
Abstract
Pulmonary fibrosis, particularly idiopathic pulmonary fibrosis, is a progressive and fatal interstitial lung disease with limited curative therapies. This narrative review provides a concise overview of emerging molecular targets and novel therapeutic strategies for pulmonary fibrosis, including mesenchymal stem cell therapy and nanomaterial-based inhalation systems, focusing on their mechanisms, preclinical evidence, and clinical potential. We highlight critical challenges in translating preclinical discoveries into clinical benefit, such as animal model limitations and patient heterogeneity, and outline future research priorities. Notably, this review is not a systematic review; it does not include formal meta-analysis or a comprehensive literature search following Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines, which may have introduced selection bias. Thus, findings should be interpreted with appropriate caution.
Keywords
Introduction
Pulmonary fibrosis (PF) is a common imaging feature observed during the progression and outcome of various pulmonary diseases. This includes its presence as a characteristic feature throughout the course of interstitial lung disease (ILD) as well as a secondary manifestation following the chronic, protracted stages of diseases such as chronic obstructive pulmonary disease (COPD), pulmonary tuberculosis, and coronavirus disease 2019 (COVID-19). PF progressively affects the lung interstitium, leading to impaired gas exchange, dyspnea, reduced quality of life, and ultimately, respiratory failure and death. 1 PF encompasses multiple clinical subtypes with distinct etiologies, including secondary PF (e.g. associated with autoimmune diseases, environmental exposures, or drug toxicity) and idiopathic pulmonary fibrosis (IPF). Etiological factors may include exposure to allergens, toxins, or medications as well as underlying autoimmune diseases, although in many cases the cause remains unknown or is classified as idiopathic. 2 PF is a multicellular process involving impaired alveolar epithelial regeneration and persistent activation of the matrix, including fibroblasts and immune cells. 3 Due to its increasing incidence, it has become a significant focus of medical research. Importantly, the approved therapies are specifically indicated for IPF and have shown inconsistent efficacy in other forms of PF, highlighting the critical need to distinguish between PF subtypes when evaluating novel targets and therapeutic strategies. The increased incidence of PF presents substantial challenges for patients, healthcare providers, and researchers. 4 Although numerous compounds have demonstrated efficacy in alleviating PF in animal models, only a few have shown beneficial effects in clinical trials. 5 Furthermore, with a rise in the number of patients recovering from severe COVID-19, it is becoming increasingly evident that a considerable proportion exhibit signs of post-inflammatory PF. 6 Consequently, research on the mechanisms, pathways, and targeted therapies for PF continues unabated.
