
Abstract
Context
Deficiency of 17β-hydroxysteroid dehydrogenase type 3 (17β-HSD3) is a rare autosomal recessive disorder of sex development affecting individuals with a 46, XY karyotype. It is caused by pathogenic variants in the HSD17B3 gene that impair the conversion of Δ4-androstenedione to testosterone.
Objective
To evaluate the clinical presentation and management of a female patient who exhibited masculinization during puberty and assess her long-term life satisfaction during follow-up.
Methods
A female patient who exhibited masculinization during puberty underwent comprehensive endocrinological and genetic assessments. Subsequently, she underwent sex reassignment surgery and follow-up investigation.
Results
Hormonal findings showed a baseline testosterone/androstenedione ratio of 1.77, which decreased to 1.32 after human chorionic gonadotropin stimulation; these findings are atypical for the diagnosis. Subsequently, compound heterozygous mutation p.I60 T(c.179T > C) and exon 1 deletion were identified. After extensive counseling, the patient’s parents decided to reverse the patient’s sex to male. Orchidopexy and two-stage urethroplasty were performed; histological evaluation demonstrated no malignancies of the testicular tissue. Follow-up after 1 year indicated that the patient was satisfied with their sex assignment.
Conclusions
The management of pediatric patients with rare 17β-HSD3 deficiency remains challenging. For patients diagnosed early, premature removal of the gonads is not recommended as the risk of malignancy is very low. Any decision must be carefully discussed within a multidisciplinary disorders of sex development team, together with the patient and their parents. Long-term follow-up is required to assess the impact of the chosen option on future quality of life.
Introduction
Disorders of sex development (DSD) represent a complex group of congenital conditions wherein the sex chromosome, gonads, or anatomical sex are inconsistent with the typical embryonic developmental pathway. 1 In 2006, the Chicago Consensus Meeting classified these disorders into three major categories: (a) sex chromosome DSD; (b) 46, XX DSD; and (c) 46, XY DSD. 2 Individuals with 46, XY DSD may present with a wide spectrum of clinical manifestations, ranging from typical female external genitalia to undervirilized male genitelia. 3
Deficiency of 17 β-hydroxysteroid dehydrogenase type 3 (17β-HSD3) is a rare autosomal recessive 46, XY DSD 4 in Western countries; however, it is relatively common among certain Arab populations. 5 17-β-HSD3, encoded by the HSD17B3 gene, is an enzyme primarily expressed in the testes where it converts Δ4-androstenedione (A) to the active androgen testosterone (T). 6 Patients may present with female genitalia or ambiguous features and are often raised as females. Significant virilization during puberty depends on higher levels of Δ4-A, which may be partially converted to T through the residual activity of 17β-HSD3 or via other isoenzymes. 7 Diagnosis involves hormone level assessment, with a T/A ratio <0.8 after the human chorionic gonadotropin (hCG) stimulation test; 8 however, molecular genetic testing is required for confirmation. 9
Herein, we report a case of a pediatric Chinese patient with 17-βHSD3 deficiency who had been raised as a girl since childhood. However, she presented for medical attention due to masculinization during puberty. The patient underwent sex reassignment, and follow-up was completed.
Materials and methods
Clinical studies
The patient was assigned female sex at birth based on external genitalia and was raised as a girl until the age of 10 years. Both parents were healthy individuals of Chinese ethnicity with no consanguinity or abnormal family history. She presented at our center with clitoromegaly. The phenotype of the genitals was scored using the external masculinization score (EMS). Physical examination revealed inguinal masses and clitoral enlargement. Ultrasound imaging was subsequently performed, which revealed that the bilateral testes were located in the inguinal region and that no uterus or ovarian structures were present. Blood karyotyping confirmed a 46, XY karyotype. She was hospitalized to confirm the diagnosis. This study was approved by the Medical Ethics Committee of the Children's Hospital of Soochow University (2025CS262). Written informed consent was obtained from both parents.
Hormonal testing
Dehydroepiandrosterone (DHEA), dihydrotestosterone (DHT), A, total T, anti-Müllerian hormone (AMH), luteinizing hormone (LH), and follicle-stimulating hormone (FSH) levels were measured using commercial kits, employing the chemiluminescent method (Roche Diagnostics GmbH, ET800133). For the stimulation test, subcutaneous hCG (1500 IU/day) was administered for 3 consecutive days (GuanLong, China).
