
Abstract
Objective
This study aimed to characterize the bacterial and fungal communities in the facial skin of patients with rosacea versus healthy controls and assess their association with skin oil content.
Methods
In this prospective observational study, facial skin samples from eight individuals (six patients with rosacea and two healthy controls) across three skin oil types were analyzed using 16S rRNA and internal transcribed spacer sequencing. Analyses included alpha/beta diversity, compositional profiling, and cross-kingdom correlations.
Results
Patients with rosacea exhibited higher bacterial diversity (Shannon index: 2.26 ± 1.12) than controls (0.71 ± 0.07). Fungal communities underwent extreme restructuring with near-complete species replacement between individuals (Bray–Curtis ∼1.0). Skin oil content was a key determinant of microbial diversity. Cross-kingdom bacteria–fungi correlations were weak and nonsignificant.
Conclusions
Rosacea is associated with distinctive cross-kingdom microbiome alterations, featuring increased bacterial diversity and profound fungal reorganization. These findings challenge prevailing dysbiosis paradigms and highlight the potential for therapeutic strategies targeting both bacterial and fungal elements.
Keywords
Introduction
Rosacea is a common chronic inflammatory dermatosis, clinically characterized by persistent facial erythema, papulopustular lesions, and, in severe cases, rhinophyma, which significantly impairs patients’ quality of life owing to its persistent inflammatory nature.1–3 Although the exact pathophysiological mechanisms driving rosacea remain incompletely understood, a growing body of evidence highlights the critical role of cutaneous dysbiosis—alterations in the skin microbiome composition—in disease initiation and progression.4,5 This dysbiosis often manifests as decreased microbial diversity and is associated with inflammatory pathways involving keratinocytes, T cells, macrophages, and antimicrobial peptides such as cathelicidin LL-37, which exacerbate skin inflammation through neuro-immune interactions.1,6–8 For instance, hyperactivation of mTORC1 signaling in the epidermis suppresses adiponectin expression and amplifies inflammatory cascades, contributing to rosacea-like skin lesions. 9 Traditionally, microbiome research in rosacea has primarily focused on bacterial communities, using techniques such as 16S rRNA sequencing to identify shifts associated with skin inflammation. 5 However, this approach overlooks the complex inter-kingdom dynamics involving fungal inhabitants, which constitute an integral component of the facial skin ecosystem. 10 The human skin microbiome functions as a multifaceted system where bacteria and fungi compete for nutrients, exchange metabolites, and collectively modulate host immune responses through interactions with key immune pathways such as NLRP3 inflammasome activation and interferon-gamma–mediated barrier disruption.6,11 These multi-kingdom interactions, including those with commensals such as Demodex folliculorum mites, can drive cascading shifts toward dysbiosis and inflammation; however, they remain understudied in rosacea.5,10,12
In addition to facial involvement, demodicosis—caused by Demodex folliculorum and Demodex brevis—may present with overlapping clinical features with rosacea, including papulopustular eruptions and erythema. Interestingly, demodicosis can also occur in extrafacial regions such as the chest and ocular area, further complicating differential diagnosis.13,14 These overlapping manifestations suggest that Demodex infestation may contribute to or exacerbate rosacea-like lesions in predisposed individuals, particularly in the context of immunosuppression or topical steroid use.14,15
Additionally, the role of Demodex folliculorum and Demodex brevis mites in rosacea pathogenesis is well-established. A comprehensive meta-analysis by Chang and Huang (2017) demonstrated that patients with rosacea have a 9-fold higher odds of Demodex infestation than healthy controls (pooled odds ratio (OR) = 9.05, 95% confidence interval (CI): 4.54–18.06). 16 The prevalence of Demodex infestation in patients with rosacea varies from 64.5% to 90% based on the detection methods, with significantly higher mite densities observed in both erythematotelangiectatic and papulopustular rosacea subtypes.17,18 Demodex mites harbor endosymbiotic bacteria, most notably Bacillus oleronius (recently reclassified as Heyndrickxia oleronia), which plays a crucial role in the pathogenesis of rosacea. 19 This endosymbiotic bacterium produces antigenic proteins (particularly 62 kDa and 83 kDa proteins) that stimulate peripheral blood mononuclear cell (PBMC) proliferation in 79% of papulopustular rosacea patients compared with only 29% of controls. 20 These bacterial antigens can activate neutrophils, increase intracellular calcium signaling, and promote the release of inflammatory mediators including interleukin (IL)-1β, IL-6, and matrix metalloproteinases.
Ocular involvement in rosacea is remarkably common, affecting approximately 58%–72% of patients with cutaneous rosacea; however, reported prevalence varies widely (6%–72%) based on study methodology and clinical criteria.21,22 The most characteristic ocular manifestation is meibomian gland dysfunction (MGD), which occurs in 50%–90% of patients with rosacea and represents a key pathophysiological link between skin and eye disease. 23 Common ocular signs include lid-margin erythema and telangiectasia, posterior blepharitis, inspissated meibomian gland secretions, chalazia, evaporative dry eye, and conjunctival hyperemia. 24
Moreover, emerging evidence implicates the gut–skin axis, as dysbiosis in the gut microbiota may contribute to the pathogenesis of rosacea through immune modulation, opening avenues for probiotic interventions but failing to address direct fungal involvement.25–27
A deeper understanding of these cross-kingdom interactions is crucial, as they hold promise for developing targeted therapeutic interventions such as hydrogel-based patches that protect against environmental triggers or medications that restore microbiome balance to alleviate inflammation.28,29 However, the lack of comprehensive data on fungal communities—evaluated using ITS sequencing—and their synergistic or antagonistic relationships with bacteria represents a significant gap in rosacea research. 10 This limitation hinders the development of holistic treatments that account for the full ecosystem dynamics in facial skin, which varies across different oil content types (e.g. sebaceous-rich zones prone to pustules).30,31
To address this, our study provides the first comprehensive dual-kingdom analysis of rosacea-associated microbiome alterations by simultaneously examining bacterial (via 16S rRNA sequencing) and fungal (via ITS sequencing) communities in facial samples from patients with rosacea and healthy controls.
Materials and methods
Study participants
Facial skin samples were collected from eight participants, comprising six patients with rosacea (one severe and five moderate cases based on clinical assessment) and two healthy controls who were medically screened volunteers without facial skin lesions. Samples were categorized into three groups according to skin oil content: Excessive Oil Skin (n = 2), Oily Skin (n = 3), and Minimal Oil Skin (n = 3). All participants provided written informed consent, and this study was conducted in accordance with the principles of the Declaration of Helsinki of 1975, as revised in 2024. The inclusion criteria were as follows: 1. Patients in the case group required a diagnosis of rosacea confirmed by at least two physicians independently, fulfilling the diagnostic criteria established by the 2019 Global Rosacea Consensus Expert Committee; 20 healthy controls had no manifestations of facial skin lesions. 2. All participants agreed to refrain from washing their faces or using cosmetics for 12 h prior to skin sampling. 3. All participants voluntarily provided signed informed consent. The exclusion criteria included the following: (a) systemic use of antibiotics or antifungal medications within 28 days prior to sampling; (b) topical application of corticosteroids or antibiotics on the face within 2 weeks prior to sampling; (c) coexistence of other facial dermatoses, such as acne, systemic lupus erythematosus, or seborrheic dermatitis; (d) excessive facial hair that could interfere with sampling; (e) pregnancy or lactation; and (f) unwillingness to provide signed informed consent. All patient details have been de-identified to ensure anonymity. The reporting of this study conforms to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines for observational studies. 32
Methods
Sample collection
Sterile foam swabs moistened with physiological saline were used to collect samples from the bilateral cheek and nasal skin of participants. Each sampling area measured 2 cm × 2 cm. All designated areas were firmly rubbed 25 times with a sterile swab (12 times in one direction, followed by 13 times perpendicular to that direction). Immediately after collection, samples were placed into sterile specimen tubes, temporarily stored at −4°C, and then transferred to −80 °C for long-term storage within 24 h.