Building on recent in vitro and in vivo studies on PF, novel therapeutic agents such as pirfenidone and nintedanib have been developed, with the aim of slowing the progression of this complex disease.7,8 PF is typically observed in older patients presenting with progressive dyspnea and cough, and examination may reveal bibasilar inspiratory crackles and digital clubbing. The natural course of PF varies considerably. Although several patients experience a gradual decline in respiratory function, some remain stable, whereas others demonstrate rapid deterioration. 9
Pathogenesis of PF
In PF research, classically activated macrophages (M1) and alternatively activated macrophages (M2) have garnered significant attention. Evidence indicates that M1 macrophages are closely associated with pro-inflammatory responses, whereas M2 macrophages play a crucial role in anti-inflammatory responses. 10 When stimulated by the presence of toxic factors, M1 macrophages produce several pro-inflammatory cytokines that mediate inflammatory injury in the alveolar tissue. M2 macrophages secrete various growth factors that contribute to the pathogenesis of fibrosis and abnormal remodeling of alveolar structures. 11 The accumulation of M2 macrophages in the lungs has been identified as a primary source of many key profibrotic mediators (e.g. transforming growth factor-beta (TGFβ), connective tissue growth factor (CTGF), and CC motif chemokine ligand 18 (CCL18)). M2 macrophages are also believed to promote fibrosis through the release of factors such as tumor necrosis factor-alpha (TNFα), interleukin (IL)-1 (IL-1), IL-10, IL-13, IL-33, platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and fibronectin (Figure 1). 12
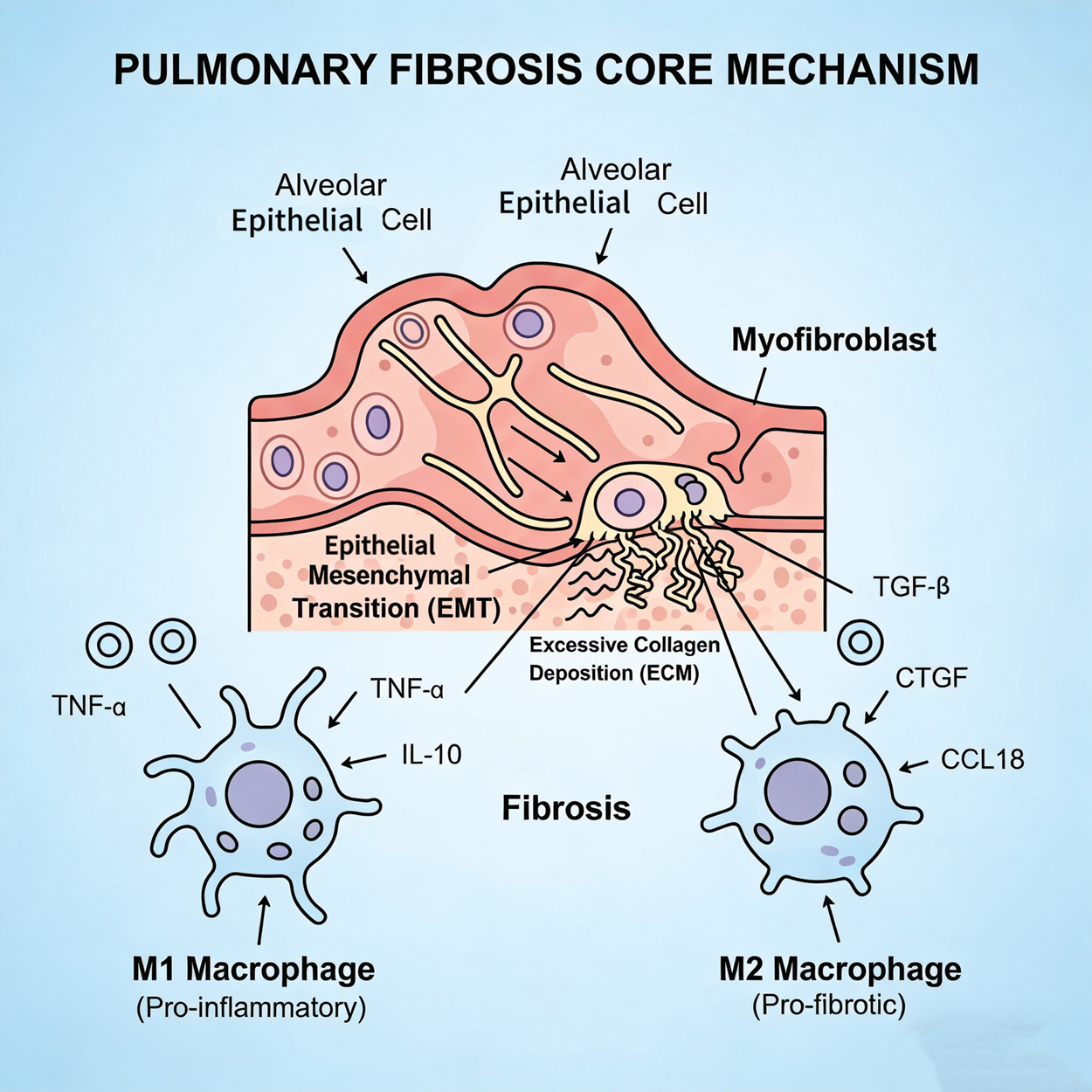
The core mechanism of pulmonary fibrosis can be conceptualized as a cascade initiated by alveolar epithelial damage. Pulmonary fibrosis is a progressive and often fatal interstitial lung disease characterized by excessive extracellular matrix deposition and architectural destruction of the lung parenchyma. The underlying pathogenesis is complex and involves a dynamic interplay between repeated alveolar epithelial cell injury, dysregulated repair mechanisms, and a persistent inflammatory and profibrotic microenvironment.
