
Abstract
Ferroptosis is an iron-dependent form of programmed cell death implicated in various pathological conditions. We investigated whether ferroptosis contributes to acute ischemic stroke using a transient middle cerebral artery occlusion model in pial collateral-deficient CB-17/Icr-+/+Jcl mice. Mice were subjected to 90 min of ischemia followed by reperfusion, and the effects of the ferroptosis inhibitor UAMC-3203 on infarct evolution and post-stroke histological changes were examined. UAMC-3203 increased the survival of CD13-positive pericytes within infarct areas and enhanced infarct reduction in the subacute phase. In vitro, the glutathione peroxidase 4 inhibitor RAS-selective lethal 3 (RSL3) induced dose-dependent cell death in pericytes but not in endothelial cells, which was suppressed by UAMC-3203 but not by inhibitors of apoptosis or necroptosis. RSL3 induced lipid peroxidation in pericytes, as evidenced by malondialdehyde accumulation. Extracellular ferrous iron (Fe2+), but not ferric iron (Fe3+) or transferrin, caused intracellular Fe2+ accumulation and cell death in pericytes, which was further enhanced by oxygen–glucose deprivation with reperfusion and suppressed by UAMC-3203. Myelin debris containing abundant Fe2+ induced ferroptosis in cultured pericytes. These findings indicate that pericytes are a major ferroptosis-vulnerable cell type during ischemia–reperfusion and may represent a therapeutic target in acute ischemic stroke.
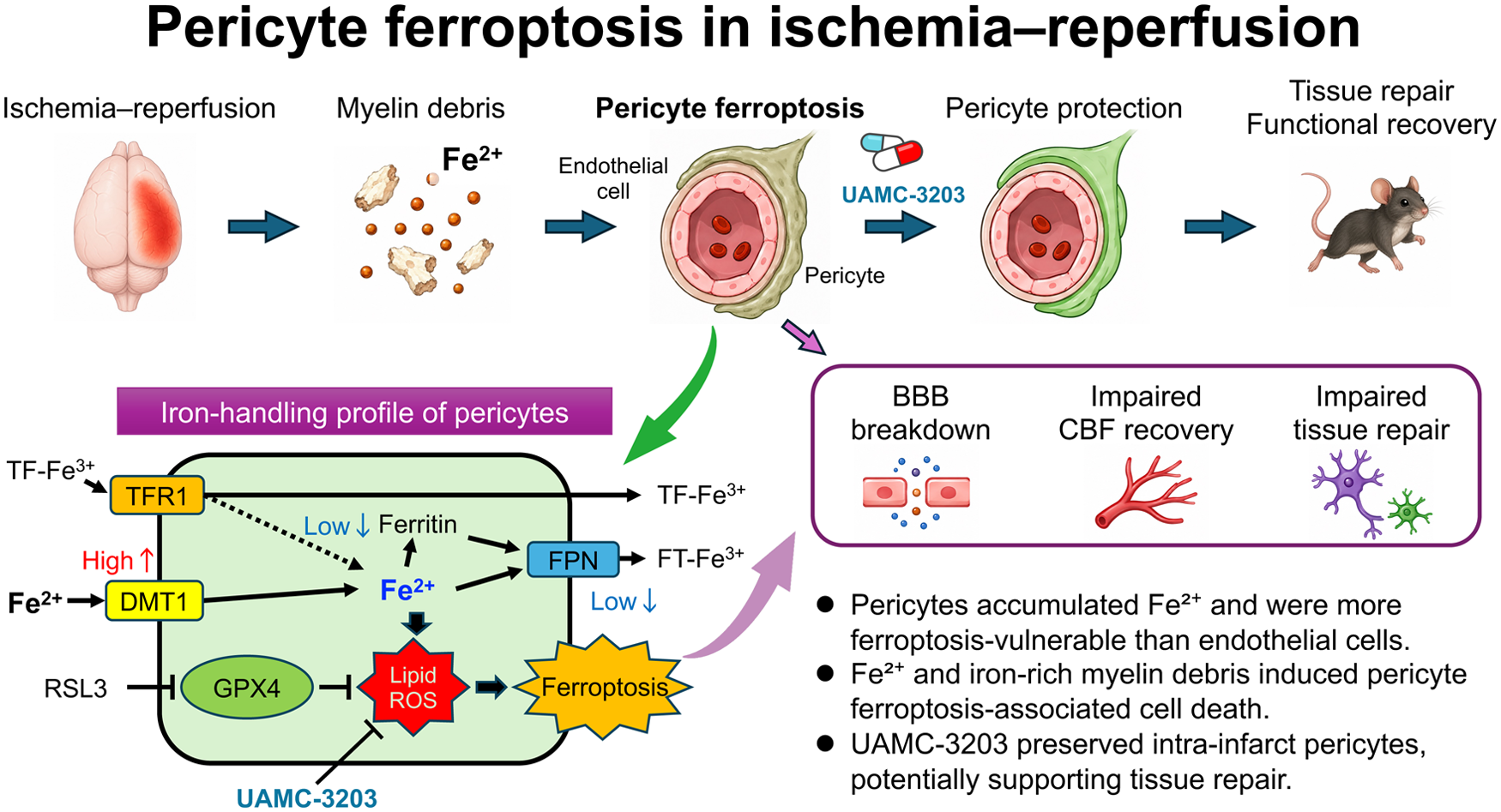
Introduction
Early reperfusion achieved by recombinant tissue-plasminogen activator infusion or endovascular thrombectomy following acute large vessel occlusion is an established therapy for rescuing neural cells in ischemic areas in patients.1,2 Moreover, it has been suggested that early reperfusion may promote functional recovery. 3 We have recently demonstrated that the intra-infarct survival rate of microvascular pericytes surrounding endothelial tubes is a crucial determinant of post-stroke intra-infarct repair leading to functional recovery in mice.4,5 While endothelial cells are considerably resistant to ischemic insults and can survive for several days even within infarct areas, microvascular pericytes are more vulnerable and readily undergo cell death or detachment from endothelial cells in proportion to the severity and duration of ischemia. Since pericyte coverage around endothelial cells is indispensable for maintaining the structure of the blood–brain barrier and supplying appropriate blood flow, pericyte damage or loss can cause intra-infarct edema or hemorrhage and a no-reflow phenomenon even after the achievement of reperfusion.5 –9 Moreover, perivascular pericytes play an important role in recruiting macrophages through chemokine production and supporting macrophage-mediated removal of myelin debris within infarct areas, while they trans-differentiate into fibroblast-like cells, thereby contributing to intra-infarct repair and reduction of infarct volume during the subacute phase.5,10 –12 Thus, rescuing intra-infarct pericytes could be a therapeutic target for promoting post-stroke functional recovery.13,14
However, reperfusion is inevitably accompanied by oxidative stresses, which can induce cellular damage and death within the reperfusion area, despite being the most effective strategy to rescue neural cells and pericytes after ischemia.15,16 Recent evidence establishes that ferroptosis, an iron-dependent, regulated necrotic cell death linked to lipid hydroperoxides, occurs in certain cell types,17,18 under conditions accompanied by increased production of reactive oxygen species (ROS), including ischemia–reperfusion. 19 Since neural cells harbor iron-containing enzymes abundantly within cells, they may be susceptible to ferroptosis and can also be iron suppliers inducing ferroptosis of neighboring cells in acute ischemic stroke.20 –23 Furthermore, recent studies suggest that ferroptosis is not restricted to neural cells but can also occur in multiple cell populations within the neurovascular unit.24,25 Therefore, we hypothesized that microvascular pericytes are vulnerable to ferroptosis during ischemia–reperfusion injury.
In the present study, we examined whether ferroptosis contributes to acute ischemic stroke pathophysiology, with a particular focus on microvascular pericytes, using a transient middle cerebral artery occlusion (tMCAO) model in mice.
Material and methods
Animals
Animal experiments were conducted according to the Guidelines for the Proper Conduct of Animal Experiments of the National Science Council of Japan. The Animal Care and Use Review Committee of Kyushu University approved all animal procedures. We used C.B-17/Icr-+/+Jcl mice (CB-17 mice, CLEA Japan, Tokyo, Japan), a mouse strain harboring few leptomeningeal anastomoses at baseline, to judge the real effect of ischemia–reperfusion.4,26,27 We used male mice aged 8–12 weeks and weighing 20–30 g. Mice were bred and housed 2–5 per cage in an animal facility at Kyushu University at 21 °C and 65% humidity with a regulated 12/12 h light/dark cycle and free access to food and water. 4 All experiments were reported according to the ARRIVE guidelines.
