
Abstract
The successful clinical treatment of acute ischemic stroke pivots on reestablishing blood flow. After attempting recanalization, the “no-reflow” phenomenon undermines the long-term benefits of clot-busting strategies, including thrombolytic and intravascular thrombectomy. A key pathological driver of this phenomenon is immunothrombosis, a specialized form of microthrombosis characterized by the aberrant aggregation of platelets with neutrophils and subsequent microvascular clot formation. The immunothrombotic process may be initiated or promoted by brain ischemia-induced endothelial inflammation. In this commentary, we elaborate on the mechanisms underlying the role of endothelial inflammation in the no-reflow cascade, and we recommend targeting immunothrombosis to mitigate no-reflow and optimize stroke management.
The special issue “Immunology of Nervous System Disorders” highlights immunothrombosis as a key pathological event in acute ischemic stroke (AIS), and addresses a critical unmet need in AIS management. Although endovascular thrombectomy achieves 70%–80% recanalization in large-vessel occlusion AIS, approximately one-third of patients have poor outcomes due to post-reperfusion “no-reflow”—a clinical challenge rooted in persistent microvascular obstruction. 1 Emerging evidence points to immunothrombosis as a contributor of no-reflow, with endothelial dysfunction and leukocyte evolution as upstream initiators of this pathological cascade. This commentary summarizes key findings from seminal studies, focusing on endothelial damage in immunothrombosis and its translational potential for overcoming futile recanalization.
Endothelial dysfunction: A contributor to immunothrombosis
The initiation of cerebrovascular endothelial inflammation is an early step in immunothrombosis pathogenesis following cerebral ischemia-reperfusion. Soon after ischemia, pro-inflammatory cytokines and danger-associated molecular patterns are elevated in the brain parenchyma, triggering endothelial inflammation and promoting microthrombus formation. Transcriptomic and proteomic analyses of post-stroke brains reveal upregulation of chemokines (e.g. CCL2, CXCL10) and adhesion molecules (e.g. E-selectin, CD146) in ischemic endothelial cells. 2 This CNS endothelial adhesion receptor response was first described by G J del Zoppo et al in non-human primate models in the 1990s. 3 This enhancement drives recruitment and infiltration of neutrophils and monocytes in perfused arterioles and venules, dysregulated lipid metabolism, and forms a vicious loop for augmented pro-inflammatory responses. 2 Uncontrolled endothelial inflammation triggers microthrombosis by downregulating Caveolin-1 and RXR-γ expression, relieving inhibitory control of pro-inflammatory adhesion molecules (e.g. ICAM-1 and VCAM-1) and pro-thrombotic factors (e.g. vWF and PAI-1). 4 This pathway disrupts the vascular anti-thrombotic balance and amplifies immune cell recruitment, promoting microthrombotic formation. From a neurovascular unit perspective, endothelial inflammation disrupts its homeostasis, such as impairing pericyte-mediated capillary tone regulation, which strengthens the link between endothelial dysfunction and cerebral hypoperfusion.
Notably, endothelial inflammation is modulated by endogenous protective molecules that safeguard vascular homeostasis and intercept pathological factors, providing additional therapeutic potential. Peroxiredoxin-4 (Prx4), an endothelial-specific antioxidant protein, is one such vascular protectant. 5 Endothelial-targeted Prx4 deletion exacerbates blood-brain barrier (BBB) disruption post-stroke, upregulates pro-inflammatory genes in vasculature, and promotes neutrophil infiltration. 5 Conversely, Prx4 overexpression inhibits endothelial inflammation and preserves BBB integrity after stroke. 6 These studies support the role of endothelial inflammation and BBB disruption in mediating ischemic brain injury. Notably, BBB disruption directly contributes to microvascular perfusion failure and worsens no-reflow by inducing tissue edema, increasing vascular resistance, and causing capillary compression, establishing a critical link between BBB integrity and the physiological mechanism of no-reflow.
Immunothrombosis: Obstacle to microvascular reperfusion
No-reflow is a well-characterized experimental and clinical phenomenon that occurs after arterial occlusion and reperfusion, leading to complex microcirculatory failure. Immunothrombosis, a specialized form of microthrombosis, has recently emerged as an important underlying contributor alongside classical drivers (capillary constriction, pericyte dysfunction, glycocalyx injury, and microvascular collapse). Post-ischemia vascular obstruction evolves temporally: endothelial activation/platelet adhesion within minutes, neutrophil recruitment at 30–60 min. 7 Neutrophil extracellular traps (NETs) have been detected in large-vessel thrombi, while direct evidence for NET-mediated occlusion in capillaries and post-capillary venules during no-reflow remains limited in stroke models. This no-reflow, as primarily demonstrated in primate models, is most prevalent in the striatum (basal ganglia) and less in the cerebral cortex, emerging acutely at ischemia onset and worsening post-reperfusion. Unlike regular thrombi, these immunothrombi are inherently resistant to conventional antithrombotic agents such as alteplase, as NET-derived chromatin shields fibrin from plasmin, and elevated plasminogen activator inhibitor-1 (PAI-1) in the thrombotic microenvironment further impairs fibrinolysis, rendering standard thrombolytic therapies less effective.
