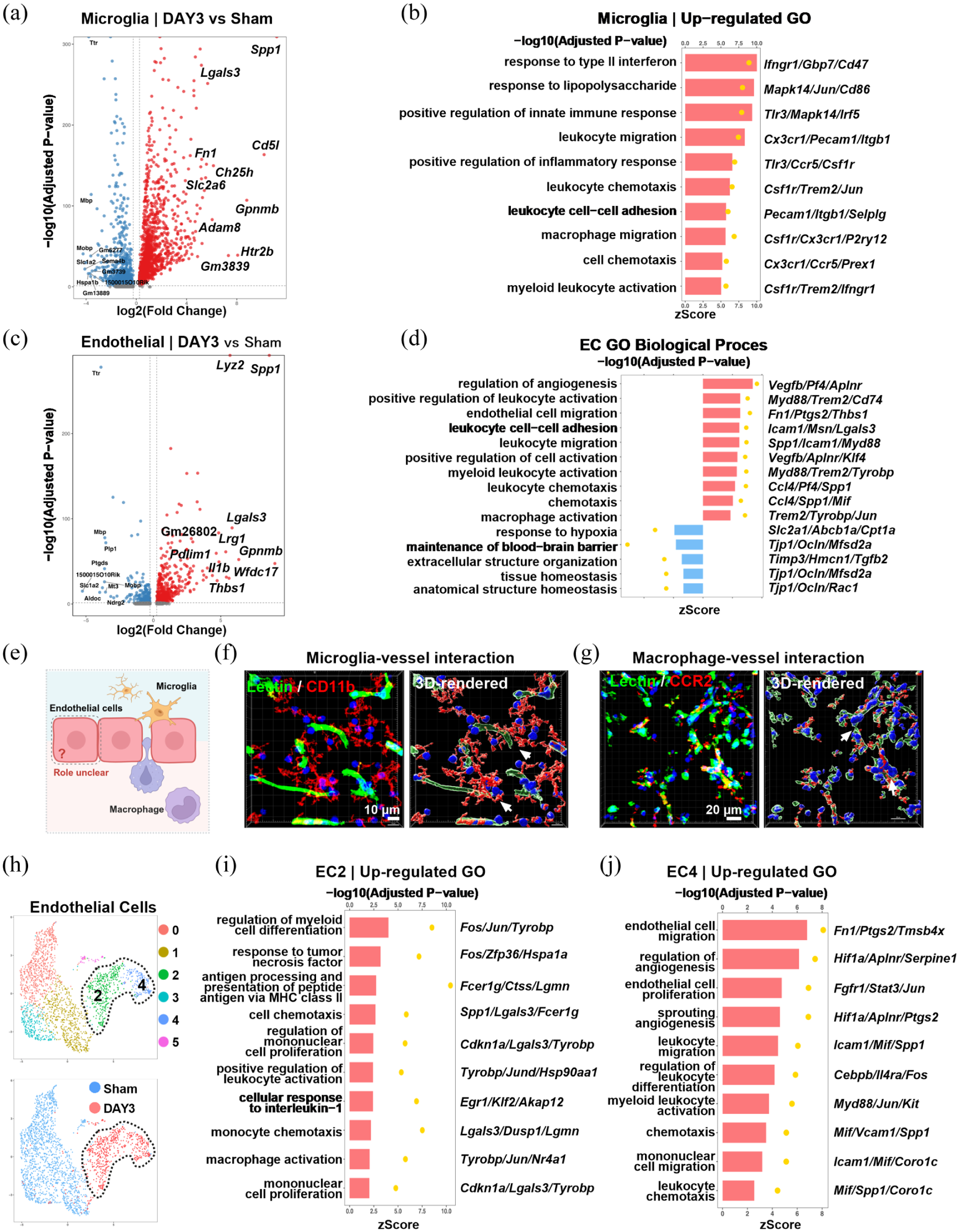
Cerebral endothelial cells acquire chemotactic and pro-inflammatory signatures with concurrent BBB dysfunction following tMCAO: (a) volcano plot of differentially expressed genes in microglia from sham and tMCAO mice (3 days post‑tMCAO), (b) GO enrichment analysis of upregulated genes in microglia, (c) volcano plot of differentially expressed genes in endothelial cells from sham and tMCAO mice (3 days post‑tMCAO), (d) GO enrichment analysis of upregulated genes in endothelial cells, (e) schematic diagram illustrating the anatomical position of cerebral endothelial cells at the interface between the central nervous system and the circulation, (f) immunofluorescence and 3D reconstruction showing microglial processes (Iba1+) contacting cerebral vessels (Lectin+) at 3 days after tMCAO, (g) immunofluorescence and 3D reconstruction of CCR2+ macrophages interacting with CD31+ endothelial cells at 3 days after tMCAO, (h) UMAP visualization of endothelial subclusters (EC0–EC5) from sham and tMCAO mice, (i) GO enrichment analysis of the EC2 subcluster (3 days post‑tMCAO), and (j) GO enrichment analysis of the EC4 subcluster (3 days post‑tMCAO). Bar = 10 μm in (f) and bar = 20 μm in (g).
Further analysis of endothelial cells, which are anatomically positioned at the critical interface between the central nervous system and circulating immune cells, revealed an unexpectedly broad immunomodulatory response following tMCAO. Notably, this profile spanned the entire cascade of myeloid cell recruitment and activation, from chemokine signaling and leukocyte adhesion to migration and activation, with particularly strong enrichment for macrophage activation pathways (Figure 2(c) and (d)). By contrast, gene sets associated with BBB integrity and tissue homeostasis were markedly underrepresented, suggesting concurrent impairment of BBB function (Figure 2(d)).