Stroke model
Phosphate-buffered saline (PBS) containing 3.75% DMSO (control, n = 28) or UAMC-3203, a specific inhibitor of ferroptosis (#S8792; Selleck Chemicals, Houston, TX, USA) dissolved in 3.75% DMSO (2.5 μmol/kg body weight; n = 24), was injected intraperitoneally into CB-17 mice 24 and 2 h before tMCAO and every 24 h after tMCAO until sacrifice (Figure 1(a) and Supplementary Table I). Mice were randomly assigned to treatment or vehicle groups prior to surgery, anesthetized by inhalation of 2% isoflurane in air, and maintained under anesthesia with 1.5% isoflurane. The MCA was transiently occluded with a 6–0 monofilament nylon suture, distal to the site where it crossed the olfactory tract. The suture was rotated 180° clockwise, horizontally with the artery, to completely interrupt blood flow. Mice were returned to their cages and kept without anesthesia until reperfusion. After occlusion for 90 min, the mice were anesthetized, and cerebral blood flow was restored by returning the nylon suture to its original position by counterclockwise rotation. 4 Interruption and restoration of cerebral blood flow in the MCA territory during ischemia–reperfusion was confirmed by laser speckle flowmetry (OMEGAZONE OZ-2; OMEGAWAVE, Inc., Tokyo, Japan). 5 Animals were excluded from the analysis only when predefined technical or biological exclusion criteria were met, including failure of sample preparation, early death, intracerebral hemorrhage, or insufficient ischemia; in total, seven mice were excluded (Supplementary Table I).
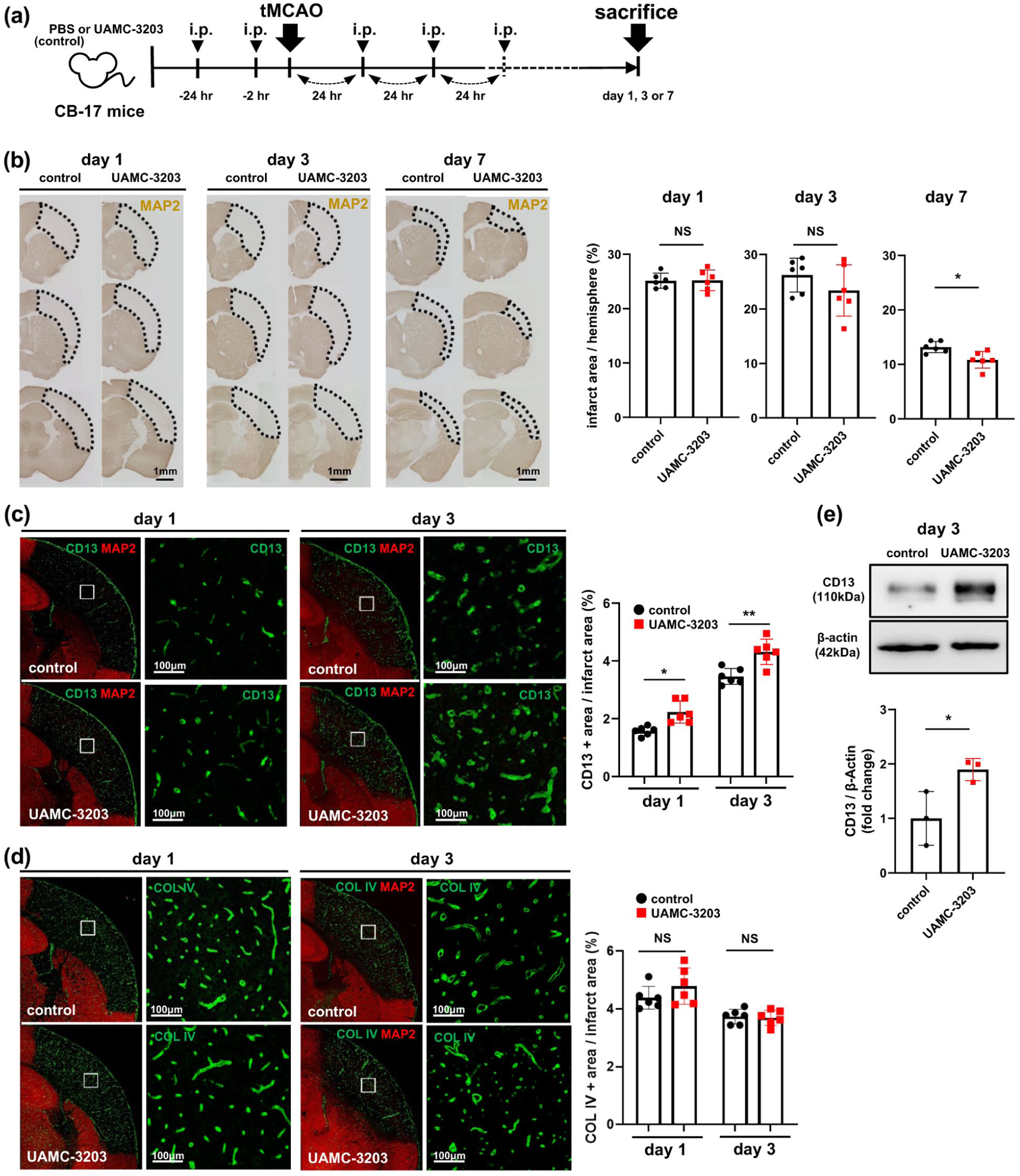
Effects of UAMC-3203 on infarct volume and survival of CD13-positive pericytes within infarct areas following tMCAO for 90 min in CB-17 mice: (a) experimental scheme: either PBS (control) or UAMC-3203 (2.5 μmol/kg body weight) was injected i.p. into CB-17 mice 24 and 2 h before tMCAO and every 24 h after tMCAO until sacrifice (on days 1, 3, or 7), (b) effects of UAMC-3203 on infarct volume, as assessed by immunohistochemistry with MAP2, on days 1, 3, and 7 following tMCAO for 90 min (n = 6, each group). Representative images (left; scale bar, 1 mm) and quantification of infarct volumes are shown to the right, (c) representative immunofluorescent double staining with CD13 and MAP2 on days 1 and 3 after tMCAO for 90 min (n = 6, each group; left; scale bar, 100 µm). Quantification data are shown to the right, (d) representative immunofluorescent double staining with COL IV and MAP2 on days 1 and 3 after tMCAO for 90 min (n = 6, each group; left; scale bar, 100 µm). Quantification data are shown to the right, and (e) effects of UAMC-3203 on CD13 expression in the ischemic hemisphere on day 3 after tMCAO for 90 min, as assessed by immunoblot using brain homogenates (n = 3). Data are shown as mean ± standard deviation.