DNA extraction and sequencing
Thawed samples were aseptically transferred to lysis matrix B tubes. Enzymatic digestion was performed using lysozyme, mutanolysin, proteinase K, and lysostaphin, followed by mechanical disruption via bead beating. DNA extraction and purification were conducted using the TruSeqTM DNA Sample Prep Kit (Illumina, USA) according to the manufacturer’s instructions. The integrity of the extracted DNA was assessed via 1% agarose gel electrophoresis to ensure quality. Qualified DNA samples were subjected to polymerase chain reaction amplification of the V3–V4 hypervariable regions of the bacterial 16S rRNA gene, followed by sequencing on the MiSeq PE300 platform (Illumina, USA). 33
Bioinformatics analysis
Following primer removal, the processing of bacterial 16S rRNA gene and fungal ITS region sequences followed separate bioinformatics pipelines. For bacterial sequences, clean reads were processed using the UNOISE algorithm within USEARCH (v11.2.64) to perform denoising and generate a high-resolution table of zero-radius operational taxonomic units (ZOTUs). This involved quality filtering (-fastq_maxee 1), dereplication, denoising with unoise3 (minsize = 8), and mapping clean reads to ZOTU representative sequences to construct the abundance table. Taxonomy was assigned using the SINTAX classifier against the RDP 16S training set v18 database with a confidence threshold of 0.8, and ZOTUs classified as chloroplasts or mitochondria were removed. For fungal ITS sequences, the DADA2 algorithm (v1.16) was used to correct errors, merge paired-end reads, and remove chimeras, resulting in a table of amplicon sequence variants (ASVs). Taxonomic assignment for fungal ASVs was performed using the UNITE database. The resulting ZOTU and ASV tables were rarefied to the minimum sequencing depth per sample prior to downstream analysis.
Microbial diversity within samples (alpha diversity) was calculated using observed richness (ZOTUs or ASVs), Shannon diversity, Simpson diversity, and Pielou’s evenness. Differences in community composition between samples (beta diversity) were quantified using Bray–Curtis dissimilarity and visualized via hierarchical clustering and principal coordinate analysis (PCoA). Krona charts were utilized to visualize taxonomic compositions for individual samples.
Statistical analysis
Statistical comparisons employed Mann–Whitney U tests for group differences and Spearman correlations for association analysis. Statistical significance was set at p < 0.05. All analyses were performed using Python (pandas, scipy, and scikit-learn) and visualized using matplotlib and seaborn.
Results
Dataset overview
The bacterial dataset comprised 742 ZOTUs across 8 samples with total reads ranging from 72,539–86,926 per sample (mean =110.21 reads per OTU). The fungal dataset contained 1137 OTUs (ASV) with 78,550–102,453 reads per sample (mean =78.84 reads per OTU). All samples successfully passed quality control metrics (Supplementary Excel Files 1 and 2).
Microbial (bacteria + fungi) diversity analysis
Alpha diversity analysis revealed patterns that contrast with conventional dysbiosis models. Notably, we observed bacterial hyperdiversity in rosacea, which manifested as significantly higher richness and diversity in patients compared with those in healthy controls. This finding contradicts the typical microbial patterns observed in many other inflammatory diseases. Specifically, patients with moderate rosacea exhibited the highest bacterial diversity (Shannon: 2.26 ± 1.12, Richness: 386.0 ± 269.2 OTUs) relative to healthy controls (Shannon: 0.71 ± 0.07, Richness: 124.0 ± 4.2 OTUs). In contrast, fungal diversity patterns varied markedly, with healthy controls maintaining higher diversity (Shannon: 2.13 ± 0.01, Richness: 83.5 ± 3.5 OTUs). The pronounced reduction in fungal diversity observed in the severe rosacea case (P11; Shannon: 1.28, Richness: 47 OTUs) suggests the presence of distinct ecological pressures affecting the mycobiome. Furthermore, substantial individual variation was noted throughout the dataset, particularly within the bacterial communities (Figure 1).
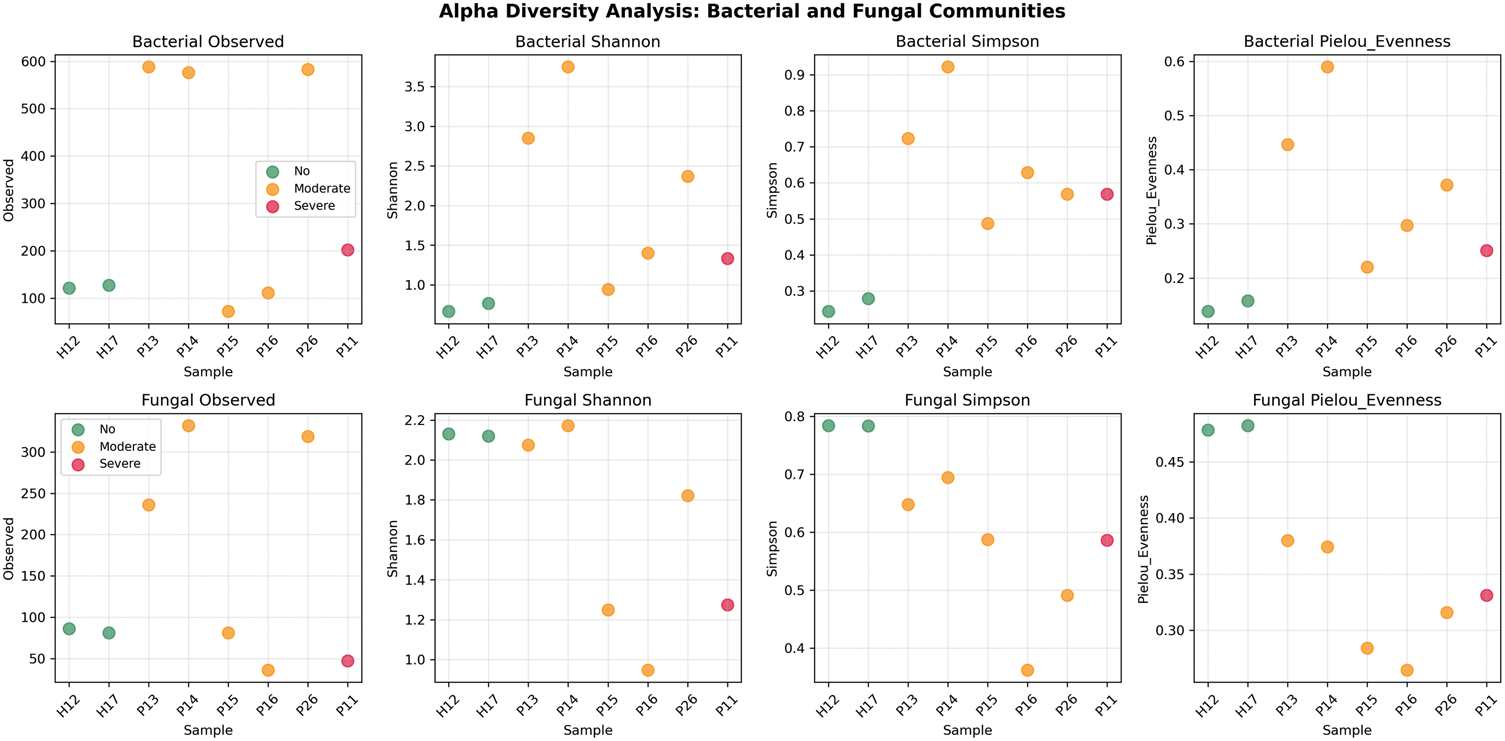
Alpha diversity analysis of bacterial and fungal communities. Comprehensive alpha diversity metrics (Observed richness, Shannon diversity, Simpson diversity, and Pielou’s evenness) for bacterial (top row) and fungal (bottom row) communities across all samples. Points are colored by rosacea status: green (healthy), orange (moderate), red (severe). Error bars represent standard error.