Consistent with their anatomical location (Figure 2(e)), cerebral endothelial cells exhibited enhanced interactions with both resident microglia and infiltrating macrophages after tMCAO. Immunofluorescence and 3D reconstruction analyses revealed that resident microglia established close contacts with cerebral vessels (Figure 2(f)), while infiltrating CCR2+ macrophages formed extensive direct interactions with endothelial cells (Figure 2(g)). Together, these findings support a model in which endothelial cells act as central mediators of crosstalk between central and peripheral immune responses in the ischemic brain.
To further dissect the endothelial heterogeneity underlying these responses, subclustering analysis identified six distinct endothelial subpopulations (EC0–EC5), with notable shifts in abundance between sham and tMCAO mice (Figure 2(h)). Among these, EC2 and EC4 subclusters were selectively expanded post-tMCAO and exhibited complementary functional signatures. EC2 was exclusively enriched in pro-inflammatory pathways associated with myeloid cell recruitment and activation, including monocyte chemotaxis, mononuclear cell proliferation, and macrophage activation (Figure 2(i)). In contrast, EC4 displayed a dual functional signature, combining vascular remodeling processes, such as angiogenesis, sprouting angiogenesis, endothelial cell proliferation/migration, and leukocyte infiltration, including leukocyte chemotaxis, migration, differentiation, and activation (Figure 2(j)). These findings suggest that specialized endothelial subpopulations cooperatively coordinate myeloid cell recruitment and vascular adaptations within the ischemic microenvironment.
Collectively, these data suggest that microglia act as early initiators, while cerebral endothelial cells function as central mediators that translate local signals into systemic leukocyte recruitment, driving robust macrophage infiltration, alongside impaired BBB homeostasis post-stroke.
Cerebral endothelial-specific ACKR1 overexpression alleviates tMCAO-triggered macrophage infiltration
Building on our observation that endothelial cells act as central mediators of myeloid cell recruitment after tMCAO, we next sought to identify key regulators governing this process. Given that atypical chemokine receptors (ACKRs, including ACKR1–4) are predominantly expressed on endothelial cells and are known to modulate local chemokine bioavailability, we profiled their expression in our endothelial cell scRNA-seq dataset.
Among the ACKR family, Ackr1 emerged as the only member robustly upregulated in endothelial cells following tMCAO. This induction was most prominent in the pro-angiogenic, pro-inflammatory EC4 subcluster (Figure 3(a)), while expression was nearly undetectable in sham controls. In contrast, Ackr2 and Ackr4 showed negligible expression, whereas Ackr3, although broadly expressed, was downregulated following ischemia (Supplementary Figure 1). Immunofluorescent staining further confirmed upregulation of ACKR1 in lectin-labeled cerebral endothelial cells in the ischemic brain (Figure 3(b) and Supplementary Figure 2), in agreement with our transcriptomic data. Together, these findings identify ACKR1 as a distinct and inducible endothelial receptor that we hypothesize may regulate immune cell recruitment after ischemia.
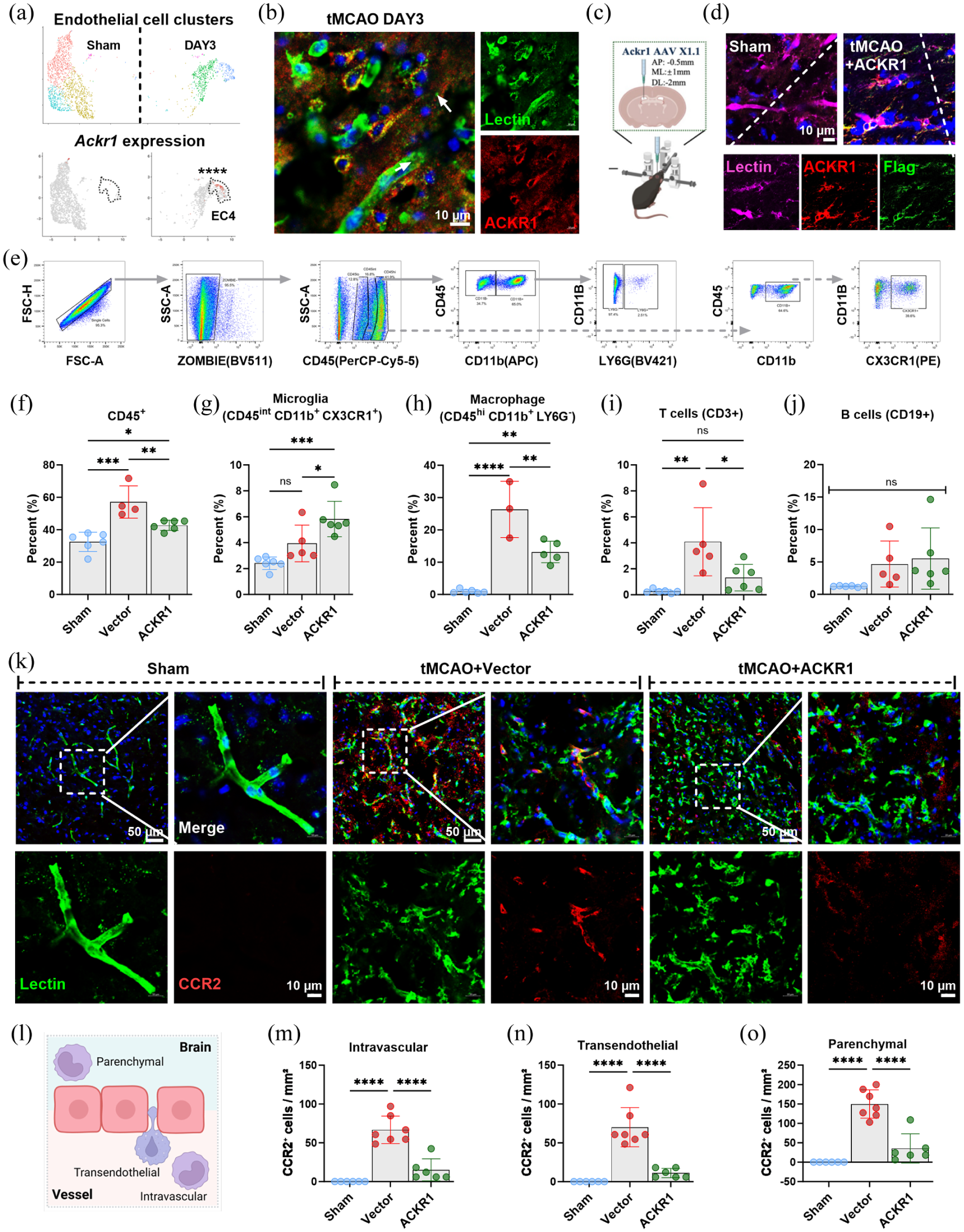