Genetic analysis
Whole blood samples were collected for genetic analysis. Whole-exome sequencing (WES) was performed using the genomic DNA of the patient. Mutations in the HSD17B3 gene were verified using Sanger sequencing for the patient and their parents. The pathogenicity of mutations was evaluated using four algorithms: (a) Sorting Intolerant from Tolerant (SIFT, http://sift.jcvi.org/); (b) Polymorphism Phenotyping version 2 (PolyPhen2; http://genetics.bwh.harvard.edu/pph2/); (c) MutationTaster (http://www.mutationtaster.org/); (d) and Mutpred2 (https://mutpred.mutdb.org/)). We categorized the variants according to international standards established by the American College of Medical Genetics and Genomics (ACMG).
In silico protein structure analysis
In silico protein structures were predicted for the HSD17B3 variants. Initially, AlphaFold2 was used to model the three-dimensional structures of wildtype proteins encoded by HSD17B3. Based on the predicted wildtype protein structures, SWISS-MODEL2 was subsequently utilized to generate structural models of the variant proteins, with the resulting architectures visualized using PyMOL (http:// www.pymol.org).
Quantitative real-time polymerase chain reaction (qPCR) analysis
qPCR assays were performed to validate the genomic deletion of the HSD17B3 gene. Genomic DNA was extracted from whole blood using the TGuide S32 Blood Genomic DNA Extraction Kit (Tiangen Biotech). No reverse transcription step was performed. Gene expression levels were quantified using the SYBR Green assay. The data were normalized to β-actin expression. To determine the exact breakpoint of the deletion, conventional PCR was performed using primers flanking the suspected deletion region (HSD17B3_E2-seq), followed by Sanger sequencing analysis. The primer sequences are listed in Table S1.
Hematoxylin and eosin (HE) and immunohistochemical (IHC) staining
Testis tissue samples were collected from the patient for histological analysis. After fixation, these testis samples were dehydrated, paraffin embedded, and cut into 5-mm thick sections. The sample sections were then deparaffinized and rehydrated for HE and IHC staining. For IHC staining, tissue sections are boiled in 10 mmol/L citrate buffer for 30 min to extract antigens. Sheep serum was used to block non-specific binding sites. The tissue sections were incubated overnight with the primary antibody at 4°C. Subsequently, 3% hydrogen peroxide (H2O2) was used to block endogenous peroxidase activity. The sections were then washed and incubated with the secondary antibody at 37°C for 30 min and finally stained with hematoxylin. The primary antibodies utilized for IHC were as follows: (a) alpha-fetoprotein (AFP, ZM-0009, ready-to-use, ZSGB-BIO; (b) hCG (GA023102, ready-to-use, Shanghai Gene); (c) cluster of differentiation 117 (CD117; ZA-0523, ready-to-use, ZSGB-BIO); and (d) placental alkaline phosphatase (PLAP, GM719102, ready-to-use, Shanghai Gene). The results were analyzed using a digital pathological slide scanner (Magscanner, KF-PRO-005).
Results
Clinical and hormonal characteristics of the patient
The patient was born with fully developed female genitalia and was raised as a girl. Upon examination, the patient was found to have a 2.5-cm penis with both urethral and vaginal openings at the base. Palpation uncovered bilateral gonads in the inguinal region. The EMS score was recorded as 2 (Figure 1(a)).