Immunohistochemistry
Mice were sacrificed on the indicated day after tMCAO by intraperitoneal administration of pentobarbital (150 mg/kg body weight) and transcardially perfused with 20 ml saline, followed by 20 ml 4% paraformaldehyde (PFA) in PBS at 4 °C. Whole brains were fixed with 4% PFA in PBS for 24 h. PFA-fixed 2-mm-thick coronal slices were embedded in paraffin and sectioned at 4 μm. The sections were then deparaffinized, rehydrated using a graded series of ethanol solutions, and washed in PBS. For frozen sections, transcardially fixed whole brains were fixed with 4% PFA in PBS for 1 h, sliced to 2-mm thickness, and cryoprotected in 30% sucrose in PBS for 24 h. Sliced tissues in OCT compound (Leica Biosystems Richmond, Inc. #3801481, Richmond, IL, USA) were frozen in hexane-dry ice coolant and sectioned at 5 μm. Sections that were deparaffinized or removed from the OCT compound by washing with PBS were blocked with a solution of 5% skimmed milk for 30 min at room temperature and then incubated with primary antibodies: anti-microtubule-associated protein 2 (MAP2, 1:1000; Sigma–Aldrich #M4403, St. Louis, MO, USA), anti-CD13 (1:200; R&D Systems #AF2335, Minneapolis, MN, USA), anti-collagen type IV (COL IV, 1:1000; Sigma–Aldrich #AB769), anti-PDGFRβ (1:100; Cell Signaling Technology (CST) #3169, Danvers, MA, USA), anti-F4/80 (1:100; Abcam #ab6640, Cambridge, UK), anti-GFAP (1:200; CST #3670), anti-OLIG2 (1:200; R&D Systems #AF2418), and anti-malondialdehyde (MDA, 1:100; Abcam #ab27642) at 4 °C overnight. After washing with PBS/Triton X-100, the sections were incubated with the appropriate secondary antibodies conjugated to Alexa Fluor dyes (Thermo Fisher Scientific, #A10037, #A11055, #A21202, #A21441, Waltham, MA, USA), FerroOrange (RhoNox-4, 10 μmol/L; Goryo Chemical #GC904-01, Sapporo, Japan), or stained with 3,3′-diaminobenzidine (DAB) using an appropriate kit (Nichirei, Tokyo, Japan). For DAB staining, endogenous peroxidase was inactivated with 0.3% hydrogen peroxide for 30 min before blocking with a skimmed milk solution. For Nissl staining, deparaffinized and rehydrated sections were stained with cresyl violet solution, differentiated and dehydrated through a graded ethanol series, cleared in xylene, and mounted. The sections were observed under a BIOREVO BZ-9000 microscope (Keyence, Osaka, Japan) or a Nikon A1R confocal microscope (Nikon, Tokyo, Japan), and assessed by a blinded investigator who was not involved in animal procedures. Nissl-positive cells were quantified within manually defined regions of interest in the contralateral and ipsilateral cerebral cortex after 8-bit grayscale conversion, background subtraction, fixed-threshold segmentation, and watershed separation. CD13 was used as the primary pericyte/perivascular cell marker based on our previous studies evaluating intra-infarct pericytes and pericyte-mediated tissue repair after ischemic stroke.5,28,29
Cell culture
Human brain microvascular pericytes were purchased from ScienCell Research Laboratories (#1200, Carlsbad, CA, USA) and cultured on poly-l-lysine (PLL)-coated dishes (AGC TECHNO GLASS #4000-101, Tokyo, Japan) with pericyte medium (ScienCell Research Laboratories #1201). Human umbilical vein endothelial cells were purchased from ScienCell Research Laboratories (#8000) and cultured on COL I-coated dishes (AGC TECHNO GLASS #4000-010) with endothelial cell medium (Lonza #CC3162, Basel, Switzerland). Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air. RAS-selective lethal 3 (RSL3; Selleck Chemicals #S8155) as an inhibitor of glutathione peroxidase 4 (GPX4); UAMC-3203; Z-VAD-FMK (Selleck Chemicals #S7023), an inhibitor of apoptosis; necrostatin-1 (Selleck Chemicals #S8037), an inhibitor of necroptosis; ammonium iron(II) sulfate (Fe2+; Fujifilm WAKO #091-00855, Osaka, Japan); ferric ammonium citrate (Fe3+; Fujifilm WAKO #097-01815); and holo-transferrin (Fujifilm WAKO #11096-37-0) were used. Acrolein (Fujifilm WAKO, #513-66081) and l-glutamic acid (Fujifilm WAKO, #072-00501) were used as potential inducers of ferroptosis. Myelin debris was prepared from the brains of 10–12-week-old mice by sucrose density gradient centrifugation and used to stimulate cultured cells at a concentration of 1 mg/mL of myelin protein. 12 As indicated, pericytes were cultured under oxygen-glucose deprivation (OGD), that is, 5% CO2 and 95% N2 in glucose-free medium, for 3 or 6 h and then replaced under normal culture conditions for 24 h (OGDR).
Immunocytochemistry
The cells were fixed with 4% PFA in PBS for 30 min. After blocking with a solution of 2% albumin and 0.1% Triton X-100 for 30 min at room temperature, they were incubated with primary antibodies: anti-malondialdehyde (1:300; Abcam #ab27642) and Hoechst33342 (1:500; Dojindo Laboratories #346-07951, Kumamoto, Japan) overnight. After washing with PBS, the cells were incubated with secondary antibodies conjugated to Alexa Fluor dyes (Thermo Fisher Scientific #A21441) for 30 min and observed under a BIOREVO BZ-9000 microscope. To detect intracellular free ferrous iron (Fe2+), FerroOrange (1 μmol/L) and Hoechst 33342 were directly added to living cells for 30 min after washing twice with Hanks’ balanced salt solution containing magnesium and calcium (HBSS) twice.
SYTOX Green-based cell death assay
SYTOX Green (Thermo Fisher Scientific #S7020), a membrane-impermeable DNA dye that enters cells with compromised plasma membranes, was used to assess cell death. 30 Nuclei were co-stained with Hoechst 33342. Pericytes and endothelial cells were seeded in 96-well plates (AGC TECHNO GLASS #4860-040 or #4860-010) at 5 × 103 cells/well. After the indicated treatments, 20 µL of SYTOX Green (1.4 µL/mL) was added to each well containing 100 μL of culture medium, and fluorescence intensity was measured using a microplate reader (Perkin Elmer ARVO X3, Waltham, MA, USA) with excitation/emission wavelengths of 485/535 nm. Fluorescence intensity was measured again after the addition of 20 μL of 0.1% Triton X-100 to each well for 2 h. The fluorescence ratio before and after Triton X-100 addition was used as an index of cell death.
Alternatively, an LDH release assay was performed when myelin debris was added to the culture medium, because myelin debris contained abundant nucleic acids that interacted with SYTOX Green. The reaction mixture (20 μL; Cytotoxicity Detection Kit PLUS, Roche #4744934001, Basel, Switzerland) was added to the cultured cells and incubated in 96-well plates containing 100 μL of culture medium. After incubation for 30 min, absorbance was measured at 490 nm, with background correction at 650 nm.
PCR and qPCR
Total RNA was prepared from cultured cells using TRIzol (Thermo Fisher Scientific #15596018). A total of 1 μg total RNA was reverse-transcribed using a ReverTra Ace qPCR RT kit (Toyobo #FSQ-101, Osaka, Japan). Using the reverse transcription product as a template, polymerase chain reaction (PCR) was performed with primers specific for the target genes (Supplementary Table II). After pre-incubation at 94 ℃ for 5 min, PCR was performed using 30 cycles of denaturation at 94 ℃ for 30 s, annealing at 55 ℃ for 30 s, and elongation at 72 ℃ for 30 s. Quantitative PCR (qPCR) was performed in duplicate using a LightCycler (Roche #05815916001). Reaction volumes of 20 μL included 10 μL KOD SYBR qPCR mix (Toyobo #QKD-201), 0.25 μmol/L primers, and 1 μL cDNA. The mRNA copy number was normalized to that of 18S ribosomal RNA as an internal control. 5
Immunoblot analysis
After the mice were sacrificed, ischemic hemispheres on day 3 were extracted, frozen in liquid nitrogen, and stored at −80 ℃ until use. The hemispheres or cultured cells were homogenized in RIPA lysis buffer (50 mmol/L Tris–HCl, pH 7.5, 150 mmol/L NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, 5 mmol/L EDTA, 10 mmol/L Na4P2O7, 0.1 mmol/L Na3VO4, 1 mmol/L NaF, and protease inhibitors cocktail (Thermo Fisher Scientific #78410)). Protein concentration was determined using a BCA Protein Assay Kit (Thermo Fisher Scientific #23228). Samples were subjected to SDS-PAGE (15 μg/lane) and transferred onto PVDF membranes. Membranes were incubated for 1 h with ECL Prime blocking reagent (Cytiva #RPN418, Tokyo, Japan) at room temperature and probed overnight at 4 °C with the following primary antibodies: anti-β-actin (1:2000; Sigma–Aldrich #A5441), anti-CD13 (1:1000; R&D Systems #AF2335), anti-GPX4 (1:1000; Abcam #ab125066), and anti-COX2 (1:1000; CST #4842). The membranes were then washed and incubated with secondary antibodies (1:50,000; CST #7074 or #7076; Santa Cruz Biotechnology #2020, Dallas, TX, USA) for 60 min at room temperature. Blots were developed using an ECL Advance Western Blotting Detection Kit (Fujifilm Wako, #292-69903). Quantification was performed densitometrically on ImageJ. 5
Measurement of intracellular glutathione
Intracellular glutathione (GSH) levels were measured using a GSSG/GSH Quantification Kit II (Dojindo Laboratories #G263). Cell lysates were prepared using acid extraction with sulfosalicylic acid, and glutathione levels were determined by an enzymatic recycling assay based on 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB). Absorbance was measured at 405 nm, and concentrations were calculated from standard curves. Reduced GSH levels were obtained by subtracting twice the oxidized glutathione (GSSG) concentration from total glutathione and normalized to cell number.
Statistical analysis
Statistical analyses were performed using Student’s t-test (for two-group comparisons) and one-way ANOVA, followed by a post-hoc Bonferroni’s comparison test (for comparison of multiple groups) using Prism 8 (GraphPad Software, La Jolla, CA, USA). The Fe2+ dose–response experiment was analyzed by linear regression with comparison of slopes between pericytes and endothelial cells. Results are presented as mean ± standard deviation; p < 0.05 was considered significant.
Sample size estimation was performed based on prior studies using the same mouse strain and tMCAO model. Assuming a 25% difference in infarct volume at day 7 between groups, with an estimated standard deviation of 20%, a minimum of six mice per group was required to achieve 80% power at a two-sided α level of 0.05.