Endothelial-specific ACKR1 overexpression attenuates tMCAO-induced macrophage recruitment into the brain: (a) UMAP plots showing Ackr1 expression levels in single-cell transcriptomics data from sham versus tMCAO groups at 3 days post-occlusion, (b) representative immunofluorescence staining of ACKR1 at 3 days after tMCAO, (c) schematic diagram of stereotaxic injection for Ackr1 overexpression, (d) representative immunofluorescence staining of ACKR1 following exogenous Ackr1 overexpression, (e) flow cytometry gating strategy for evaluating brain immune cells at 3 days after stroke, (f–j) quantification of immune cell population changes in each group: (f) CD45+ cells, (g) microglia (CD45+CD11b+CX3CR1+), (h) macrophages (CD45+CD11b+Ly6G−), (i) T cells (CD3+), (j) B cells (CD19+), (k) representative immunofluorescence images of CCR2+ cells in each group at 3 days after tMCAO, (l) schematic diagram illustrating the localization of macrophages relative to cerebral vessels: intravascular (within the vessel lumen), transendothelial (crossing the vessel wall), and parenchymal (within brain tissue), (m–o) quantification of CCR2+ cell density in the intravascular (m), transendothelial (n), and parenchymal (o) compartments for each group. Data are presented as mean ± SD (n = 4–6); statistical significance was determined by one-way ANOVA followed by Tukey’s post hoc test. Bar = 10 µm in (b) and (d), bar = 50 µm and 10 µm in (k).
To explore its functional role, we overexpressed ACKR1 specifically in endothelial cells using an AAV-X1.1-mediated approach (Figure 3(c)). Enhanced expression in the brain endothelium was validated by ACKR1 and Flag-tag staining (Figure 3(d)). The overexpression efficiency of ACKR1 was satisfactory, elevating the proportion of ACKR1-positive endothelial cells in the ischemic penumbra from approximately 7% to about 40%. Moreover, the overexpression was endothelial-specific, as no co-localization of ACKR1 with NeuN (neurons), CD11b (microglia/macrophages), or GFAP (astrocytes) was detected in the ACKR1-overexpressing group (Supplementary Figure 3). Flow cytometric analysis of cells isolated from the infarcted hemisphere (Figure 3(e)–(j)) revealed that the tMCAO-induced increase in total CD45+ immune cells was significantly reduced in mice overexpressing ACKR1 (Figure 3(e) and (f)). This reduction was most notable in infiltrating CD45hiCD11b+LY6G− macrophages. CD3+ T lymphocytes were also decreased, while resident CD45intCD11b+CX3CR1+ microglia displayed a compensatory increase (Figure 3(g)–(j)).
Given the pronounced effect of macrophage infiltration, we assessed their spatial distribution within the ischemic penumbra using in situ immunofluorescence. CCR2+ macrophages were quantified across intravascular (within the vessel lumen), perivascular/at the vessel interface (on the endothelial surface or adjacent to vessels), and parenchymal (within the brain tissue proper) compartments. ACKR1 overexpression markedly reduced tMCAO-triggered monocyte/macrophage infiltration in all three locations (Figure 3(k)–(o)).
Overall, these results indicate that cerebral endothelial-specific ACKR1 overexpression potently limits peripheral macrophage infiltration in to the ischemic brain, supporting its role as a key modulator of leukocyte trafficking in acute ischemic stroke.
Cerebral endothelial-specific ACKR1 overexpression preserves blood–brain barrier integrity in tMCAO mice
Building on our finding that ACKR1 overexpression potently inhibits macrophage infiltration, we next investigated its impact on the BBB, the primary physical and functional barrier regulating immune cell entry into the central nervous system. Following ischemia, BBB breakdown occurs early, creating a permissive gateway for circulating macrophages to infiltrate the brain parenchyma. Importantly, this early BBB disruption is driven, in part, by elevated local chemokine gradients. Although ACKR1 modulates chemokine bioavailability, it remains unclear whether it also influences this early phase of vascular destabilization.