Beyond macrophage activation, genetic evidence reveals that the lung epithelium plays a pivotal role in disease susceptibility and progression. Furthermore, epithelial–mesenchymal interactions are considered a central factor driving epithelial remodeling and extracellular matrix (ECM) expansion during the pathological process of PF.13,14
Emerging therapeutic targets for PF
Triggering receptor expressed on myeloid cells-2 (TREM-2)
TREM2 is a cell surface receptor that is expressed on myeloid cells and belongs to the TREM family. 15 Recent studies highlight its critical role in the pathogenesis of Alzheimer’s disease (AD), influencing amyloid pathology, microglial function, inflammation, and metabolism.16,17 Its role in IPF is also emerging. Transcriptomic analysis has confirmed upregulated TREM2 gene expression in bronchoalveolar lavage (BAL) cells from patients with IPF compared with those from healthy donors. 18 Studies have shown that TREM2 knockout or knockdown, even in the absence of an identified ligand, promotes macrophage apoptosis, abrogates the anti-apoptotic effects of sphingomyelins on macrophages, and protects mice from lung fibrosis. 19 Future research should further explore the mechanisms of TREM2 in PF and develop effective therapies targeting this molecule. Although it exhibits antifibrotic potential, its bidirectional regulatory role in myeloid cells remains unclear, warranting caution against potential systemic immunosuppression.
Cluster of differentiation (CD) 36 (CD36)
CD36, also known as scavenger receptor B2, is a multifunctional receptor widely distributed across various organs. 20 Located on the cell membrane, it responds to dietary fats and transports fatty acids. 21 Recent research has elucidated its crucial role in CD36 signaling, regulating phospholipase activation for Ca2+ mobilization. This process involves the release of arachidonic acid (AA) from membrane phospholipids, remodeling of membrane long-chain fatty acid composition, and generation of bioactive eicosanoids.22–24 Type II alveolar epithelial cell (AEC2) injury/apoptosis and monocyte/macrophage accumulation/activation are pivotal in progressive PF.25,26 Researchers have discovered CD36 involvement in the conversion of latent transforming growth factor-beta1 (TGF-β1) to active TGF-β1 in alveolar macrophage cultures obtained after bleomycin (BLM)-induced lung injury. 27 TGF-β1 plays a central role in fibrosis progression by activating multiple signaling pathways, including the Smad, mitogen-activated protein kinase (MAPK), and extracellular signal-regulated kinase (ERK) pathways.28–30 During PF development, CD36 activation in macrophages triggers inflammatory responses. Therefore, upregulating CD36 expression or enhancing its function represents a novel therapeutic strategy aimed at promoting macrophage clearance of apoptotic cells, thereby mitigating lung inflammation and fibrosis. As a lipid metabolism hub, modulating CD36 may impact systemic energy homeostasis, making tissue-selective targeting a future challenge.
Fos-related antigen-2 (Fra-2)
Fra-2, the newest member of the Fos family, 31 is intricately linked not only to skeletal development, metabolism, the immune system, and eye development but also to the progression of respiratory diseases such as lung cancer, asthma, PF, and COPD.32,33 Elevated Fra-2 expression has been documented in several chronic lung diseases, including COPD and asthma. Infection in Fra-2 transgenic (TG) mice led to increased levels of pro-inflammatory cytokines and chemokines (including IL-1β, IL-6, TNF-α, and CXC motif chemokine ligand 1 (CXCL1)) in BAL supernatant and plasma. 34 Studies have indicated that Fra-2 inactivation attenuates BLM-induced PF, whereas fibrosis is exacerbated in Fra-2t bone marrow chimeras. 35 This phenotype is characterized by pulmonary vascular remodeling and occlusion, closely associated with osteopontin expression. Osteopontin plays multifaceted roles in health and disease, promoting wound healing and maintaining bone homeostasis. 36 However, in disease states, it can exert detrimental effects.37,38 Osteopontin mediates complex processes that coordinate fibroblast differentiation into myofibroblasts, thereby promoting fibrosis.39,40 Fra-2 represents a potential therapeutic target, offering a new perspective for developing novel treatments for PF to slow or even reverse its progression.