Microbial community clustering analysis
Beta diversity analysis revealed distinct clustering patterns between healthy controls and patients with rosacea (Figures 2 and 3). Healthy controls maintained consistent microbiome signatures, clustering tightly with high similarity (Bray–Curtis distance: 0.030 for bacteria, 0.004 for fungi). In contrast, patients with rosacea demonstrated individualized community structures, exhibiting greater dispersion in the ordination space. PCoA demonstrated clear separation of bacterial communities, with PC1 explaining 69.1% and PC2 explaining 11.8% of variance, where sample P14 exhibited the most divergent bacterial profile. Notably, fungal communities exhibited more extreme alterations than bacterial communities, showing even greater differentiation with most sample pairs reaching maximum dissimilarity (Bray–Curtis ∼1.0), which indicates near-complete species turnover in the mycobiome of patients with rosacea.
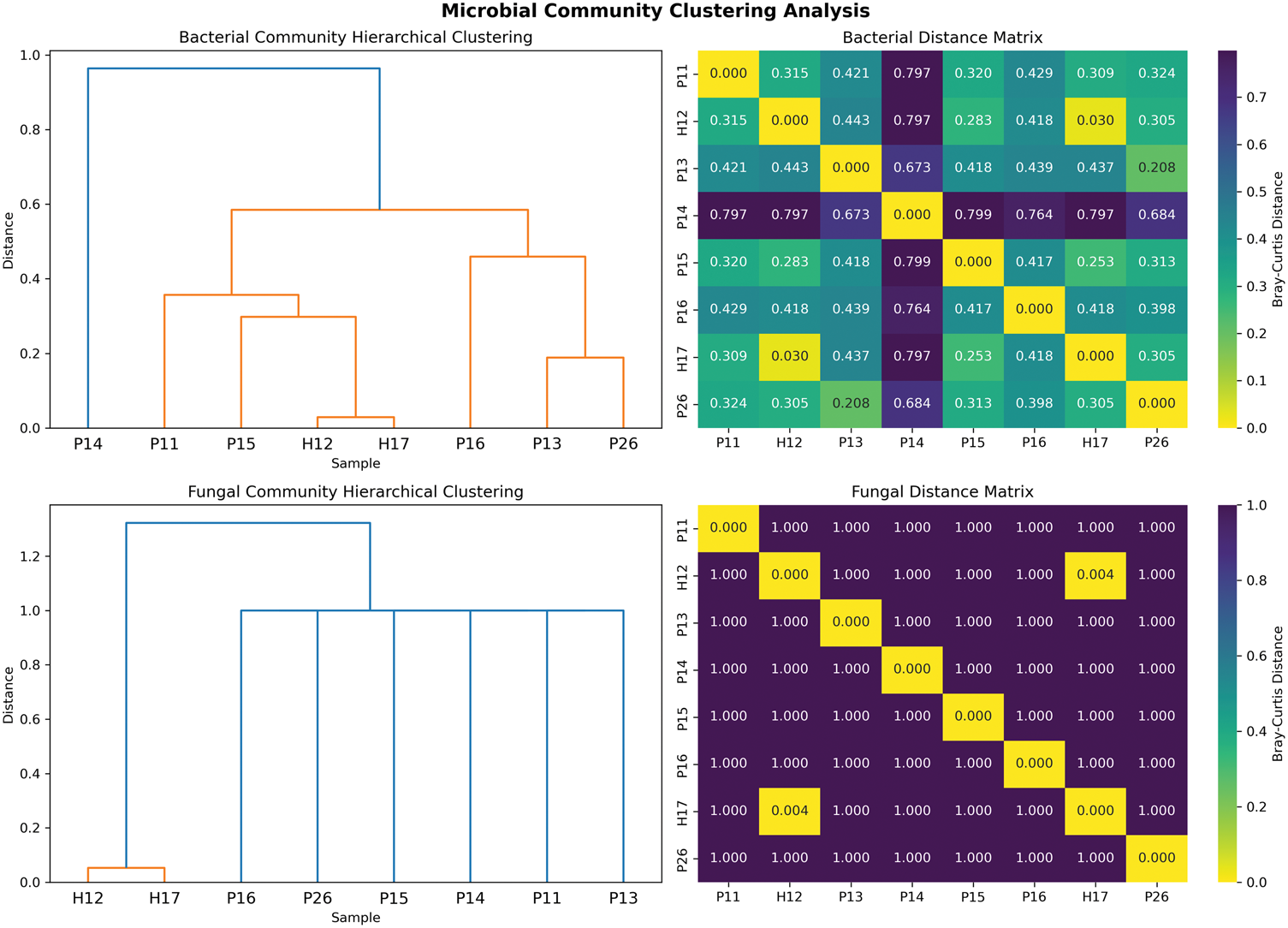
Community clustering analysis. Hierarchical clustering dendrograms and Bray–Curtis distance matrix heatmaps for bacterial (UP) and fungal (DOWN) communities. Dendrograms show Ward linkage clustering, with distance matrices displaying pairwise Bray–Curtis dissimilarities between samples.

Principal component analysis of microbial communities. Principal component analysis ordination plots for bacterial (left) and fungal (right) communities. Points are colored by rosacea status and labeled with sample IDs. Percentage of variance explained by each principal component is indicated on axis labels.