To address this gap, we assessed the impact of ACKR1 overexpression on the structural and functional integrity of the BBB 1 day post-tMCAO. These structural stainings were performed in the ischemic penumbra. Co-staining key BBB components, including the endothelial tight-junction protein ZO-1, the pericyte marker PDGFRβ, and the astrocytic endfoot protein AQP4, with the vascular marker Lectin (Figure 4(a)) revealed severe BBB disruption post-tMCAO. This disruption was evident through fragmented ZO-1, reduced pericyte, and astrocytic endfoot coverage. ACKR1 overexpression significantly preserved the structural integrity of the above three indicators (Figure 4(b)–(f)). Moreover, correlation analyses revealed that the integrity of these BBB components (ZO-1, AQP4, and PDGFRβ colocalization with lectin) positively correlated with neurological function scores and negatively correlated with infarct volume, further supporting the protective role of ACKR1 (Supplementary Figures 4–6).
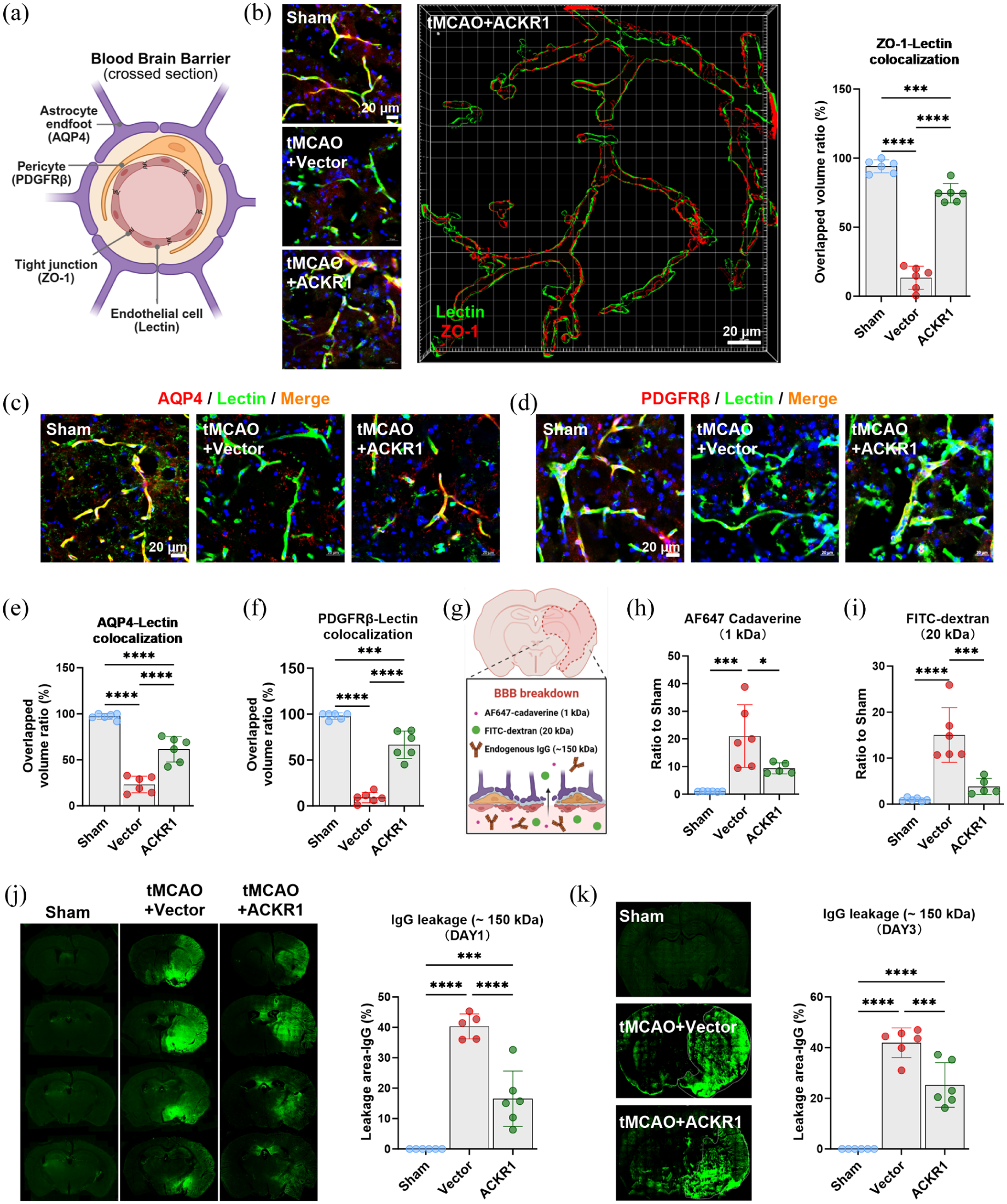
Endothelial ACKR1 overexpression maintains blood–brain barrier structural and functional integrity in the acute phase of tMCAO: (a) schematic diagram illustrating the assessment of BBB structural integrity, (b) representative immunofluorescence images, 3D reconstructions, and quantification of ZO-1 expression in each group at 1 day after tMCAO, (c) representative immunofluorescence images of PDGFRβ expression in each group at 1 day after tMCAO, (d) representative immunofluorescence images of AQP4 expression in each group at 1 day after tMCAO, (e) quantitative analysis of PDGFRβ+ pericyte coverage on cerebral vessels at 1 day after tMCAO, (f) quantitative analysis of perivascular AQP4 polarity at 1 day after tMCAO, (g) schematic diagram illustrating the assessment of BBB functional integrity, (h) leakage index (ratio to sham) of AF647-conjugated cadaverine in each group at 1 day after tMCAO, (i) leakage index (ratio to sham) of FITC-conjugated dextran in each group at 1 day after tMCAO, (j) representative images and quantification of endogenous IgG extravasation area in each group at 1 day after tMCAO, and (k) representative images and quantification of endogenous IgG extravasation area in each group, 3 days after tMCAO. Data are presented as mean ± SD (n = 6); statistical significance was determined by one-way ANOVA followed by Tukey’s post hoc test. Bar = 20 µm in (b–d).