Results
UAMC-3203 preserved CD13-positive perivascular cells within infarct areas in a tMCAO stroke model in CB-17 mice
To elucidate whether ferroptosis could be involved in the pathophysiology of acute ischemic stroke, we treated CB-17 mice intraperitoneally with either PBS (control) or UAMC-3203 and then subjected them to tMCAO for 90 min followed by reperfusion (Figure 1(a)). tMCAO produced a brain infarct in the entire MCA territory on day 1 in both groups (Figure 1(b)). Infarct volume on days 1 and 3 was not significantly different between control mice and those treated with UAMC-3203 but was slightly smaller on day 3 in UAMC-3203-treated mice, without statistical significance (Figure 1(b)). However, we found that the infarct volume was significantly smaller on day 7 in the UAMC-3203-treated mice than in the control mice (Figure 1(b)). This implies that ferroptosis inhibition promotes intra-infarct fibrotic repair, which is mainly mediated by intra-infarct surviving pericytes.4,5 Therefore, we compared the survival rates of perivascular pericytes within infarct areas. Immunofluorescence double staining demonstrated that the number of CD13-positive pericytes within MAP2-negative infarct areas was significantly greater in UAMC-3203-treated mice than in control mice on days 1 and 3 (Figure 1(c)). In contrast, the density of COL IV, a major endothelial cell-derived extracellular matrix protein localized in the basal membrane, 31 within MAP2-negative areas was not significantly different between groups (Figure 1(d)). Immunoblot analysis using brain homogenates confirmed that the expression levels of CD13 within infarct areas were significantly higher in UAMC-3203-treated mice than in control mice (Figure 1(e)). These findings indicate that UAMC-3203 preferentially preserves CD13-positive perivascular cells within infarct areas following tMCAO, thereby potentially contributing to post-stroke intra-infarct repair.
To characterize the cellular components within the ischemic lesion, we performed additional histological analyses. Nissl staining showed a marked reduction in Nissl-positive cell density in the ipsilateral cerebral cortex, without obvious preservation by UAMC-3203 (Supplementary Figure I(a) and (b)). GFAP- and OLIG2-positive cells were also markedly reduced within the ischemic lesion on day 1, without obvious differences between groups (Supplementary Figure I(c)). These findings suggest that UAMC-3203 did not clearly preserve neurons, astrocytes, or oligodendrocyte-lineage cells in the early ischemic lesion.
We next examined whether CD13-positive cells reflected pericyte/perivascular cell populations rather than infiltrating macrophages/microglia. F4/80-positive macrophages/microglia were sparse on day 1 and increased by day 3, with limited overlap with CD13-positive cells (Supplementary Figure I(d)). In contrast, CD13 immunoreactivity was mainly localized to perivascular cells and largely overlapped with PDGFRβ-positive cells, particularly on day 3 (Supplementary Figure I(e)). These findings support the interpretation that CD13-positive perivascular cells represent a major cell population preserved by UAMC-3203, although indirect effects mediated through changes in the peri-infarct milieu or inflammatory responses cannot be excluded.
We also examined lipid peroxidation by MDA immunostaining using high-resolution confocal microscopy. On day 3 after tMCAO, MDA immunoreactivity was detectable around CD13-positive perivascular structures and appeared more closely associated with CD13-positive cells than with CD31-positive endothelial structures. MDA immunoreactivity appeared lower in UAMC-3203-treated mice than in control mice (Supplementary Figure I(f)).
Ferroptosis can occur in cultured pericytes, but not in endothelial cells
To test whether pericytes undergo ferroptosis, we treated the cultured cells with RSL3. The treatment with RSL3 induced cell death of cultured pericytes, but not of endothelial cells, in a dose-dependent manner, as assessed by SYTOX Green (Figure 2(a)). The RSL3-induced pericyte death was significantly suppressed by pretreatment with UAMC-3203 (Figure 2(a)). In contrast, neither Z-VAD-FMK nor necrostatin-1 suppressed RSL3-induced pericyte death (Figure 2(a)). These findings indicated that RSL3 induces ferroptosis-associated cell death in pericytes. Immunocytochemistry confirmed that treatment with RSL3 induced the accumulation of malondialdehyde in cultured pericytes in a time-dependent manner, which was completely suppressed by pretreatment with UAMC-3203 (Figure 2(b)). We further demonstrated that treatment with RSL3 induced the expression of COX2 whereas UAMC-3203 inhibited the RSL3-mediated upregulation of COX2 (Figure 2(c)).
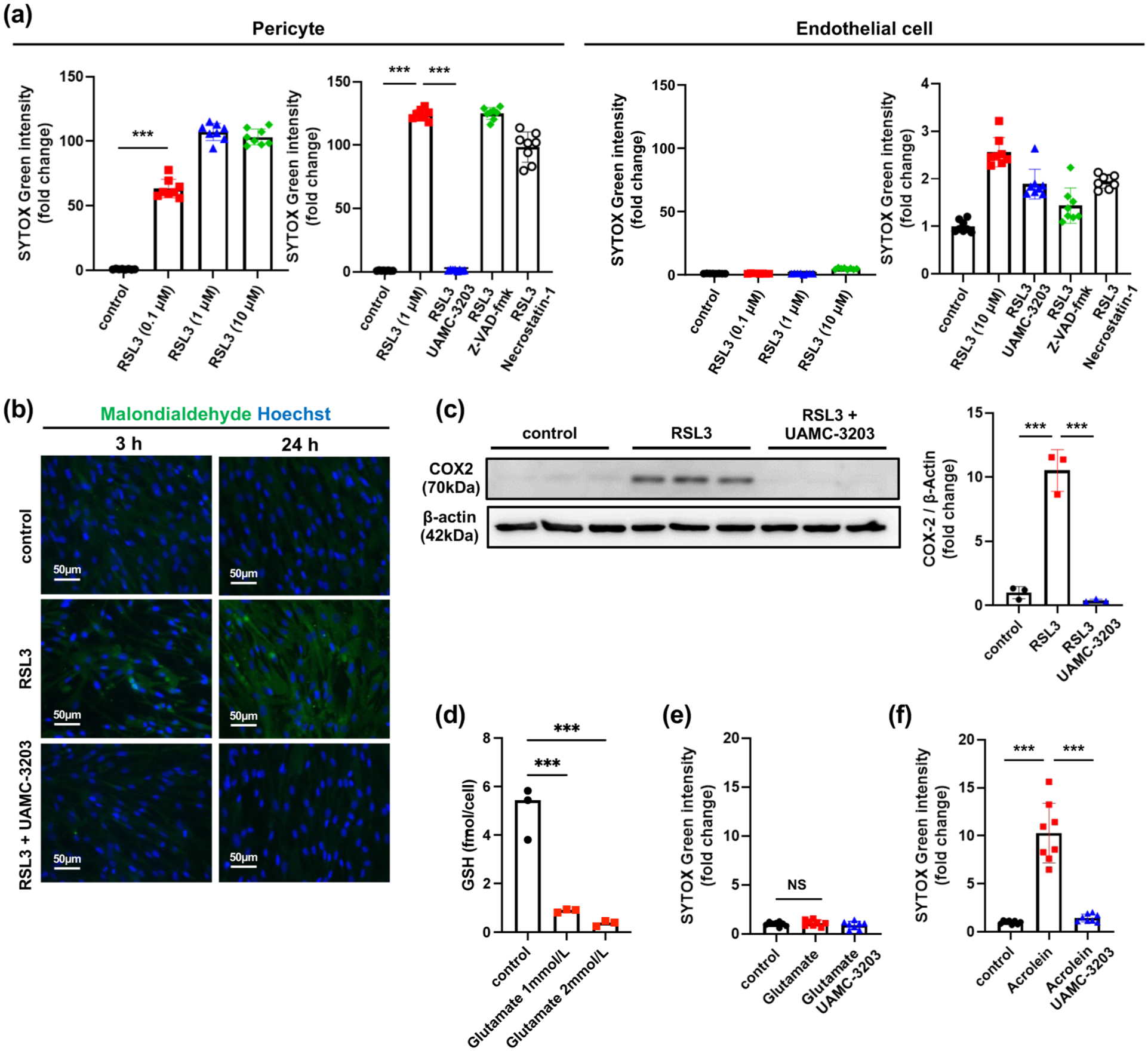
RSL3 induces ferroptosis-associated cell death in pericytes, but not in endothelial cells: (a) dose-dependent effects of RSL3 (0.1, 1, and 10 µmol/L) on cell death in cultured pericytes and endothelial cells, as assessed by SYTOX Green (left; n = 8). Effects of UAMC-3203 (1 µmol/L), Z-VAD-FMK (50 µmol/L), and necrostatin-1 (10 µmol/L) on RSL3-induced cell death in pericytes (RSL3, 1 µmol/L) and endothelial cells (RSL3, 10 µmol/L) are shown (right; n = 8), (b) effects of RSL3 (1 µmol/L), in the presence or absence of UAMC-3203 (1 µmol/L), on malondialdehyde accumulation in cultured pericytes, as assessed by immunocytochemistry (n = 3). Representative images are shown (scale bar, 50 µm), (c) effects of RSL3 (1 µmol/L), in the presence or absence of UAMC-3203 (1 µmol/L), on COX2 expression in cultured pericytes, as assessed by immunoblotting (n = 3), (d) effects of glutamate on intracellular reduced GSH levels in cultured pericytes (n = 3), (e) effects of glutamate (1 mmol/L), in the presence or absence of UAMC-3203 (1 µmol/L), on cell death in cultured pericytes, as assessed by SYTOX Green (n = 8), and (f) effects of acrolein (50 µmol/L), in the presence or absence of UAMC-3203 (1 µmol/L), on cell death in cultured pericytes, as assessed by SYTOX Green (n = 8). Data are shown as mean ± standard deviation.