Methyl-CpG-binding domain protein 2 (MBD2)
MBD2 is a member of the MBD protein family that binds to methylated promoters and functions as a methylation-dependent transcriptional repressor. 41 Studies have shown significantly elevated MBD2 expression in a mouse model of neutrophil-dominant severe asthma, suggesting its potential as a novel biomarker for identifying severe asthma and airway inflammation types. 42 Beyond inflammation, MBD2 is involved in executing apoptosis.43,44 In a mouse alveolar epithelial cell line, LPS induces MBD2 expression and apoptosis in a dose- and time-dependent manner. Short hairpin ribonucleic acid (shRNA)–mediated MBD2 knockdown markedly reduces apoptosis, whereas MBD2 overexpression enhances LPS-induced apoptosis. 45 Research has demonstrated that MBD2 deficiency protects mice from BLM-induced PF, reducing TGF-β1 production and M2 macrophage accumulation in the lungs. Mechanistically, macrophages in fibrotic lung tissue exhibit a DNA hypermethylation signature.46,47 The MBD2 protein binds to methylated CpG DNA sequences in the promoter region of the Ship gene, inhibiting its expression. This binding enhances phosphoinositide 3-kinase (PI3K)/Akt signaling pathway activity, promoting macrophage polarization toward the M2 phenotype. Reducing MBD2 protein levels helps alleviate BLM-induced lung injury and fibrosis in mice. 48 As a methylation-dependent transcriptional repressor, MBD2 acts as a key disease driver and potential therapeutic target in severe asthma and PF by promoting apoptosis and driving M2 macrophage polarization.
Oligopeptide/histidine transporter solute carrier family 1 member 3 (SLC15A3)
The oligopeptide/histidine transporter SLC15A3 is a lysosomal peptide transporter that transports free histidine from the lumen to the cytosol. 49 Its relationship with macrophages during inflammatory responses has been revealed. Toll-like receptors-2 (TLR2) ligands in macrophages promote SLC15A3 upregulation at both mRNA and protein levels. Moreover, SLC15A3 knockdown or overexpression alters the expression of TLR4-triggered pro-inflammatory cytokines.50,51 In BLM- or radiation-induced PF models, macrophage accumulation leads to increased pulmonary SLC15A3 levels. However, SLC15A3 deficiency paradoxically protects mice from PF. 52 The observation that macrophage accumulation coincides with elevated SLC15A3 levels in PF models; however, its absence is protective, suggests that SLC15A3 is an important molecule driving fibrotic progression and could be a therapeutic target for PF.
Peroxiredoxin 1 (PRDX1)
PRDX1, a member of the peroxiredoxin family, regulates intracellular reactive oxygen species (ROS) levels and participates in various other physiological processes by acting as a chaperone, playing a crucial role in disease onset and development.53,54 PRDX1 deficiency leads to elevated ROS levels in lung epithelial cells and promotes epithelial–mesenchymal transition (EMT) via the PI3K/Akt and c-Jun N-terminal kinase (JNK)/Smad signaling pathways. PRDX1 deletion significantly enhances TGF-β secretion, ROS production, and cell migration in primary lung fibroblasts. 55 ROS accumulation is a common feature of IPF, 56 and excessive ROS levels further promote PF progression. PRDX1 regulation may represent a potential therapeutic target for PF.