Functionally, BBB permeability was evaluated using exogenous tracers of varying molecular weights and measuring endogenous IgG leakage (Figure 4(e)). These functional assessments were conducted in the ipsilateral hemisphere. One day post-tMCAO, robust extravasation of both small (1 kDa AF647 Cadaverine) and large (20 kDa FITC–dextran) tracers was detected, indicating widespread barrier disruption. ACKR1 overexpression markedly reduced the leakage of both tracers, confirming improved functional BBB integrity at this early time point (Figure 4(h) and (i)). Consistent with the tracer results, endogenous IgG leakage on day 1 following tMCAO was also significantly attenuated in mice overexpressing ACKR1 (Figure 4(j)). To further assess the longevity of this protection, we evaluated endogenous IgG extravasation in the subacute phase (day 3), which revealed that the BBB-preserving effect of ACKR1 overexpression persisted, with markedly reduced IgG leakage compared to sham (Figure 4(k)).
Collectively, these data reveal that cerebral endothelial-specific ACKR1 overexpression preserves both structural and functional BBB integrity in the acute phase of ischemic stroke. By reducing early BBB disruption driven by chemokines, ACKR1 stabilizes vascular homeostasis and prevents circulating macrophages from infiltrating the brain, reinforcing its role in regulating both vascular protection and macrophage recruitment in the ischemic brain.
Cerebral endothelial-specific ACKR1 overexpression improves neurological function and mitigates brain injury in tMCAO mice
Given that endothelial ACKR1 overexpression preserves early BBB integrity and reduces macrophage infiltration, we investigated whether these protective effects translated into improved functional outcomes after tMCAO. Three days after MCAO, we evaluated neurological function, sensorimotor, and motor coordination using several behavioral tests (Figure 5(a)).
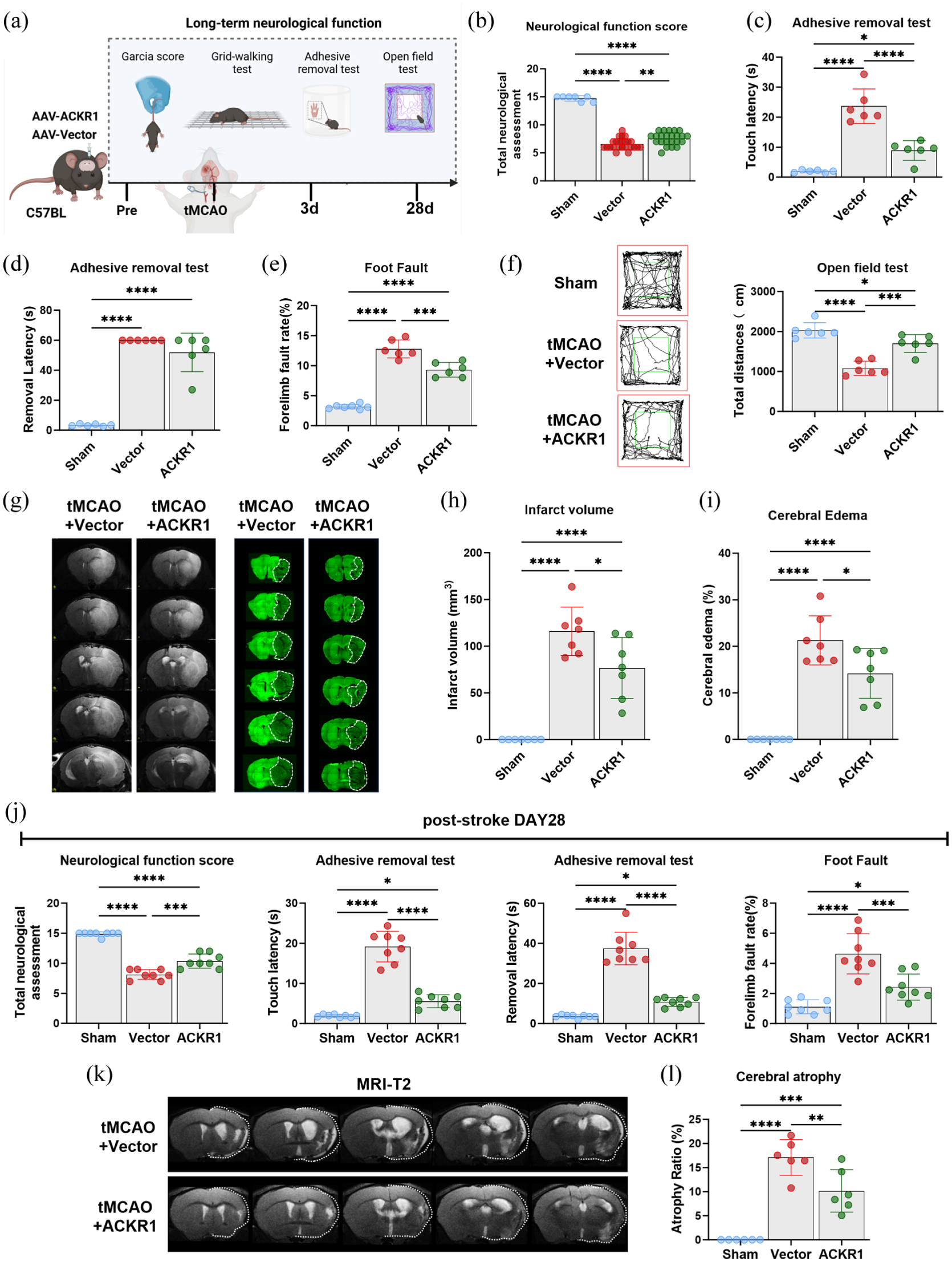
Endothelial-specific ACKR1 overexpression ameliorates neurological deficits and reduces brain injury after tMCAO: (a) schematic diagram illustrating the assessment of long-term neurological function after tMCAO, (b) neurological function assessed by the modified Garcia score (0–15, higher score indicates better function) at 3 days after tMCAO, with schematic diagrams (left) and statistical analysis (right), (c–e) sensorimotor function evaluation: (c) quantification of touch latency (adhesive removal test), (d) quantification of removal latency (adhesive removal test), (e) quantification of forelimb foot fault rate (foot fault test), (f) representative traces of the open field test (left) and quantification of total distance traveled (right) at 3 days after tMCAO, (g, h) infarct volume assessment: (g) representative T2-weighted MRI coronal images, (h) quantification of total infarct volume, (i) quantification of brain edema percentage at 3 days after tMCAO. Data are presented as mean ± SD (n = 6–24 per group), (j) quantitative analysis of neurological function at 28 days after tMCAO, including modified Garcia score, adhesive removal test, and foot fault test (n = 8 per group), and (k, l) representative T2-weighted MRI images (k) and quantitative analysis (l) of brain atrophy in each group at 28 days after tMCAO (n = 6 per group). Statistical significance was determined by one-way ANOVA followed by Tukey’s post hoc test.