We next examined whether glutamate affects intracellular glutathione levels in pericytes. Glutamate treatment reduced intracellular GSH levels compared with control conditions (Figure 2(d)). Despite this reduction in GSH, glutamate did not induce pericyte cell death (Figure 2(e)). In contrast, acrolein, a known ferroptosis inducer, 32 induced pericyte death, which was prevented by pretreatment with UAMC-3203 (Figure 2(f)). These findings suggest that depletion of intracellular GSH alone is insufficient to trigger ferroptosis in pericytes and that additional pro-ferroptotic stimuli, such as iron-dependent lipid peroxidation or direct GPX4 inhibition, may be required for ferroptotic cell death.
Expression of iron transport-related molecules in pericytes and endothelial cells
To elucidate why pericytes tend to undergo ferroptosis, we examined the expression of molecules that regulate intracellular iron homeostasis in cultured pericytes and endothelial cells. The expression of divalent metal transporter 1 (DMT1), which transports free ferrous iron (Fe2+) into cells, was significantly higher in pericytes than in endothelial cells (Figure 3(a)). In contrast, the expression of the iron exporter ferroportin (FPN) was significantly higher in endothelial cells than in pericytes (Figure 3(b)). Furthermore, the ratio of ferritin heavy chain (FTH) to ferritin light chain (FTL), which reflects ferritin-related iron-handling properties, was significantly higher in endothelial cells (Figure 3(c)). The expression of GPX4 was slightly higher in pericytes than in endothelial cells (Figure 3(d)). At the mRNA level, these results suggest that pericytes are characterized by a basal iron-handling profile favoring Fe2+ uptake and limited iron export/storage responses compared with endothelial cells.
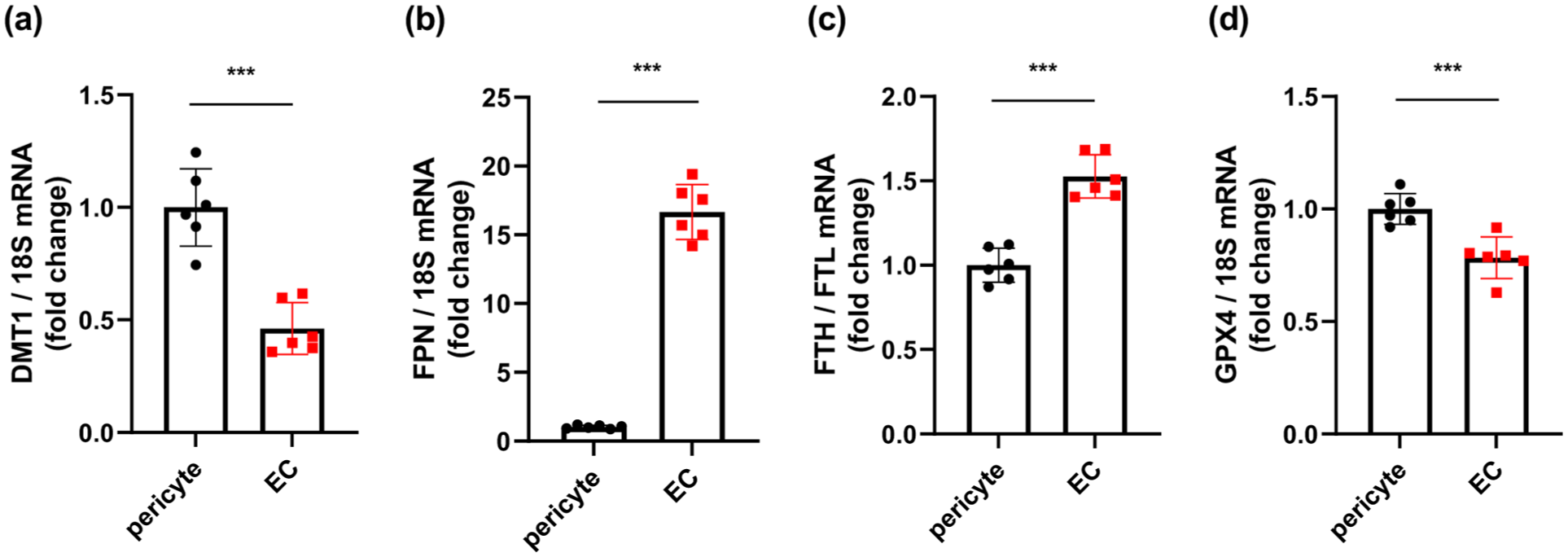
Basal expression of iron-handling molecules in pericytes and EC. qPCR analysis of (a) DMT1, (b) FPN, (c) the ratio of FTH to FTL, and (d) GPX4 in cultured pericytes and EC (n = 6 per group). Data are shown as mean ± standard deviation.
We further examined the responses of iron-handling molecules to extracellular Fe2+ exposure. Fe2+ treatment reduced TFR1 expression in pericytes, consistent with iron-dependent feedback regulation (Supplementary Figure II(a)). However, FPN expression remained low in pericytes even after Fe2+ loading, whereas endothelial cells showed markedly higher FPN expression under both control and Fe2+-treated conditions (Supplementary Figure II(b)). In addition, FTH/FTL remained higher in endothelial cells than in pericytes after Fe2+ exposure (Supplementary Figure II(c)). These findings suggest that pericytes can sense iron loading but have limited iron export and ferritin-associated buffering responses compared with endothelial cells.
Extracellular Fe2+ induces ferroptosis-associated cell death in pericytes
We treated cultured pericytes and endothelial cells directly with ferrous (Fe2+) or ferric iron (Fe3+). Intracellular accumulation of free Fe2+, as assessed by FerroOrange, 33 was readily detected when pericytes were treated extracellularly with Fe2+ (Figure 4(a)). Treatment with Fe2+ led to pericyte death, which was rescued by pretreatment with UAMC-3203 (Figure 4(b)). To further evaluate the concentration dependence of Fe2+-induced cell death, we performed dose–response analyses using increasing concentrations of extracellular Fe2+. Linear regression analysis showed that the Fe2+ concentration–cell death relationship was significantly steeper in pericytes than in endothelial cells (Supplementary Figure III), providing supportive evidence that pericytes are more sensitive to extracellular Fe2+ loading. In contrast, treatment with Fe2+ did not induce intracellular accumulation of Fe2+ or cell death in cultured endothelial cells (Figure 4(a) and (b)). These findings indicate that extracellular Fe2+ tends to induce ferroptosis in pericytes rather than in endothelial cells. Furthermore, the intracellular accumulation of free Fe2+ increased under OGD (Figure 4(c)). OGD significantly decreased the expression of FPN, while it increased that of DMT1, in cultured pericytes (Figure 4(d)). Furthermore, transient OGD for 3 and 6 h followed by normoxia and normal glucose levels (OGDR) reduced the expression of GPX4 at protein levels in pericytes (Figure 4(e)). Consistently, OGDR-induced pericyte death was attenuated by UAMC-3203 (Figure 4(f)). These findings suggest that extracellular Fe2+ increases susceptibility to ferroptosis in pericytes, particularly during reperfusion following ischemia.