Octyl itaconate. 4-octyl itaconate (4-OI), a cell-permeable derivative of itaconate, exhibits antioxidant and anti-inflammatory properties.57,58 In inflammatory diseases, activated macrophages shift from oxidative phosphorylation to aerobic glycolysis. Research finds that 4-OI modulates macrophage function by alkylating cysteine residue 22 of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), reducing its activity, downregulating aerobic glycolysis, and exerting anti-inflammatory effects. 59 4-OI exerts immunomodulatory effects by modifying target protein cysteines. Strict control of nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome cleavage is crucial to prevent excessive inflammation. 60 Beyond its prominent anti-inflammatory and antioxidant effects on inflammation-induced fibrosis, 4-OI improves BLM-induced PF via a nuclear factor erythroid 2–related factor 2 (Nrf2)-dependent mechanism. Its role in alleviating PF is partly attributed to the direct inhibition of EMT and partly to the indirect suppression of M2 macrophage-mediated EMT.61,62 These findings reveal 4-OI as a novel antifibrotic agent with significant clinical potential for treating PF.
Sparganii Rhizoma (SR)
SR has shown anticancer potential in various malignancies and is clinically used to promote blood circulation and resolve blood stasis.63,64 Furthermore, SR is widely used in clinical practice to treat organ fibrosis and inflammation. 65 Curcumae Rhizoma–Sparganii Rhizoma (CR-SR) is an ancient Chinese herbal formula. Ultra-performance liquid chromatography and quadrupole time-of-flight mass spectrometry (UPLC-Q/TOF-MS) identified 57 chemical components in CR-SR, of which 27 were associated with liver fibrosis protein targets. 66 Studies have demonstrated that SR significantly alleviates BLM-induced experimental PF in mice by inhibiting fibroblast differentiation and the EMT process. 67 SR is a promising candidate traditional Chinese medicine for treating PF, although its specific active components and molecular mechanisms require further elucidation.
Histone deacetylase 3 (HDAC3)
HDAC3 is a key epigenetic modifying enzyme that influences chromatin structure and transcriptional regulation, making it a compelling therapeutic target. It is also widely investigated as a potential treatment option for organ fibrosis in clinical applications.68,69 HDAC3 inhibitors are emerging as potential key drugs for treating acute lung injury and fibrotic diseases. HDAC3 plays a role in regulating inflammatory gene expression in macrophages, a process crucial for PF development. 70 In PF models, HDAC3 promotes increased expression of EMT markers, activates the neurogenic locus notch homolog protein 1 (Notch1) and signal transducer and activator of transcription 1 (STAT1) signaling pathways, and upregulates inflammasome components absent in melanoma 2 (AIM2) and apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC). 71 Nrf2 is a key redox regulator closely linked to IPF pathogenesis. 72 Studies in IPF patients and BLM-treated mouse models have shown HDAC3 upregulation and Nrf2 suppression. Research has demonstrated that HDAC3 binds to FOXM1 and deacetylates the Nfe2l2 promoter, inhibiting Nrf2 transcription. The HDAC3 inhibitor RGFP966 can restore Nrf2 function and reduce PF, suggesting that inhibiting HDAC3 to maintain Nrf2 levels prevents PF.73,74
Although the aforementioned emerging targets have opened critical molecular avenues for PF treatment, several targeted agents face inherent challenges, including insufficient in vivo stability, low lung accumulation, and significant systemic adverse effects, which hinder their optimal therapeutic efficacy. In this context, integrating these targeted regulatory strategies with advanced delivery technologies, such as novel delivery systems, stem cell therapy, and nanomaterial-based inhaled formulations, enables targeted drug delivery, controlled release, and efficient lung enrichment. This integration not only enhances the precision and therapeutic outcomes of target intervention but also provides key technical support for the clinical translation of these promising targets.
Novel therapeutic approaches for PF
Pirfenidone and nintedanib are the primary antifibrotic drugs currently used in clinical practice. Studies have confirmed their ability to slow the decline of forced vital capacity in patients with IPF and reduce the risk of acute exacerbations.75,76 Current treatment strategies primarily rely on these antifibrotic agents. Although they can delay disease progression, their significant associated adverse effects adversely impact patients’ quality of life. 77 Therefore, developing novel and more effective therapeutic strategies, whether as monotherapies or in combination with existing drugs, has become particularly urgent.