Immunothrombosis exhibits marked spatial and temporal heterogeneity. Spatial heterogeneity involves diverse cellular/extracellular components (e.g. platelets, NETs, necrotic debris) within thrombi, and temporal heterogeneity includes post-stroke periods, circadian rhythms, and aging-related variations. For example, single-cell RNA-seq of human stroke thrombi identifies functionally distinct acute stage neutrophil subsets, with dominant NET-rich prothrombotic subsets and minor fibrinolytic pro-resolving subsets. 8 Chemoattractant regulation involves the CXCL8-CXCR1/2 non-classical monocyte-neutrophil axis, which induces a pro-resolving phenotype and promotes thrombus resolution, thereby establishing the concept of immunothrombolysis. 8 Circadian regulation may shape immunothrombosis pathogenicity. Recent work shows that the clock gene BMAL1 drives peaks in NET-related biomarkers, correlating with denser immunothrombosis, and poorer microcirculatory perfusion. 9 This supports circadian regulation of immunothrombosis, as stroke onset during the inactive phase correlates with a larger infarct. These observations may explain the variable efficacy of recanalization therapies by intervention timing, highlighting the potential to target circadian-dependent immunothrombosis and no-reflow. Among the upstream regulators, pyruvate kinase M2 (PKM2) activation drives immunothrombosis post-stroke via the STAT3-NFκB cascade, impairing cerebral microvascular perfusion. 10 Preclinically, the PKM2 inhibitor ML265 reduces thromboinflammation and restores long-term neurological outcomes. 6
Notably, suppressing immunothrombosis clears intraluminal mechanical obstruction, thereby supporting capillary reperfusion. However, effective microcirculatory recovery also requires addressing additional dysfunctions, such as pericyte-mediated capillary constriction, capillary stasis, and dysregulation of microvascular tone, necessitating integration of the broader microcirculatory system rather than focusing solely on intraluminal processes.
In summary, immunothrombosis contributes to microvascular reperfusion failure due to its resistance to standard therapies, cellular/temporal specificity, and inflammatory regulatory effects. By integrating immune and coagulation pathologies, it perpetuates microvascular obstruction beyond macrovascular recanalization, prioritizing it as a target for overcoming no-reflow in AIS.
Bridging endothelial inflammation to counteract immunothrombosis
The notion that immunothrombosis contributes to post-reperfusion no-reflow may reshape the therapeutic landscape for AIS. Given the dynamic evolution of immunothrombosis post-stroke, temporal considerations are critical for clinical applicability, with two interconnected strategic directions. First, prioritize immunothrombosis as a core therapeutic target to alleviate no-reflow in recanalized brain. As a central driver of thromboinflammatory cascades, targeting immunothrombosis effectively disrupts the self-reinforcing cycle between inflammation and thrombosis. This requires elucidating the spatiotemporal and evolutionary characteristics of immunothrombosis to clarify key translational dimensions, including preventive versus rescue targeting, hyperacute versus delayed intervention windows, the relationship to reperfusion timing, and the evolution of immunothrombosis post-recanalization, laying a theoretical foundation for therapy. Here, we propose a new focus on developing treatments that tackle head-on immunothrombosis by targeting both NET formation and platelet-immune cell interactions, in conjunction with standard thrombolytic or thrombectomy therapy, to convert futile recanalization into effective microcirculation recovery in stroke patients.
Second, controlling endothelial inflammation holds promise for attenuating or preventing immunothrombosis post-stroke. By targeting pro-inflammatory endothelial signaling cascades, this approach blocks the upstream events driving immunothrombosis across spatial and temporal dimensions (Figure 1). This new strategy offers the key advantage of avoiding systemic immunosuppression, which may impair brain repair and long-term recovery.