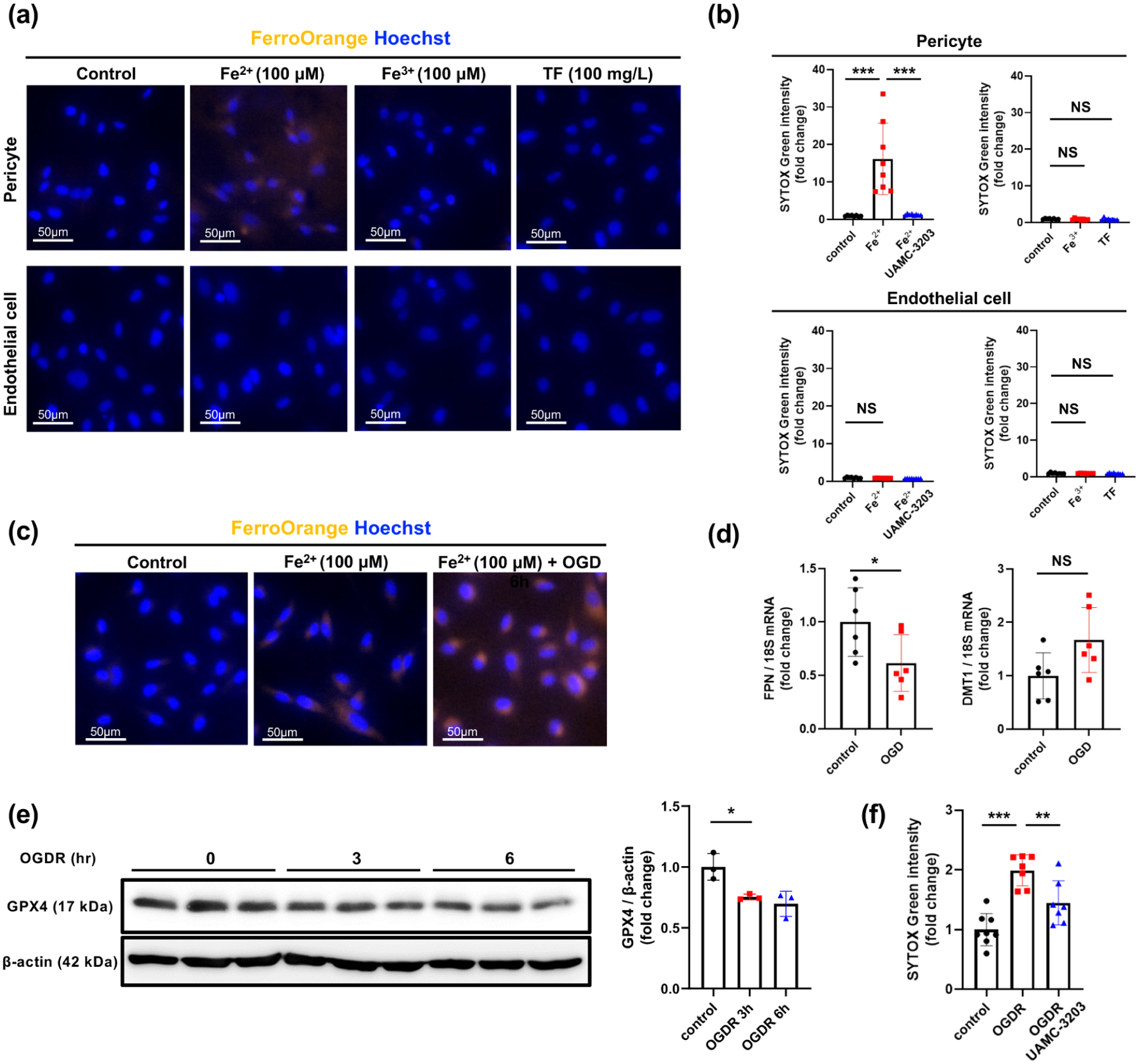
Extracellular Fe2+ induces ferroptosis-associated cell death in pericytes: (a) effects of treatment with Fe2+ (100 µmol/L), Fe3+ (100 µmol/L), or holo-TF (100 mg/L) on intracellular accumulation of free Fe2+ in cultured pericytes and EC, as assessed by immunocytochemistry with FerroOrange and Hoechst (blue, nucleus; n = 3). Representative images are shown (scale bar, 50 µm), (b) effects of treatment with Fe2+ (100 µmol/L), Fe3+ (100 µmol/L), or holo-TF (100 mg/L), in the presence or absence of UAMC-3203 (1 µmol/L), on death of pericytes or EC, as assessed by SYTOX Green (n = 8), (c) effects of OGD for 6 h on the intracellular accumulation of free Fe2+ induced by treatment with Fe2+ (100 µmol/L), as assessed by immunocytochemistry with FerroOrange and Hoechst (blue, nucleus; n = 6). Representative images are shown (scale bar, 50 µm), (d) effects of OGD for 6 h on FPN and DMT1 expression in cultured pericytes (n = 6), (e) effects of transient OGD for 3 and 6 h followed by normal culture conditions for 24 h (OGDR) on the expression of GPX4 in cultured pericytes (n = 3), and (f) effects of transient OGD for 6 h followed by normal culture conditions for 24 h (OGDR) on pericyte death in the presence or absence of UAMC-3203 (1 µmol/L; n = 8). Data are shown as mean ± standard deviation.
Intra-infarct myelin debris contains Fe2+ and can induce pericyte death
Finally, we examined how Fe2+ could be exposed to pericytes within the infarct areas following tMCAO. Immunofluorescent double labeling demonstrated that Fe2+ was easily detected within MAP2-negative infarct areas on day 1 after tMCAO by immunohistochemistry with FerroOrange, in contrast to the contralateral intact cortical areas (Figure 5(a)). We then prepared myelin debris from the brain and confirmed that it contained abundant Fe2+. Calibration curve using FerroOrange and Fe2+ estimated that 1 mg/mL of myelin debris used in this study contained approximately 40 µmol/L of Fe2+ (Figure 5(b)). We demonstrated that treatment with myelin debris (1 mg/mL) induced pericyte death but not that of endothelial cells (Figure 5(c)). Myelin debris-induced cell death in pericytes was prevented by pretreatment with UAMC-3203 (Figure 5(c)), indicating that myelin debris could directly induce ferroptosis-associated cell death in pericytes.
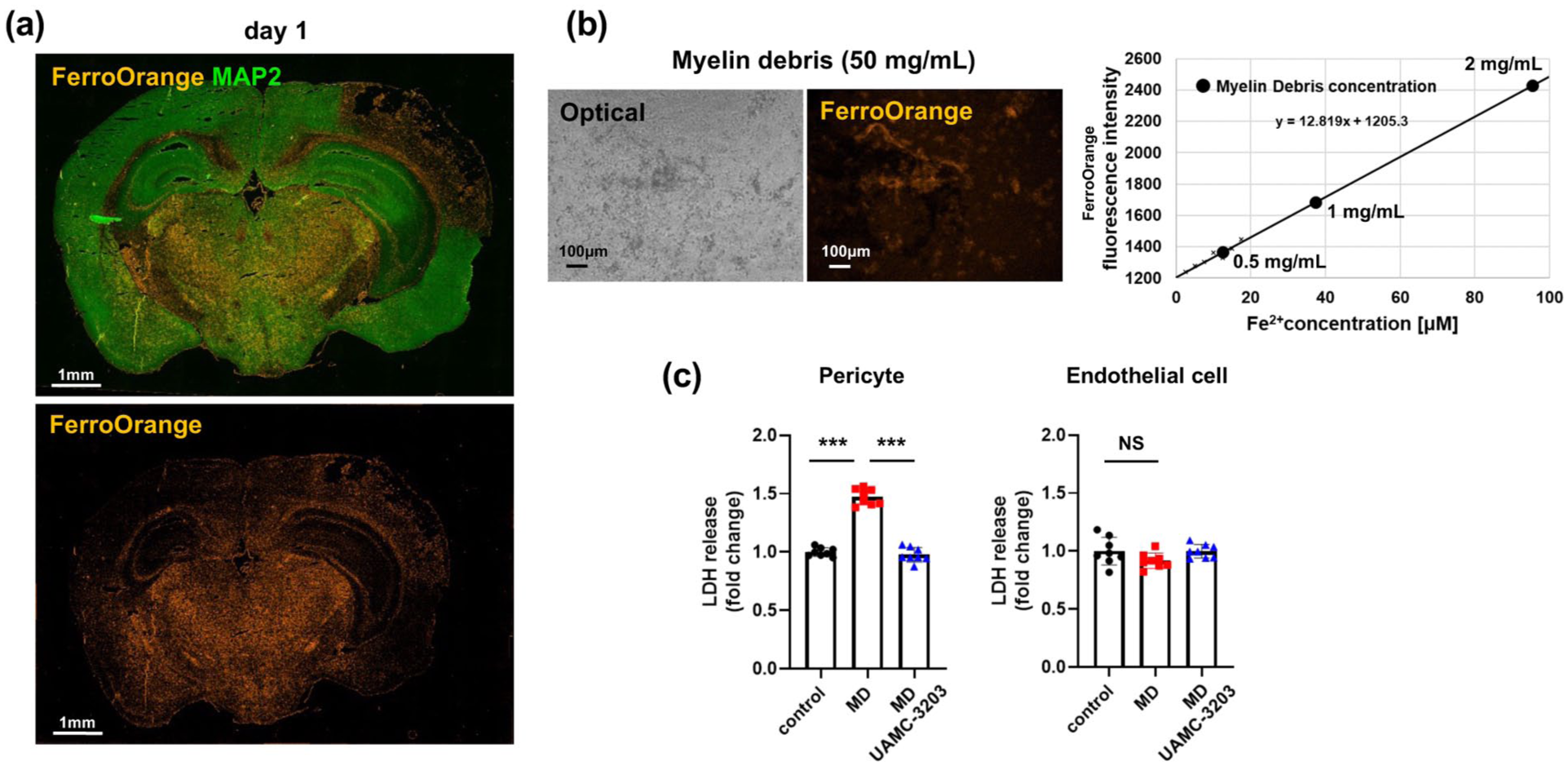
Intra-infarct myelin debris contains abundant Fe2+ and induces cell death in pericytes: (a) representative images of immunofluorescent double labeling with FerroOrange and MAP2 (green) in the whole brain on day 1 after tMCAO (n = 3; scale bar, 1 mm), (b) representative images of myelin debris (50 mg/mL) stained by FerroOrange on an optical microscope (left) or fluorescent microscopy (right; scale bar, 100 µm). Fe2+ content in myelin debris was estimated from the calibration curve using FerroOrange and Fe2+ (right), and (c) myelin debris (1 mg/mL ≈ 40 µmol/L Fe2+ equivalent)-induced cell death in pericytes (left) and endothelial cells (right) in the presence or absence of UAMC-3203 (1 µmol/L; n = 8), as assessed by LDH assay. Data are shown as mean ± standard deviation.