Total neurological function scores, assessing sensorimotor deficits, reflexes, and overall neurological status, were markedly reduced in tMCAO mice treated with Vector compared to sham controls. ACKR1 overexpression significantly improved these scores (Figure 5(b)), indicating robust early functional recovery. To further evaluate sensorimotor enhancements, adhesive removal tests were conducted. ACKR1-overexpressing mice exhibited considerably shorter touch latency and removal latency than tMCAO mice treated with Vector (Figure 5(c) and (d)). Consistently, tMCAO-induced forelimb foot fault showed a reduced error rate in the affected limb in ACKR1-overexpressing mice compared to their Vector-treated counterpart (Figure 5(e)), reflecting enhanced motor coordination. Results in the open field test indicated that tMCAO-induced decreases in spontaneous exploratory behavior were partially rescued in the ACKR1 group, with these mice traveling significantly longer total distances than tMCAO mice treated with Vector (Figure 5(f)).
Parallel to the observed functional improvements, ACKR1 overexpression substantially mitigated tMCAO-induced tissue damage. At 3 days post-tMCAO, T2-weighted MRI and MAP2 immunofluorescence staining revealed extensive infarct formation and marked cerebral edema in vector-treated tMCAO mice. Conversely, the ACKR1-overexpressing group exhibited visibly smaller lesions (Figure 5(g)). Quantitative MRI analysis confirmed that both infarct volume (Figure 5(h)) and cerebral edema (Figure 5(i)) were significantly attenuated in the ACKR1 group compared with tMCAO mice treated with Vector. Furthermore, ACKR1 overexpression provided sustained long‑term neuroprotection, as evidenced by better neurological scores, reduced adhesive removal times, and a lower foot fault rate at 28 days after tMCAO (Figure 5(j)). Meanwhile, the protective effect of ACKR1 resulted in a lower brain atrophy rate in model animals detected at the 28‑day endpoint (Figure 5(k) and (l)).
Collectively, these data demonstrate that cerebral endothelial-specific overexpression of ACKR1 exerts neurological protection after ischemic stroke. This protection is underpinned by preserved BBB integrity, a modulated immune response, functional recovery, and reduced tissue damage. By limiting early BBB disruption and subsequent infiltration of pro-inflammatory macrophages, ACKR1 confers lasting neurological benefits, highlighting its potential as a therapeutic target for ischemic stroke.
Discussion
In this study, we elucidate the protective role of endothelial ACKR1 in experimental ischemic stroke. Using scRNA-seq profiling, we identified specific upregulation of ACKR1 in cerebral endothelial cells after cerebral ischemia. Endothelial cell-specific overexpression of ACKR1 preserved BBB integrity during the early phase of ischemia, thereby reducing macrophage infiltration into the brain parenchyma, reducing infarct volume, and markedly improving neurological functional outcomes.