Analysis of microbial community diversity and structural changes
Compositional analysis of the top 20 most abundant taxa revealed dramatic structural changes in patients with rosacea (Figures 4 to 6), characterized by kingdom-specific alteration patterns. Bacterial communities revealed selective expansion of specific OTUs rather than community collapse, with substantial enrichment of ZOTU_3 (Peptoniphilus species), ZOTU_5 (Unidentified2_species), and ZOTU_6 (Acinetobacter_species) displaying >10-fold increases in rosacea samples (log2 fold changes: 15.2, 14.9, and 14.7, respectively). This selective expansion was further evidenced by the finding that 85% of top bacterial OTUs increased in patients with rosacea. The dominant bacterial OTU (ZOTU_1, Cutibacterium_acnes) comprised 404,892 total reads across samples but demonstrated variable distribution. In contrast, fungal communities exhibited extreme turnover with near-complete species replacement, dominated by Malassezia (597,004 reads) but showing extreme polarization where taxa were nearly exclusive to either healthy or rosacea samples. Notably, the appearance of various environmental fungi including Chaetomium and Aspergillus in rosacea samples suggests altered colonization resistance in the diseased skin microenvironment. Bacterial taxonomic compositions were determined based on the assigned taxonomy (see Supplementary Excel_s1.xlsx for full annotations), revealing selective expansion of specific ZOTUs in patients with rosacea.
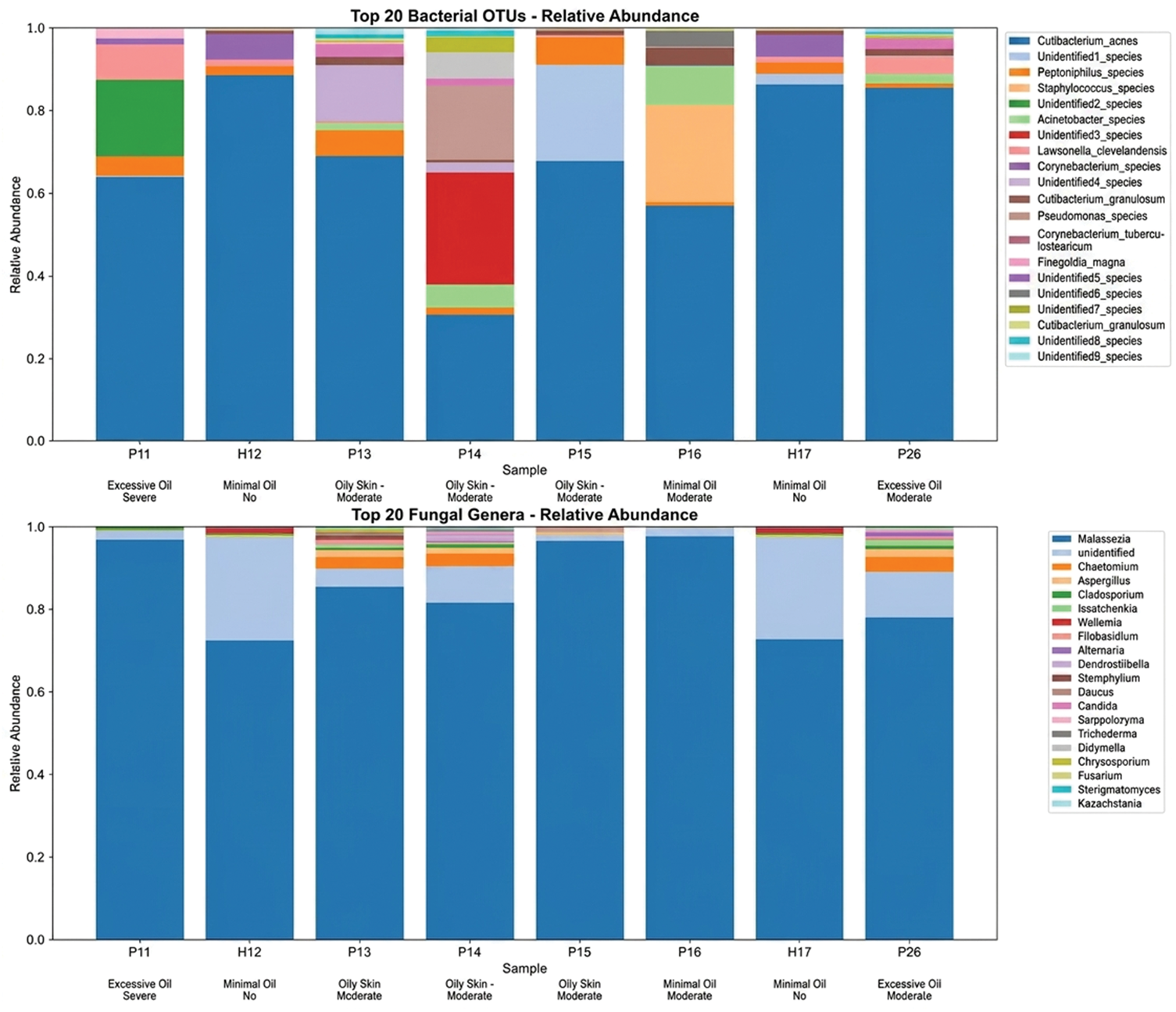
Compositional analysis of top taxa. Stacked bar charts showing relative abundance of top 20 bacterial OTUs (top) and fungal genera (bottom) across all samples. Sample metadata (skin type and rosacea status) are indicated below x-axis labels.