Discussion
We demonstrated here that CD13-positive perivascular cells within infarct areas are pharmacologically preserved by ferroptosis inhibition during reperfusion following tMCAO. Suppression of ferroptosis during ischemia–reperfusion in the brain did not affect infarct volume on day 1, that is, acute neuronal injury, but could rescue intra-infarct pericytes to some extent, thereby leading to enhanced reduction of infarct volume on day 7. We confirmed that RSL3 induced UAMC-3203-sensitive cell death in cultured pericytes, but not in endothelial cells, which was inhibited by UAMC-3203. Pericytes appeared to retain abundant iron within cells because they expressed higher amounts of DMT1 and lower amounts of FPN than endothelial cells. Simple extracellular exposure to ferrous iron (Fe2+) increased intracellular accumulation of free Fe2+ and induced ferroptosis in pericytes, an effect that was further enhanced by transient oxygen–glucose deprivation, which was associated with reduced GPX4 expression. Finally, we demonstrated that myelin debris induced ferroptosis in pericytes, consistent with the abundant presence of Fe2+ within myelin debris and its accumulation within infarct areas on day 1 after 90 min of tMCAO.
Involvement of pericyte ferroptosis during ischemia–reperfusion in the brain
Although ferroptosis occurs in a cell-specific or organ-specific manner,30,34 it has been reported that almost all neural cells, such as neurons, 20 astrocytes, 21 oligodendrocytes, 22 and microglia, 23 can undergo ferroptosis under specific conditions. Recent studies further indicate that ferroptosis represents a key pathological contributor in ischemic stroke.25,35 However, as shown in the present study, pretreatment with a ferroptosis inhibitor did not reduce infarct volume on day 1 after 90 min of tMCAO. This suggests that the severe ischemic insult in this model caused extensive acute neuronal injury that could not be sufficiently rescued by UAMC-3203 alone, even if ferroptosis contributed in part to neuronal cell death. In contrast to the ineffectiveness of ferroptosis inhibition on neural cell rescue, we found that ferroptosis inhibition could increase pericyte survival within the infarct area during ischemia–reperfusion. Notably, the involvement of ferroptosis in endothelial cells in ischemic brain injury has also been reported, 36 but sensitivity may vary between cell types depending on the pathological context and stimuli. In fact, vascular cells are more resistant to ischemic insults than neural cells; however, pericytes are more vulnerable to ischemic insults than endothelial cells, even after reperfusion. 4 The different vulnerabilities of pericytes and endothelial cells to ischemia–reperfusion may be explained in part by their susceptibility to ferroptosis.
Further histological analysis showed that macrophages/microglia were scarce at the early stage and that GFAP- or OLIG2-positive glial cell populations were markedly reduced, whereas CD13-positive perivascular cells remained detectable. These findings suggest that these non-pericyte cell populations were unlikely to be the major targets preserved by UAMC-3203 in the early ischemic lesion. Although MDA immunostaining provided supportive evidence of lipid peroxidation around CD13-positive perivascular structures, other in vivo ferroptosis-related markers were not examined in CD13-positive pericytes. Therefore, the in vivo findings should be interpreted as pharmacological and histological evidence supporting pericyte involvement, whereas our in vitro experiments provide direct evidence that pericytes are vulnerable to ferroptosis.
Mechanisms by which pericytes undergo ferroptosis within ischemic areas
We found that the expression patterns of iron importers, exporters, and storage-related molecules differed between pericytes and endothelial cells. At baseline, pericytes expressed higher levels of DMT1, a major Fe2+ importer, and much lower levels of FPN, the only known cellular iron exporter, than endothelial cells. FTH/FTL was also lower in pericytes, suggesting a lower ferritin-associated iron-buffering profile. These basal mRNA expression patterns may predispose pericytes to retain redox-active iron when extracellular Fe2+ is present.
The Fe2+-loading experiments further supported this interpretation. Fe2+ exposure reduced TFR1 expression in pericytes, consistent with iron-dependent feedback regulation, but did not induce FPN expression. In contrast, endothelial cells maintained markedly higher FPN expression under both control and Fe2+-loaded conditions, and FTH/FTL remained higher in endothelial cells than in pericytes after Fe2+ exposure. These findings suggest that pericytes can sense iron loading but have limited iron export and ferritin-associated buffering responses.
DMT1-mediated influx of Fe2+ may be affected not only by its expression levels but also by its intracellular localization and environmental pH. Fe2+ is more abundantly transported into cells through DMT1 under acidic conditions. 37 Since direct extracellular exposure to Fe2+ increased intracellular accumulation of free Fe2+ and induced ferroptosis of pericytes under normal pH in vitro, pericytes likely express functionally relevant DMT1 on the plasma membrane and can incorporate Fe2+ under normal pH. Moreover, because pH is usually reduced within infarct areas, 38 DMT1-mediated transport of Fe2+ may be increased in pericytes within infarct areas.
Extracellular iron normally exists as less harmful ferric iron (Fe3+) bound to transferrin (holo-transferrin); however, massive amounts of harmful free Fe2+ can leak abruptly from dead neural cells and be exposed to neighboring cells, including pericytes, within infarct areas until microglia/macrophage-mediated removal. Although all neural cells harbor high amounts of iron within cells, oligodendrocytes may contain up to 3 mM iron within cells, which is higher than other neural cells.39,40 Because oligodendrocytes are the most vulnerable to ischemic insults among neural cells, 41 brain infarction accompanied by massive oligodendrocyte death can produce abundant myelin debris containing high amounts of Fe2+. Thus, oligodendrocyte degeneration may act upstream of the myelin debris–iron axis proposed in this study. Recent evidence establishes that microglia/macrophage-mediated clearance of myelin debris is a crucial determinant of post-injury repair and recovery in various CNS disorders.12,42 It may also be important in terms of clearance of harmful iron species that could induce ferroptosis of intra-infarct cells, such as pericytes. 43 Since ischemia reduces the expression of GPX4 and FPN in pericytes, reperfusion following ischemia may enhance ferroptosis of pericytes through increased production of iron-mediated ROS. 15 However, although our findings were supported by functional Fe2+ imaging, the analysis of iron-handling molecules was based mainly on mRNA expression; therefore, protein-level validation of these molecules remains warranted.
The mechanisms underlying the differential ferroptosis susceptibility of pericytes and endothelial cells remain unclear. Sampilvanjil et al. demonstrated that acrolein, a major oxidant produced during acute ischemic stroke, 32 induces ferroptosis in smooth muscle cells, but not in endothelial cells, by depleting the intracellular GSH pool. 30 Consistent with this observation, we found that acrolein induced UAMC-3203-sensitive cell death in pericytes, but not in endothelial cells. These findings suggest that vascular mural cells, including pericytes and smooth muscle cells, may be more susceptible than endothelial cells to specific ferroptosis-inducing stimuli. In contrast, although glutamate reduced intracellular GSH levels, it did not induce detectable pericyte cell death under the present experimental conditions. This suggests that GSH depletion alone is insufficient to trigger ferroptosis in pericytes and that additional pro-ferroptotic stimuli, such as lipid peroxidation or iron-dependent oxidative stress, may be required. Thus, the ferroptosis-inducing ability of known inducers may differ depending on cell type and stimulus context.