A major challenge in stroke management is the post-stroke inflammatory storm, which aggravates brain damage and worsens prognosis. Chemokine signaling plays an indispensable role in initiating and amplifying this pathological process. 32 The modulation of chemokine-mediated inflammatory responses is tightly controlled by two distinct classes of chemokine receptors. 33 Unlike classical chemokine receptors, 34 which are predominantly expressed on immune cells to facilitate their migration and chemotaxis, ACKRs are highly enriched in endothelial cells. Functionally, ACKRs bind, sequester, and clear excess chemokines, thereby blunting exaggerated chemokine gradients in the tissue microenvironment.20,35,36 Our results revealed that ACKR1 is the predominant ACKR subtype upregulated in cerebral endothelial cells following ischemic stroke, and that it acts as a critical molecular regulator of inflammation. Notably, cerebral endothelial-specific ACKR1 overexpression attenuated the post-stroke inflammatory storm, thereby reducing cerebral infarction and improving neurological function in ischemic stroke mice.
BBB disruption occurs early in ischemic stroke and amplifies infiltration 37 of peripheral immune cells into the brain parenchyma. Elevated levels of chemokines trigger endothelial dysfunction, BBB instability, and increased vascular permeability. 38 Our data show that endothelial ACKR1 overexpression effectively preserved the structural integrity of vital BBB components, including tight junction proteins, pericytes, and astrocytic endfeet. This preservation alleviated BBB leakage, as demonstrated by reduced extravasation of exogenous tracers and endogenous IgG. The BBB-preserving effect of ACKR1 largely mitigated immune cell invasion induced by physical barrier disruption.
Beyond its role in maintaining BBB integrity, ACKR1-mediated modulation of chemokine concentrations also helps limit immune cell infiltration. Macrophages constitute the most prominent cell type infiltrating the brain after ischemic stroke, mainly through the CCL2–CCR2 signaling axis, 17 indicating that elevated chemokine levels in the brain act as potent chemotactic attractants. 39 By fine-tuning local chemokine concentration gradients, overexpressed endothelial-specific ACKR1 markedly diminished macrophage accumulation across intravascular, perivascular, and parenchymal compartments. This suppression of macrophage infiltration effectively halts the self-amplifying inflammatory loop, thereby alleviating subsequent brain damage.
Current research on post-stroke inflammation have largely focused on the intrinsic properties of immune cells, including their activation, phenotypic polarization, and pro-inflammatory cytokine secretion.40–42 Previous studies have often regarded endothelial cells as a passive physical barrier, with their active regulatory roles in orchestrating immune responses being underestimated.43–45 In contrast to this conventional view, our study highlights the critical role of cerebral endothelial cells in the progression of post-stroke inflammation. Our scRNA-seq results revealed profound changes in endothelial cells after stroke, including the upregulation of various immune regulatory functions related to including chemotaxis, leukocyte adhesion, migration, and activation. Furthermore, endothelial cells can produce various molecules that regulate inflammation. In this study, we focused on the role of ACKR1, an important molecule expressed in endothelial cells, and found that it coordinates BBB stability and regulates peripheral immune cell infiltration. This work highlights a previously underappreciated endothelial-centered regulatory axis in post-stroke inflammation and shifts the research focus from intrinsic immune cell regulation to endothelial-mediated immune modulation, thus providing an alternative strategy for targeted anti-inflammatory intervention in ischemic stroke.
While our study offers valuable insights, several limitations should be acknowledged. First, we only overexpressed endothelial-specific ACKR1; further validation using genetic knockout models is required to assess the impact of ACKR1 loss of function. Second, the exact chemokine subtypes bound and regulated by endothelial ACKR1 were not fully characterized in the present study. Third, while our flow cytometry analysis revealed that ACKR1 overexpression reduces the number of macrophages, this approach does not resolve the functional diversity of macrophages. The balance between pro-inflammatory and reparative subsets may also be altered, an important question that will require single-cell transcriptomic profiling in future studies. Fourth, although ACKR1 has been studied in other fields such as cardiovascular diseases, the considerable differences in disease models and pathogenic mechanisms suggest that the role of ACKR1 may differ substantially. Therefore, more detailed mechanistic studies are warranted to further elucidate the specific molecular pathways through which ACKR1 exerts its protective effects in ischemic stroke. Moreover, many aspects of brain physiology and pathophysiology have shown sex differences,46,47 and our study exclusively used male mice; whether the protective effects of endothelial ACKR1 extend to females remains to be determined.
Accumulating evidence indicates that ACKR1 exerts pro-inflammatory effects in peripheral cardiovascular diseases and tumors, while our work reveals its protective effect in acute ischemic stroke. Such functional divergence arises from several context-dependent characteristics. Firstly, ACKRs universally serve as chemokine scavengers and decoy receptors,48,49 dynamically modulating chemokine abundance to regulate immune cell migration. ACKR1 mitigates acute stroke injury by buffering excessive chemokine surges, maintaining BBB integrity and limiting harmful immune infiltration. By contrast, persistent chemokine regulation during chronic peripheral inflammation disrupts immune homeostasis and yields opposite outcomes. Secondly, distinct from peripheral vasculature, the CNS is enclosed by a specialized BBB that strictly controls molecular and cellular trafficking. 50 The compartmentalized immune microenvironment and diverse leukocyte migration patterns further distinguish central and peripheral inflammatory regulation,51,52 laying a structural basis for divergent ACKR1 functions. Furthermore, ischemic insult definitely induces substantial reprogramming in cerebral ECs. 53 Our data reveal that ACKR1 is co-upregulated with antioxidant and anti-inflammatory genes in ischemic endothelium, constituting intrinsic neurovascular protective responses. Collectively, these tissue-specific structures and injury-induced endothelial remodeling may explain why ACKR1 plays a distinct role in cerebral ischemia and peripheral disorders. Further mechanistic investigations are warranted to fully elucidate the specific regulatory pathways.