Mesenchymal stem cell (MSC) therapy
In recent years, stem cell therapy has emerged as a highly promising and innovative field of scientific research. 78 MSCs, as multipotent stem cells, exhibit protective effects against PF. First discovered by Friedenstein et al. in 1968, 79 MSCs play a crucial role in tissue regeneration and homeostasis due to their potential in osteogenesis and adipogenesis, among other functions. 80 Studies have shown that bone marrow-derived mesenchymal stem cell (BM-MSC) transplantation significantly reduces lung injury and fibrosis in animal models of BLM-induced PF. 81
Experiments have provided new evidence demonstrating that MSCs can inhibit alveolar type 2 (AT2) cell senescence by suppressing lysosome-mediated degradation of the nicotinamide phosphoribosyltransferase (NAMPT) protein. This finding partially elucidates the mechanism by which MSCs reverse AT2 cell senescence and BLM-induced PF. 82 The efficacy of MSC therapy for PF is closely linked to their ability to transfer mitochondria to damaged epithelial cells, reduce collagen accumulation, and promote tissue repair through the release of anti-inflammatory and antifibrotic factors, demonstrating their significant potential in treating PF.83,84 Some studies have investigated the role of induced pluripotent stem cells (iPSCs) and their derived MSCs (induced pluripotent stem cells–derived mesenchymal stem cells (iPSC-MSCs)) in treating BLM-induced PF. Results indicate that iPSC-MSC treatment alleviates inflammation and fibrosis caused by BLM, increases levels of the anti-inflammatory cytokine, IL-10, and demonstrates therapeutic potential through the secretion of immunosuppressive proteins. 85 A study evaluating the safety, tolerability, and efficacy of high cumulative doses of MSCs in patients with rapidly progressive moderate-to-severe IPF has shown no significant adverse events even with extremely high doses of stem cells, with only transient fever observed post-infusion. Lung function improved in the main treatment group, while it continued to decline in the placebo group. 86
Beyond conventional MSC therapy, research on MSCs from other sources for treating PF remains exploratory. Researchers investigated whether mouse gingiva-derived mesenchymal stem cells (GMSCs) could prevent BLM-induced PF. Experiments have demonstrated elevated TGF-β levels in the bronchoalveolar lavage fluid (BALF) and increased gene expression of profibrotic factors in the lung tissue after BLM instillation. However, GMSC administration inhibited these changes, demonstrating that GMSCs exert antifibrotic effects by reducing lysophosphatidic acid (LPA), LPA1, and TGF-β expressions. 87 The application of umbilical cord–derived mesenchymal stem cells (UCMSCs) in treating chronic lung injury offers new insights. Many studies have established the anti-inflammatory role of MSCs in inflammation-induced fibrosis. However, UCMSCs tend to promote M2 macrophage polarization, which can influence the fibrotic process. 88 Macrophage polarization is a major mechanism identified in PF progression. 89 UCMSCs can also alleviate PF by inhibiting CCL2 expression via suppression of the ERK1/2 signaling pathway, subsequently modulating monocyte–macrophage migration. 90 Adipose-derived MSCs inhibit TGF-β1-induced fibroblast activation by suppressing the TGF-β1/Smad signaling pathway and promote fibroblast autophagy by modulating p62 expression. 91 Most MSCs are obtained through invasive methods. However, urine-derived stem cells (USCs) can be acquired via a safe, noninvasive, and cost-effective procedure. USCs do not form teratomas in vivo or possess carcinogenic potential, making them ideal substitutes for other MSC types. USC transplantation can effectively reverse the PF phenotype and inhibit the TGF-β1-Smad2/3 pathway, a key driver in fibrogenesis.91–93