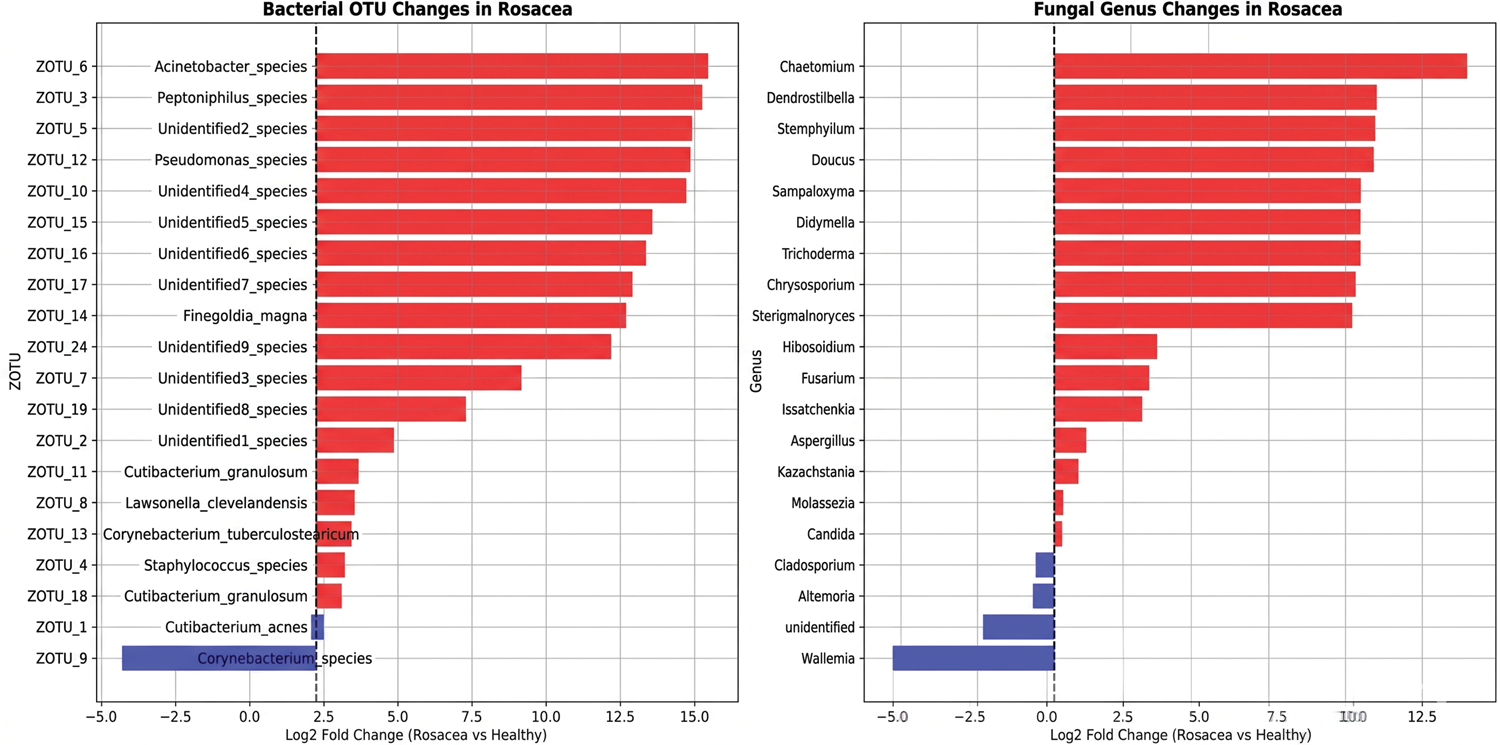
Fold change analysis. Log2 fold change comparisons between rosacea and healthy samples for top 20 bacterial operational taxonomic units (left) and fungal genera (right). Positive values (red) indicate enrichment in rosacea, negative values (blue) indicate depletion.
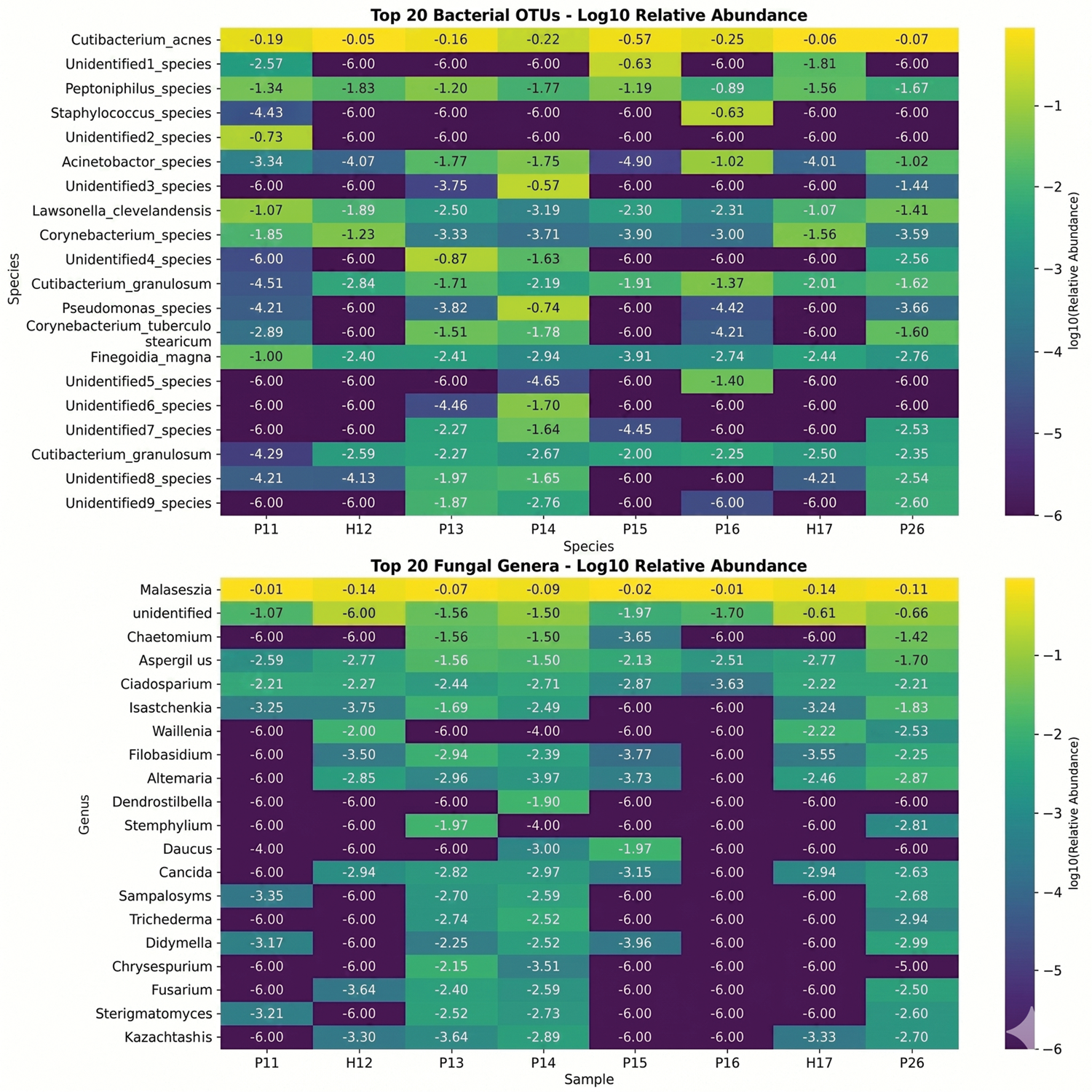
Taxonomic abundance heatmaps. Log10-transformed relative abundance heatmaps for top 20 bacterial operational taxonomic units (top) and fungal genera (bottom). Color intensity represents abundance levels, with numerical annotations for specific values.
Analysis of microbial distribution in different skin types
Skin oil content emerged as a primary driver of microbial community structure (Figure 7). Analysis across the three skin types revealed distinct ecological characteristics: Oily Skin harbored the maximum microbial diversity, predominantly observed in patients with moderate rosacea, with the highest bacterial diversity (Shannon: 2.51 ± 1.43, Richness: 412.0 ± 294.5 OTUs). Excessive Oil Skin maintained high bacterial diversity but intermediate fungal diversity, representing cases with mixed disease severity (Shannon: 1.85 ± 0.73, Richness: 392.5 ± 269.4 OTUs). In contrast, Minimal Oil Skin demonstrated the lowest overall microbial diversity for both kingdoms, a category that included both healthy controls and one moderate rosacea case (Shannon: 0.94 ±0.40, Richness: 119.7 ± 8.1 OTUs). Fungal diversity patterns, although less pronounced than bacterial trends, followed similar gradients, with Oily Skin supporting the highest fungal richness (216.3 ± 126.7 OTUs).