In addition, clinical samples are needed to verify the translational relevance of endothelial ACKR1 in human ischemic stroke. Future research targeting endothelial ACKR1 may offer promising therapeutic approaches to limit BBB breakdown and inflammatory storm in ischemic stroke.
Conclusions
In conclusion, our study found that endothelial ACKR1 is markedly upregulated after ischemic stroke. Cerebral endothelial-specific ACKR1 overexpression exerts protective effects by preserving BBB integrity and limiting macrophage infiltration, thereby alleviating brain injury and improving neurological outcomes in the ischemic stroke model (Figure 6). These findings highlight cerebral endothelial cells as a critical mediator of post-stroke inflammation, providing a novel therapeutic target for disease intervention.
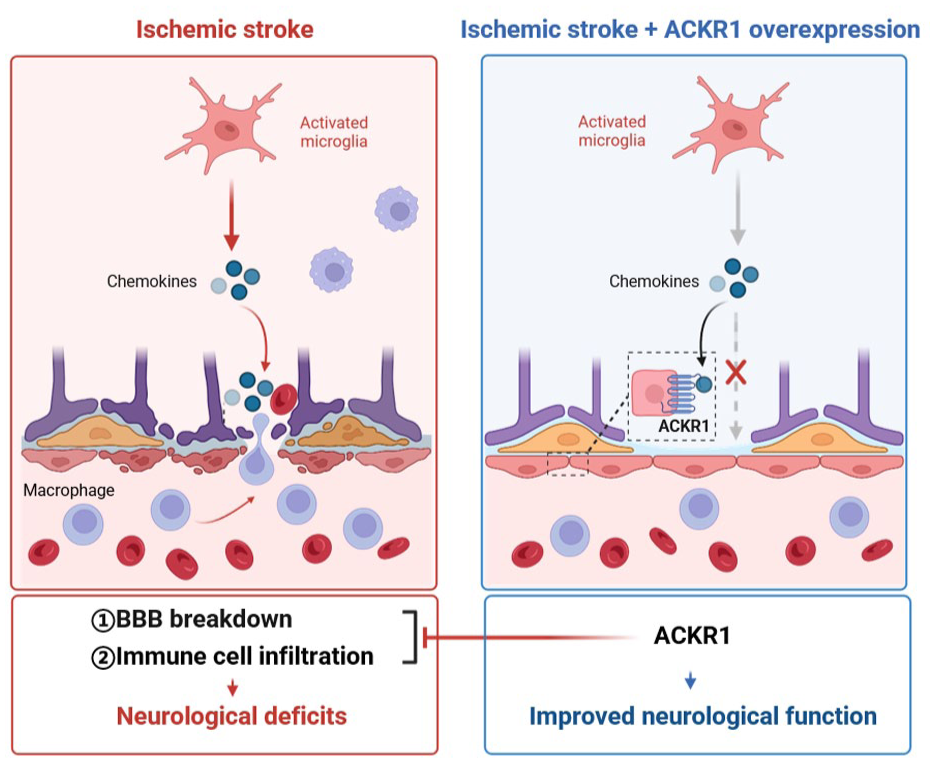
Endothelial ACKR1 preserves BBB integrity and limits neuroinflammation after ischemic stroke: (left) in the acute phase of tMCAO, ischemic injury activates resident microglia, which release large amounts of chemokines (e.g. CCL2). These chemokines disrupt BBB structural integrity, evidenced by fragmented ZO-1, reduced PDGFRβ+ pericyte coverage, and loss of perivascular AQP4 polarity. Consequent BBB leakage permits massive infiltration of CCR2+ macrophages into the brain parenchyma, leading to exacerbated infarct volume and neurological deficits and (right) cerebral endothelial-specific ACKR1 overexpression counteracts this cascade. ACKR1 sequesters excess chemokines, thereby reducing their bioavailability. This preserves BBB components (continuous ZO-1, restored pericyte coverage, and AQP4 polarity), limits both tracer extravasation and endogenous IgG leakage, and suppresses CCR2+ macrophage infiltration. Consequently, infarct volume is reduced and neurological outcomes are improved. This model highlights endothelial ACKR1 as a central regulator of post-stroke inflammation.
Supplemental Material
sj-docx-1-jcb-10.1177_0271678X261461872 – Supplemental material for Endothelial-specific ACKR1 overexpression attenuates macrophage infiltration and blood–brain barrier disruption after ischemic stroke
Supplemental material, sj-docx-1-jcb-10.1177_0271678X261461872 for Endothelial-specific ACKR1 overexpression attenuates macrophage infiltration and blood–brain barrier disruption after ischemic stroke by Mengyuan Guo, Haoyi Fang, Siyan Tang, Qianqian Shao, Jia Liu and Xunming Ji in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Author contributions
Mengyuan Guo: conceptualization, investigation, methodology, visualization, writing—original draft. Haoyi Fang: conceptualization, methodology, writing—review and editing. Siyan Tang: methodology, visualization, writing—review and editing. Qianqian Shao: methodology, supervision, writing—review and editing. Jia Liu: conceptualization, funding acquisition, supervision, writing—review and editing. Xunming Ji: conceptualization, funding acquisition, supervision, writing—review and editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (grant number: 82571493), Outstanding Youth Project of Capital Medical University (grant number: B2403), National Scientific and Technological Innovation 2030 of China-Major project (2023ZD0505300, 2023ZD0505304).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.