Different routes of administration for MSCs are also under further exploration and investigation. Mesenchymal stem cell–derived extracellular vesicles (MSC-EVs) show clinical potential in anti-aging and antifibrotic therapy.94,95 Researchers constructed CD38 antigen receptor membrane-modified MSC-EVs (CD38-ARM-MSC-EVs) targeting AT2 cells via lentiviral transfection. Compared with conventional MSC-EVs, CD38-ARM-MSC-EVs exhibited higher expression of CD38 antigen receptor and antifibrotic micro ribonucleic acids (miRNAs) in vitro and in a naturally aged mouse model. They effectively restored nicotinamide adenine dinucleotide (NAD+) levels, rejuvenated senescent cells, and alleviated PF in aged mice. This study provides a technique for designing MSC-EVs and supports CD38-ARM-MSC-EVs as a potential drug candidate against PF. 96 Researchers have developed an MSC-based nanoengineering platform for treating severe fibrosis. This platform involves bioconjugated MSCs-Lip@NCAF and type I collagenase–modified liposomes. MSCs-Lip@NCAF can migrate to fibrotic lungs and release Lip@NCAF, which ablates collagen fibers and delivers nintedanib to inhibit fibroblast hyperactivation. Concurrently, MSCs differentiate into AT2 cells to repair alveolar structure, promoting regeneration of damaged lungs in aged mice. MSCs-Lip@NCAF demonstrates significant potential for treating PF, especially in older patients. 97
Although MSC therapy shows promise for IPF treatment, its development is limited by an insufficient understanding of MSC status in vivo. Some researchers have proposed a Janus Au/mesoporous silica core/shell nanoparticle (Janus NPs) system for effective MSC therapy and real-time tracking. Janus NPs consist of an Au core and a mesoporous silica shell loaded with pirfenidone (PFD), along with two asymmetric targeting moieties: ROS-sensitive poly(ethylene glycol)-thioketal (mPEG-TK) and distearoyl phosphoethanolamine (DSPE). This asymmetric decoration enables Janus NPs to anchor onto the cell membrane long-term, facilitating the reactive release of PFD in ROS-rich environments, thereby enhancing the therapeutic effect of transplanted MSCs. After drug release, Janus NPs enter the MSCs, enabling long-term CT imaging tracking of MSCs in IPF model mice. This deepens the current understanding of cell therapy mechanisms and offers new strategies for stem cell-based IPF treatment. 98
Currently, researchers are focusing on the therapeutic effects of lung stem cell transplantation in two different PF models. Results have clearly demonstrated that lung cell transplantation exhibits significant efficacy across multiple dimensions, including histology, biochemistry, radiology, and physiology. 99 Stem cell transplantation technology shows remarkable potential in the field of lung regeneration therapy and has become a promising therapeutic strategy. The core of this technology lies in delivering healthy lung epithelial progenitor cells to the damaged lung tissue, which subsequently engraft and differentiate within the target tissue to reconstruct a functional respiratory epithelium. 100 Transplanted lung epithelial progenitor cells possess strong self-renewal capacity, offering the possibility for sustained and long-term therapeutic effects.101,102
Nanomaterial-based inhalation therapy
The inhalation route allows for noninvasive drug delivery directly to fibrotic tissues, such as from the respiratory system to the lesion site. This method offers the advantages of high efficiency, low systemic toxicity, small therapeutic doses, and stable dosage forms.103,104
An inhalable lipid nanoparticle (LNP) was specifically designed for the simultaneous delivery of two messenger ribonucleic acids (mRNAs), aiming to treat IPF by restoring surfactant production in AT2 cells and promoting alveolarization. Initially composed of dipalmitoylphosphatidylcholine mixed with a synthetic ionizable lipid, this inhalable LNP was engineered for efficient lung mucus penetration and co-delivery of two mRNAs encoding cytochrome b5 reductase 3 and bone morphogenetic protein 4. Results have shown that the mRNA-LNP system achieved high-efficiency protein expression in lung epithelial cells, significantly alleviated alveolar collapse, and extended the survival time of fibrotic mice, offering a potential clinical treatment strategy for IPF.105–107