Skin type analysis. Violin plots showing distribution of alpha diversity metrics across skin types (Excessive Oil, Minimal Oil, and Oily Skin) for bacterial (top row) and fungal (bottom row) communities. Black dots represent individual samples.
Bacteria–fungi correlation analysis
Cross-kingdom correlation analysis revealed strikingly weak associations between bacterial and fungal communities (Figures 8 and 9). Overall, no significant cross-kingdom correlations were detected (all p > 0.05). The strongest—although nonsignificant—correlation was observed for species richness (observed OTUs: r = 0.659, p = 0.076), suggesting that samples supporting high bacterial diversity may also support higher fungal diversity. In contrast, Shannon diversity showed minimal correlation (r = 0.071, p = 0.867), and Simpson diversity and Pielou’s evenness exhibited weak negative correlations (r = −0.381, p = 0.352; r = −0.286, p = 0.493, respectively), indicating that diversity and evenness indices are largely independent between kingdoms. Furthermore, sample-level within-sample correlations could not be reliably calculated due to extreme fungal community dissimilarity across samples, reinforcing that bacterial and fungal communities likely respond to distinct ecological pressures.
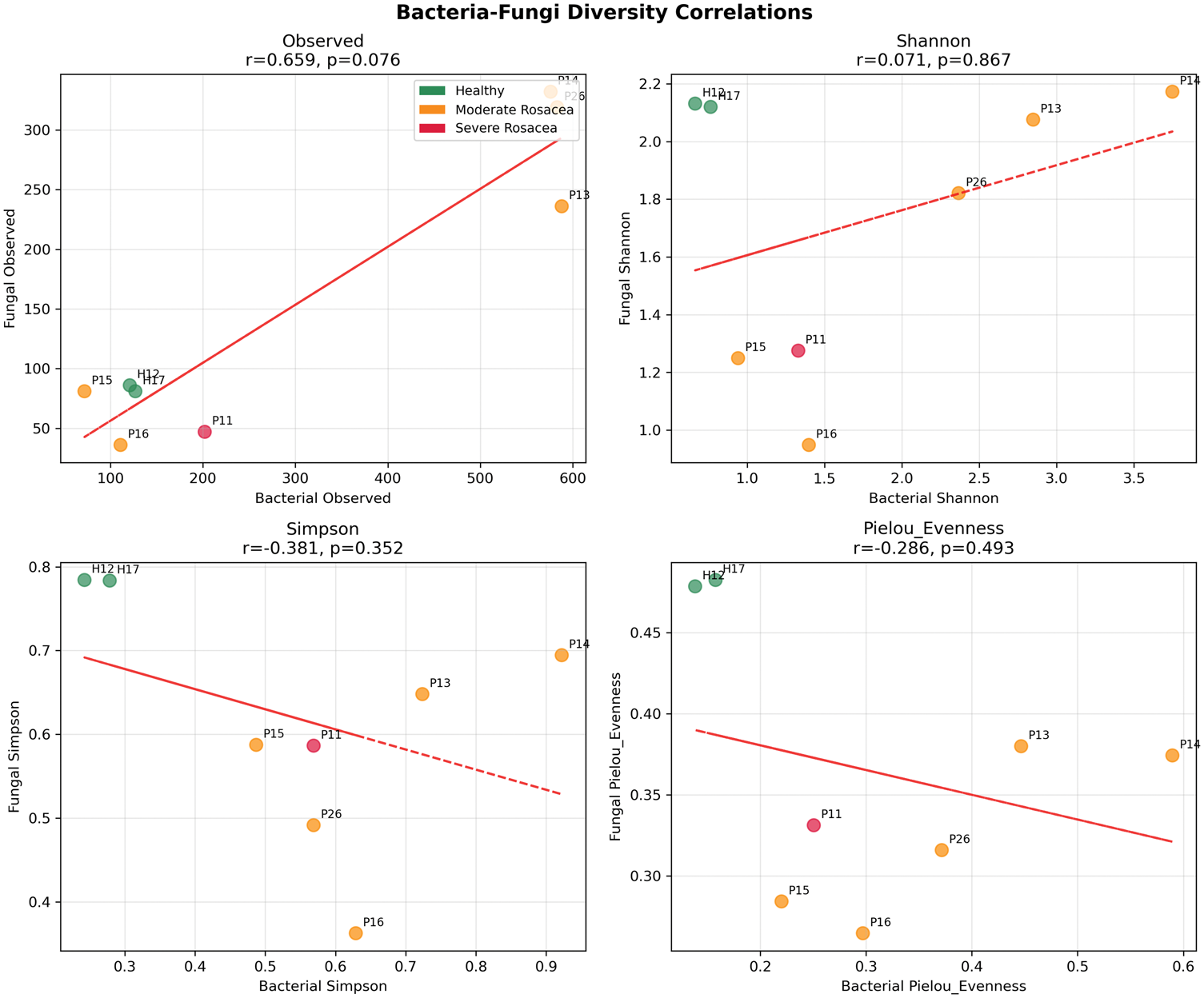
Bacteria–fungi correlation analysis. Scatter plots showing correlations between bacterial and fungal diversity metrics. Points are colored by rosacea status, with trend lines and correlation statistics displayed. Sample labels are provided for reference.
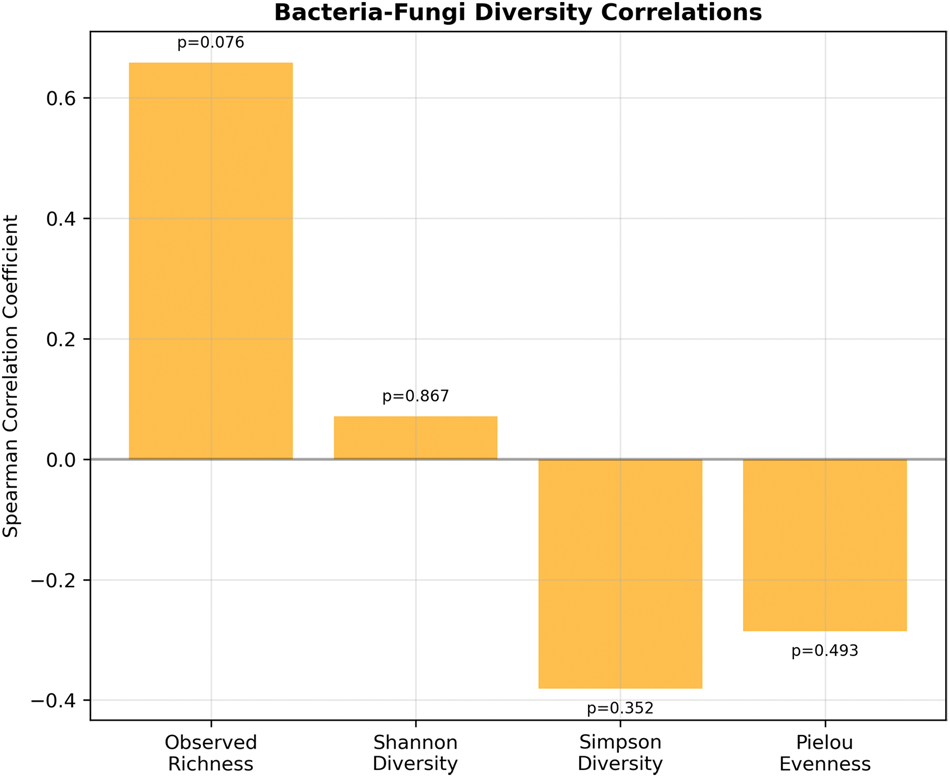
Correlation summary. Summary of bacteria-fungi correlations showing diversity metric correlations with significance indicators.