Significance of pericyte ferroptosis in cerebrovascular diseases
Intra-infarct survival of pericytes is one of the key factors determining post-stroke tissue repair leading to functional recovery through maintaining the structure of the blood–brain barrier and intra-infarct blood flow and recruitment of blood-derived macrophages that remove intra-infarct debris.4,8,12 Survival rate of intra-infarct pericytes would decrease in proportion to ischemic strength and duration; therefore, early reperfusion is required for pericyte rescue even after the development of brain infarction. 4 However, since reperfusion often causes excessive production of ROS associated with ferroptosis, ferroptosis inhibition may be a promising therapy to rescue pericytes during ischemia–reperfusion injury in the brain.14,19,25,44,45 Consistent with this concept, recent studies have demonstrated that suppression of ferroptosis can ameliorate blood–brain barrier disruption after cerebral ischemia–reperfusion. 46 In this context, because great amounts of iron are released from the extravasated broken red blood cells, the iron-related ferroptosis can also contribute to neuronal and oligodendrocytic death in intracerebral hemorrhage.22,47 Moreover, peri-hematomal edema and dysregulation of blood flow, often found in intracerebral hemorrhage, may also result from pericyte ferroptosis induced by Fe2+ released from the red blood cells.48,49
However, the functional significance of pericyte death after cerebral ischemia remains debated. In particular, acute microvascular failure may reflect not only pericyte loss but also persistent dysfunction or constriction of surviving pericytes, 50 and some studies have assigned a larger role in ischemic flow regulation to arteriolar smooth muscle cells than to capillary pericytes. 51 In addition, although capillary pericytes can influence cerebral blood flow, this effect may occur with relatively slow kinetics, suggesting that their contribution depends on timing and vascular context. 52 Moreover, post-stroke repair is a multicellular process involving endothelial, inflammatory, and glial responses. 53 Therefore, our findings should not be taken to indicate that pericyte death is the sole determinant of no-reflow or tissue repair, but rather that ferroptosis-vulnerable CD13-positive perivascular cells represent one modifiable component of ischemia–reperfusion injury in the present model.
Limitations
First, we only used a tMCAO stroke model with reperfusion to test the potential involvement of ferroptosis in acute ischemic stroke. Because intra-infarct pericytes would be nearly dead simply through ischemic damage in permanent MCAO model in CB-17 mice, 4 we chose the tMCAO model, which has a chance of being rescued by ferroptosis inhibition, which is an oxidative stress-related phenomenon. Ischemia–reperfusion, rather than permanent occlusion, could be a better target for ferroptosis.19,44
Second, we started treatment with a ferroptosis inhibitor before the tMCAO procedure. This pretreatment protocol was chosen as a mechanistic proof-of-concept approach to determine whether ferroptosis inhibition could affect the survival of neural or vascular cells during ischemia–reperfusion injury. However, this design does not mimic the clinical setting of acute ischemic stroke, in which treatment is usually initiated after ischemia onset or at the time of reperfusion. Therefore, the present study does not demonstrate the efficacy of post-reperfusion ferroptosis inhibition. Nevertheless, these findings provide a rationale for future studies to test whether post-reperfusion ferroptosis inhibition can preserve pericytes within a clinically relevant therapeutic window.
Third, our in vivo quantification of pericytes was primarily based on CD13 immunoreactivity. Although additional double immunostaining supported that CD13-positive cells mainly represented PDGFRβ-positive perivascular cells with limited overlap with F4/80-positive macrophages/microglia, CD13 is not entirely specific for pericytes. Moreover, changes in CD13-positive area may reflect altered marker expression or cell detachment under acute ischemic injury. Thus, the limitations of single-marker-based quantification cannot be completely eliminated.
Fourth, we used only young male mice. This design was chosen as an exploratory proof-of-concept approach to evaluate whether pericyte ferroptosis may be involved in ischemia–reperfusion injury under conditions that allow reproducible infarct formation and stable between-group comparisons. However, the exclusive use of young male mice is an important limitation. Ferroptosis sensitivity and stroke outcomes may differ according to sex, and pericyte function as well as iron handling may change with aging. 54 Indeed, iron deposition in pericytes, often found in cerebral microbleeds in older people, 55 may reflect an age-related response. Therefore, the present findings cannot be directly generalized to female or aged animals. Future studies should examine whether sex and aging modify pericyte susceptibility to ferroptosis and the therapeutic efficacy of ferroptosis inhibition after ischemia–reperfusion.
Fifth, we used HUVECs as a standardized endothelial model in vitro; responses may differ in brain microvascular endothelial cells, which should be addressed in future studies.
Finally, we could not demonstrate whether the increased survival of pericytes brought about by ferroptosis inhibition could contribute to better functional recovery. Because functional recovery following tMCAO for 90 min is relatively good in CB-17 young male mice without any treatments, 4 it is possible that we could not differentiate the additive effects of ferroptosis inhibition on functional recovery in the present experimental setting. In this context, we should mention that infarct volume following MCAO in CB-17 is quite reproducible due to the low variance in pial collateral blood flow among individuals, which contributed to the reduction in the number of mice used in the experiments.4,26,27
In conclusion, pericytes are a major ferroptosis-vulnerable perivascular cell population during ischemia–reperfusion and are preserved by ferroptosis inhibitors in the brain. As pericyte survival is one of the key factors contributing to post-stroke tissue repair and functional recovery, ferroptosis inhibition targeting pericyte rescue may represent a promising therapeutic concept during ischemia–reperfusion in the brain.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X261463955 – Supplemental material for Ferroptosis of microvascular pericytes contributes to ischemia–reperfusion injury in mice
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X261463955 for Ferroptosis of microvascular pericytes contributes to ischemia–reperfusion injury in mice by Hayato Takaki, Kuniyuki Nakamura, Masamitsu Takashima, Yuichi Ozaki, Fumitaka Yoshino, Masaoki Hidaka, Kei Yamanaka, Tomoya Shibahara, Takuya Kiyohara, Yoshinobu Wakisaka, Takanari Kitazono and Tetsuro Ago in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-pdf-2-jcb-10.1177_0271678X261463955 – Supplemental material for Ferroptosis of microvascular pericytes contributes to ischemia–reperfusion injury in mice
Supplemental material, sj-pdf-2-jcb-10.1177_0271678X261463955 for Ferroptosis of microvascular pericytes contributes to ischemia–reperfusion injury in mice by Hayato Takaki, Kuniyuki Nakamura, Masamitsu Takashima, Yuichi Ozaki, Fumitaka Yoshino, Masaoki Hidaka, Kei Yamanaka, Tomoya Shibahara, Takuya Kiyohara, Yoshinobu Wakisaka, Takanari Kitazono and Tetsuro Ago in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Acknowledgements
We thank Naoko Kasahara, Hideko Noguchi, and Mikio Munakata for technical support, and Keiko Hirano and Mikiko Nakashima for secretarial assistance, all from Kyushu University. We also acknowledge the technical assistance provided by the Research Support Center, Research Center for Human Disease Modeling, and the Autonomous Medical Research Center, Kyushu University Graduate School of Medical Sciences. The Research Support Center is partially supported by the Mitsuaki Shiraishi Fund for Basic Medical Research. We thank Editage (![]() ) for English language editing.
) for English language editing.
Author contributions
HT designed and performed the experiments, analyzed the data, and wrote the manuscript. YO, TS, KY, MT, MH, and FY provided the technical assistance. KN, YW, T Kiyohara, T Kitazono, and TA designed the experiments, provided general technical support, edited the manuscript, and directed the study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by Grants-in-Aid for Scientific Research (B 20H03791 to T Kitazono and TA; B 24K02556 and C 20K09373 to TA and KN; C 22K09209 and 25K12344 to YW; C 22K09236 and C 26K11974 to KN), Grant-in-Aid for Early-Career Scientists (24K19551 to TS), and Grant-in-Aid for Research Activity Start-up (21K20693 to TS; 24K23415 to KY; 23K19593 to MT) from the Japan Society for the Promotion of Science; a grant from the Smoking Research Foundation (TA); a grant from Mochida Memorial Foundation for Medical and Pharmaceutical Research (KN); a grant from SENSHIN Medical Research Foundation, Japan (TA, KN, TS, and YW); and research grants from Daiichi Sankyo, Boehringer Ingelheim, Kyowa-Kirin, Eisai, and Bayer (TK, TA, and YW). This research was also supported by AMED under Grant Number 25ym0126811j0004 to KN, and Center for Clinical and Translational Research of Kyushu University.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
ORCID iDs
Data availability statement
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.