Inhalable liposomal microparticles are also being investigated as a novel inhalation therapy. 108 Cryptotanshinone (CTS) has been shown in vitro to alleviate PF by modulating macrophage polarization. 109 CTS was loaded into mannose-modified liposomes (Man-lipos) and embedded into mannitol microparticles (M-MPs) via spray-drying technology for efficient lung delivery. Compared with the positive control drug PFD, full-course treatment with CTS showed comparable efficacy, primarily via inhibition of the NLRP3/TGF-β1 pathway in the inflammatory process and modulation of the matrix metalloproteinase-9 (MMP-9)/tissue inhibitor of metalloproteinases-1 (TIMP-1) balance in fibrotic development. 110 Other researchers developed and evaluated CTS-loaded modified liposome-chitosan (CS) microspheres SM (CT-lipo) and liposome-exosome hybrid biomimetic vesicle-CS microspheres SM (LE). SM (CT-lipo) and SM (LE) achieve specific targeting to lung myofibroblasts through the CREKA peptide, which specifically binds to fibronectin, and the homing effect of exosomes to parent cells, respectively, promoting efficient delivery of antifibrotic drugs to pulmonary lesions. Furthermore, compared with daily administration of conventional microspheres SM (NC) and the positive control drug pirfenidone, inhalation of SM (CT-lipo) and SM (LE) every two days still demonstrated similar efficacy, exhibiting excellent sustained drug release capability. 111
Summary and future perspectives
PF is a severe respiratory disease with an increasing incidence which significantly impacts patients’ quality of life and poses a life-threatening risk. Its pathogenesis is complex, involving M1 macrophage-mediated pro-inflammation, M2 macrophage–driven profibrotic processes, and intricate epithelial–mesenchymal interactions. Emerging therapeutic targets are continuously being identified. For instance, TREM2 knockout has been shown to alleviate PF, whereas modulation of CD36 may reduce lung inflammation and fibrosis by regulating macrophage function. Targets such as Fra-2, MBD2, SLC15A3, and PRDX1 also offer new therapeutic directions. Furthermore, agents such as 4-OI, SR, and HDAC3 inhibitors have demonstrated potential in PF treatment.
Novel therapeutic approaches are under active exploration. MSC therapy holds considerable promise. MSCs derived from various sources (e.g. the bone marrow, gingiva, umbilical cord, adipose tissue, and urine) exert antifibrotic effects through diverse mechanisms, with ongoing innovations in administration routes. Nano-inhalation therapy enables efficient drug delivery to pulmonary lesions, utilizing systems such as inhalable lipid nanoparticles and liposomal microparticles, thereby offering new avenues for PF treatment.
For newly discovered targets and pathways, efforts should be accelerated to translate basic research findings into clinical applications. Large-scale clinical trials are essential to rigorously validate their efficacy and safety. Concurrently, existing treatment regimens need optimization, including improving drug delivery systems to enhance pulmonary drug accumulation and efficacy as well as increasing the stability and therapeutic effectiveness of stem cell therapies and minimizing associated risks. Moreover, the discovery of biomarkers for early disease diagnosis deserves high priority. Integrating multimodal approaches, such as imaging and molecular biology techniques, can facilitate the early diagnosis and intervention of PF. Early intervention is crucial for controlling disease progression, improving patient prognosis, reducing mortality and disability rates, enhancing quality of life, and advancing the field of PF diagnosis and treatment.
Footnotes
Author Contributions
All authors contributed to the conception and literature collection of this review. Li Li drafted the manuscript; all authors revised the article critically and approved the final submitted version.
Data Availability Statement
All data cited in this review derive from publicly available published studies, which can be retrieved from corresponding original literatures.
Declaration of conflicting interest
The authors declare that there are no conflicts of interest.
Ethical Statement
This article is a narrative review based on published literatures; no original human or animal experiments were conducted, so ethical approval and informed consent are not applicable.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.