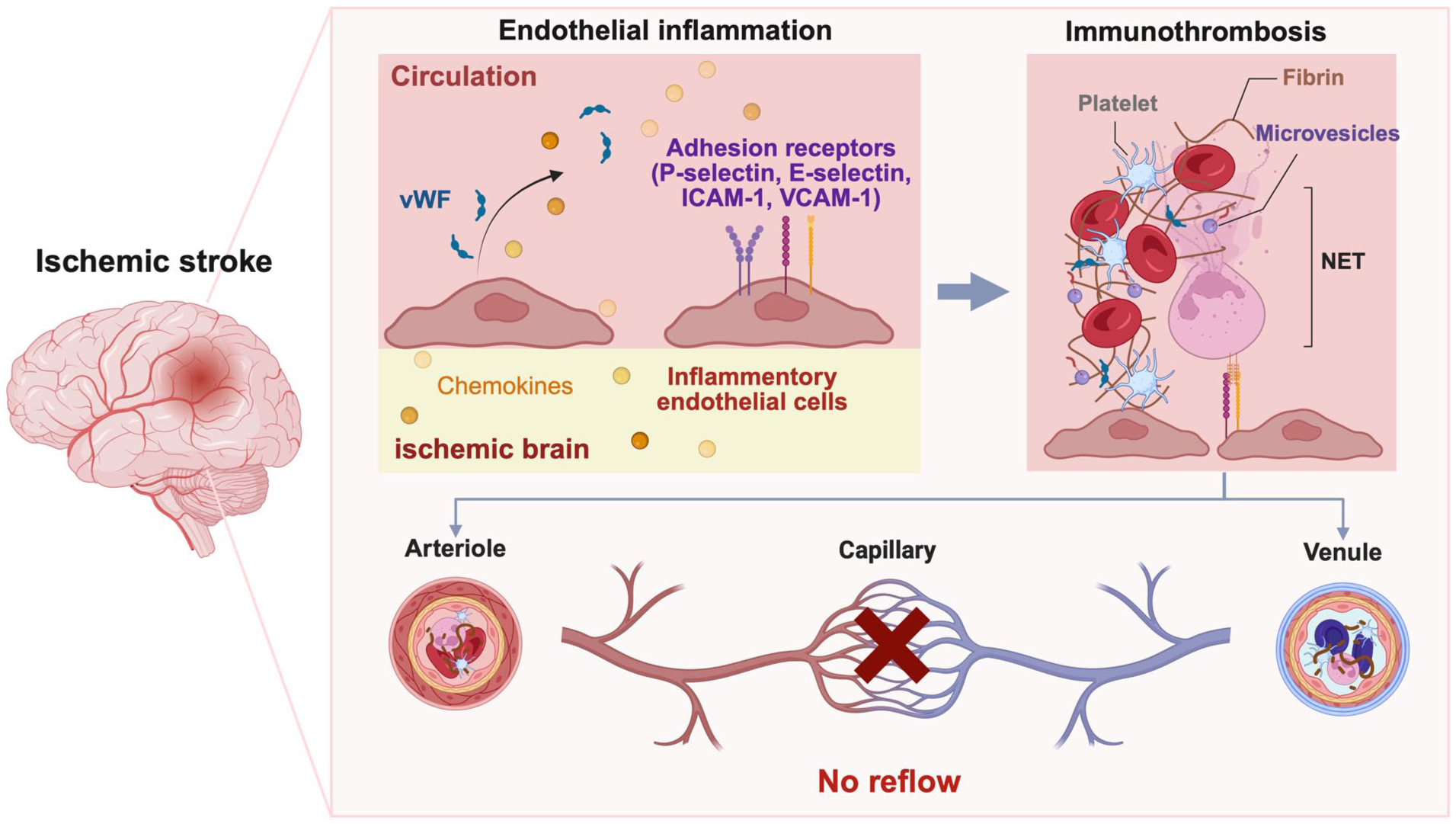
Mechanistic connection between endothelial inflammation and post-reperfusion no-reflow via immunothrombosis. Stroke induces endothelial inflammation in cerebral arterioles and venules, characterized by von Willebrand factor (vWF) release and expression of adhesion receptors (P-selectin, E-selectin, ICAM-1, VCAM-1). This inflammation-coagulation signaling cascade triggers platelet aggregation, neutrophil extracellular traps (NETs) formation, and fibrin deposition within arteriolar/venular lumens to form immunothrombi, leading to post-reperfusion no-reflow after ischemic stroke.
Here, we argue that (1) targeting immunothrombosis prevents post-reperfusion no-reflow, and (2) inhibiting endothelial inflammation is a reasonable entry point. If successful, these approaches may transform AIS clinical management, moving beyond large-vessel recanalization to restore microcirculation, which is crucial for long-term neurological recovery.
Footnotes
Acknowledgements
We acknowledge BioRender (biorender.com) for the creation of Figure 1.
Abbreviations
AIS: Acute Ischemic Stroke
BBB: Blood-Brain Barrier
BMAL1: Brain and Muscle ARNT-Like 1
CCL2: C-C Motif Chemokine Ligand 2
CXCL8: C-X-C Motif Chemokine Ligand 8
CXCL10: C-X-C Motif Chemokine Ligand 10
CXCR1/2: C-X-C Motif Chemokine Receptor 1/2
ICAM-1: Intercellular Adhesion Molecule 1
NETs: Neutrophil Extracellular Traps
NF-κB: Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells
PAI-1: Plasminogen Activator Inhibitor-1
PKM2: Pyruvate Kinase M2
Prx4: Peroxiredoxin-4
RXR-γ: Retinoid X Receptor Gamma
STAT3: Signal Transducer and Activator of Transcription 3
VCAM-1: Vascular Cell Adhesion Molecule 1
vWF: von Willebrand Factor
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Chinese Institutes for Medical Research (Grant number: CX23YQ01) and the Outstanding Youth Project of Capital Medical University (B2403).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.