Discussion
Rosacea-associated microbiome paradox
Our findings suggest a potential paradox in rosacea-associated microbiome alterations: we observed increased bacterial diversity, which appears to contrast with the typical dysbiosis pattern reported in some inflammatory skin conditions. However, given the limited sample size and lack of statistical significance, these observations should be interpreted with caution and require confirmation in future studies with larger populations. This hyperdiversity may reflect the complex pathophysiology of rosacea, where chronic inflammation creates multiple microenvironmental niches supporting diverse bacterial communities. The expansion of specific OTUs (ZOTU_3: Peptoniphilus_species, ZOTU_5: Unidentified2_species, ZOTU_6: Acinetobacter_species) alongside maintained community richness suggests targeted bacterial proliferation rather than broad-spectrum dysbiosis. Additionally, the absence of Demodex assessment in our study limits the interpretation of these microbiome findings. Demodex mites can significantly influence both bacterial and fungal communities through direct mechanical effects on follicular structures and by serving as vectors for bacterial translocation. 34 The paradoxical bacterial hyperdiversity we observed in patients with rosacea (Shannon index: 2.26 ± 1.12 vs. 0.71 ± 0.07 in controls) may be partially attributable to Demodex-mediated disruption of normal skin barrier function and introduction of follicular bacteria to the skin surface. 35 Furthermore, when Demodex mites die or are mechanically disrupted, their endosymbiotic bacteria (e.g. Heyndrickxia oleronia) are released into the follicular environment and can migrate to the skin surface. 36 This mechanism could contribute to the paradoxical increase in bacterial diversity, as endosymbiotic bacteria become part of the detectable surface microbiome. The pathogenic pathway involving Demodex endosymbionts likely involves multiple immune mechanisms relevant to our findings: 1. Pattern recognition receptor (TLR2) activation by both mite chitin and bacterial components leads to upregulation of kallikrein-5 (KLK5) and increased processing of cathelicidin to proinflammatory LL-37 peptides. 12 2. Bacterial nucleic acids and antigens promote plasmacytoid dendritic cell activation and type I interferon responses. 37 3. Inflammasome activation contributes to IL-1β production and neutrophil recruitment. 26 These mechanisms may explain why our bacterial community shifts were correlated with disease severity and why cross-kingdom correlations were weak—the inflammatory environment created by endosymbiont antigens may override normal bacteria–fungi interactions.
Fungal dysbiosis as a primary feature of rosacea
The most striking finding was the near-complete fungal community turnover in patients with rosacea, with Bray–Curtis dissimilarities approaching maximum values (1.0) between most sample pairs. This extreme restructuring suggests fundamental alterations in fungal colonization patterns that may be more profound than bacterial changes.
Our most striking finding was the significant compositional difference in fungal communities between patients with rosacea and healthy controls (PERMANOVA: R2 = 0.407, p = 0.035), while bacterial communities demonstrated nonsignificant trends (R2 = 0.375, p = 0.277). This pattern suggests that mycobiome dysbiosis may represent a more consistent or pronounced feature of rosacea than bacterial alterations. Recent studies have increasingly recognized the importance of fungi in skin health and disease. 38 Malassezia species, the dominant fungi in healthy skin, have been implicated in various inflammatory dermatoses including seborrheic dermatitis, atopic dermatitis, and folliculitis. 39 Our data extend these findings to rosacea, where fungal dysbiosis may contribute to chronic inflammation through multiple mechanisms. The patient with severe rosacea in our cohort exhibited markedly reduced fungal diversity (47 ASVs, Shannon = 1.275) compared with healthy controls (mean: 83.5 ASVs, Shannon =2.126), representing a 44% reduction in richness. This pattern aligns with the “Anna Karenina principle” in microbiome research, which posits that dysbiotic communities tend to be more variable and less diverse than healthy communities. 40 Loss of fungal diversity may impair community resilience and enable opportunistic pathogens to proliferate. Interestingly, patients with moderate rosacea showed increased but heterogeneous fungal diversity (mean: 201.0 ASVs), possibly reflecting the following: (a) compensatory colonization by environmental fungi during barrier dysfunction; (b) variable disease subtypes within the “moderate” category with distinct mycobiomes; or (c) effects of topical treatments that selectively alter fungal communities. 41
The mechanisms linking fungal dysbiosis to rosacea inflammation remain incompletely understood but likely involve multiple pathways. Fungal cell wall components, particularly β-glucans and mannans, activate pattern recognition receptors (PRRs) including Dectin-1, Dectin-2, and Toll-like receptor 2 (TLR2) on innate immune cells. 42 In rosacea-prone skin with impaired barrier function, increased fungal antigen penetration may trigger excessive PRR signaling, leading to proinflammatory cytokine release (IL-1β, IL-6, IL-8, and TNF-α) and recruitment of neutrophils and mast cells. 43 Additionally, Malassezia species produce lipases that hydrolyze sebum triglycerides into free fatty acids, some of which (e.g. arachidonic acid and oleic acid) possess direct proinflammatory and comedogenic properties. 44
Role of Demodex mites and their endosymbiotic bacteria in the pathogenesis of rosacea
The role of Demodex mites in the pathogenesis of rosacea is further supported by the identification of endosymbiotic bacteria such as Bacillus oleronius (recently reclassified as Heyndrickxia oleronia), which have been isolated from Demodex mites in patients with papulopustular rosacea. 45 These bacteria produce antigenic proteins that stimulate the activation of peripheral blood mononuclear cells and neutrophils, leading to the release of proinflammatory cytokines and matrix metalloproteinases.45,46 Other Bacillus species, including B. cereus, B. simplex, and B. pumilus, have also been isolated from Demodex mites in patients with rosacea-like dermatitis, suggesting a diverse bacterial microbiome associated with mite infestation.14,15,46 These findings highlight the potential role of mite–bacteria interactions in modulating local immune responses and disease severity.
Sebum-mediated microbiome modulation
The strong association between skin oil content and microbial diversity provides mechanistic insights into sebaceous–microbiome–inflammation pathways. Sebum creates lipid-rich microenvironments that select for specialized microbes through lipase-mediated metabolism. The observation that oily skin types supported maximum diversity for both kingdoms supports sebum’s role as a primary ecological driver. This relationship has therapeutic implications, as sebum-modulating treatments may indirectly influence microbial communities. However, such approaches should consider complex effects on both bacterial and fungal populations. Interestingly, we observed that Oily Skin harbored the highest bacterial and fungal diversity, a finding that contrasts with some previous reports indicating lower microbial diversity in sebaceous sites of healthy individuals.47,48 This discrepancy may be attributed to several factors. First, the majority of Oily Skin samples in our study were from patients with rosacea, in whom chronic inflammation and barrier disruption could create heterogeneous microenvironments that accommodate a wider array of microorganisms. Second, the small sample size and high interindividual variability limit the generalizability of our observations. Third, methodological differences, such as the inclusion of fungal ITS sequencing and the use of high-resolution ZOTU/ASV pipelines, may capture more diversity than earlier studies. Finally, sebum composition, rather than quantity alone, might be a key determinant of microbial diversity; certain lipid profiles could support a broader microbial community. Future studies with larger cohorts and integrated metabolomic profiling are needed to clarify the relationship between sebum, microbial diversity, and disease status.
Independent cross-kingdom dynamics
The weak correlations between bacterial and fungal communities contrast with the expectations of strong cross-kingdom interactions in skin microbiomes. This independence may reflect several factors: (a) bacterial and fungal communities occupy distinct ecological niches; (b) they respond to different host factors or environmental pressures; or (c) interactions occur through metabolic or chemical pathways not captured by diversity metrics. The extreme individualization of fungal communities may obscure underlying correlation patterns that could be revealed with larger sample sizes or functional analysis approaches.
Clinical and therapeutic implications
These findings have important implications for rosacea treatment strategies. The bacterial hyperdiversity observed suggests therapies aimed at “restoring normal communities” may need to focus on rebalancing rather than reducing bacterial loads. Broad-spectrum antimicrobials might be counterproductive if they further disrupt already altered communities. The extreme fungal restructuring indicates that antifungal approaches deserve greater consideration in rosacea management, particularly for severe cases. However, the individualized nature of fungal alterations suggests that personalized approaches may be necessary. Understanding the cross-kingdom microbiome alterations we identified may inform treatment approaches for both cutaneous and ocular rosacea. For instance, oral tetracyclines (such as doxycycline at anti-inflammatory doses of 40–100 mg daily) are effective for both skin lesions and ocular symptoms, potentially acting through modulation of the microbiome as well as direct anti-inflammatory effects.49,50 Similarly, topical treatments that restore microbiome balance might benefit both cutaneous and ocular manifestations.
Beyond these specific therapeutic adjustments, emerging microbiome-targeted approaches warrant exploration. Probiotics utilizing beneficial commensals (e.g. Roseomonas mucosa and Staphylococcus epidermidis) have shown promise in atopic dermatitis trials and might similarly benefit rosacea patients with dysbiotic communities. 51 Postbiotics—metabolites and cell components from beneficial microbes—could provide therapeutic benefits without the risks associated with live organism administration. 52 Fecal microbiota transplantation, explored for gut–brain–skin axis modulation in acne, may also have relevance for rosacea. 53 Finally, precision prebiotics designed to selectively nourish beneficial taxa while starving pathogens represent an attractive strategy, although optimal formulations remain to be determined. 54
Study limitations
Several limitations should be acknowledged. The small sample size (n = 8) limited statistical power for detecting significant differences. The cross-sectional design prevented assessment of temporal dynamics in microbiome–disease relationships. Our study focused exclusively on bacterial (16S rRNA) and fungal (ITS) communities while not assessing the Demodex mite population, which represents a critical oversight. Standardized skin surface biopsy is considered the gold standard for quantifying Demodex density, with ≥5 mites/cm2 commonly used as a pathological threshold.55,56 Future studies should incorporate Demodex quantification alongside microbiome analyses to provide a more comprehensive understanding of the facial skin ecosystem in rosacea.
Additionally, although our current analysis characterized microbial community composition, it lacked functional profiling to elucidate the underlying mechanisms. Future studies incorporating functional analyses—such as metagenomics, metatranscriptomics, or metabolomics—would be essential to uncover the specific metabolic interactions driving the observed shifts in microbial populations. Such approaches could clarify whether these microbial changes lead to functional consequences, such as increased production of proinflammatory molecules or disruption of the skin barrier, thereby bridging the gap between taxonomic alterations and clinical disease manifestations.
Conclusions
This comprehensive analysis demonstrates that rosacea involves complex cross-kingdom microbiome alterations characterized by bacterial hyperdiversity and extreme fungal community restructuring. These findings challenge conventional dysbiosis paradigms and highlight the importance of considering both bacterial and fungal components in rosacea pathogenesis. The strong influence of sebum production on microbial diversity patterns provides mechanistic insights, while the independence of bacterial and fungal community dynamics suggests complex ecological relationships requiring further investigation. These insights provide a foundation for developing more targeted, microbiome-informed therapeutic approaches for this challenging inflammatory skin condition. The paradoxical increase in bacterial diversity, coupled with dramatic fungal turnover, suggests that rosacea represents a unique form of dysbiosis requiring novel therapeutic paradigms that address both kingdoms of the skin microbiome. Future therapeutic development should consider these dual-kingdom alterations and the potential for personalized treatments based on individual microbiome profiles.
Supplemental Material
sj-xlsx-1-imr-10.1177_03000605261454625 - Supplemental material for A pilot study investigating the bacterial and fungal community shifts in facial skin in rosacea
Supplemental material, sj-xlsx-1-imr-10.1177_03000605261454625 for A pilot study investigating the bacterial and fungal community shifts in facial skin in rosacea by Xiaobin Tang, Zhiyong Shen, Bin Li and Jiang Yuan in Journal of International Medical Research
Supplemental Material
sj-xlsx-2-imr-10.1177_03000605261454625 - Supplemental material for A pilot study investigating the bacterial and fungal community shifts in facial skin in rosacea
Supplemental material, sj-xlsx-2-imr-10.1177_03000605261454625 for A pilot study investigating the bacterial and fungal community shifts in facial skin in rosacea by Xiaobin Tang, Zhiyong Shen, Bin Li and Jiang Yuan in Journal of International Medical Research
Footnotes
Acknowledgments
We thank all study participants for their contribution to this research and also acknowledge the technical support provided for sample processing and sequencing.
Author contributions
Zhiyong Shen and Xiaobin Tang contributed to data analysis and manuscript draft preparation. Bin Li and Jiang Yuan contributed to investigative efforts and critical review of the work.
Data availability
All data supporting the conclusions of this article are included within the article and its supplementary materials. Raw sequencing data and analysis scripts are available upon reasonable request.
Declaration of conflicting interests
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval
This study was conducted in accordance with institutional ethics guidelines, with appropriate informed consent obtained from all participants. The ethics approval number is NSFYEC-KY-2025039, approved on 9 March 2025.
Funding
This work was supported by the Shenzhen Nanshan District Technology R&D and Creative Design Project Sub-fund (Grant No. NS2025012) and the Hunan Provincial Natural Science Foundation of China (Grant No. 2026JJ80491).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.