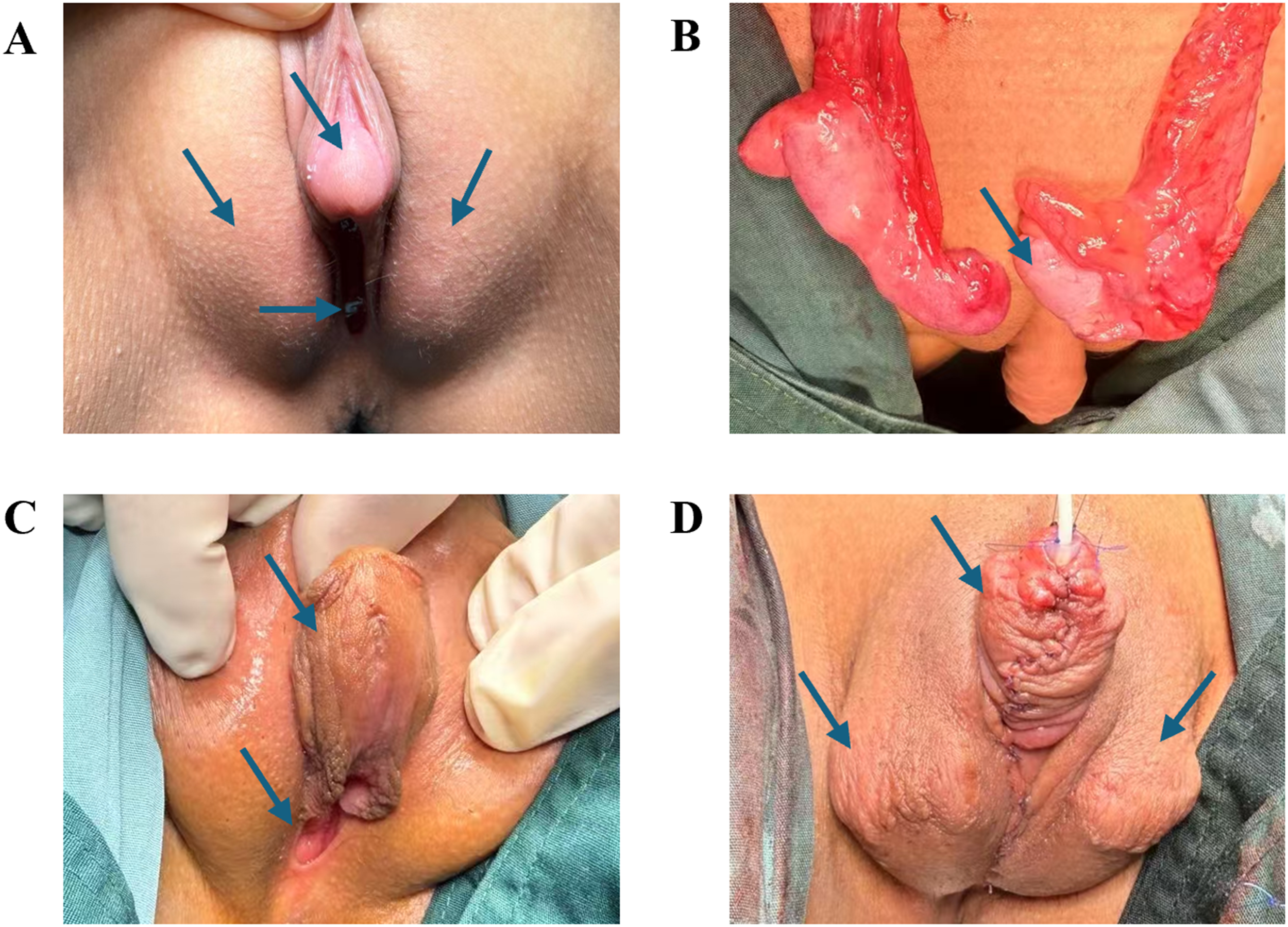
Genital morphology and surgical treatment. (a) Inguinal masses and clitoris enlargement, with both urethral and vaginal openings located at the base; (b) bilateral testicles with a generally normal shape observed during orchidopexy; (c) the penis is straightened, and the urethral opening was located at the base of the penis after the first-stage urethroplasty; (d) complete formation of the penis and bilateral testes located in the scrotum.
Hormone levels of the patient were examined. Results exhibited normal T synthesis after the hCG stimulation test but a decreased T/A ratio, indicating relative defects in T synthesis. Detailed hormone findings for the patient are listed in Table 1.
Hormonal profile of the patient before and after hCG stimulation.

hCG: human chorionic gonadotropin; T: testosterone; A: androstenedione.
After 6 months, the patient underwent corrective surgery for the undescended testicle and hypospadias to address the anatomical discrepancies identified during the clinical evaluation (Figure 1(b) to (d)). This surgical intervention aimed to enhance both function and appearance.
Detection of HSD17B3 mutation
Sequence analysis of the HSD17B3 gene unveiled heterozygous mutations in the patient, and the parents were confirmed to be carriers. WES suggested heterozygous missense variants: HSD17B3 p.I60 T (c.179T > C) and exon 1 deletion. Quantitative PCR (qPCR) indicated that exon 1 expression was approximately 50% of the normal level (Figure 2).
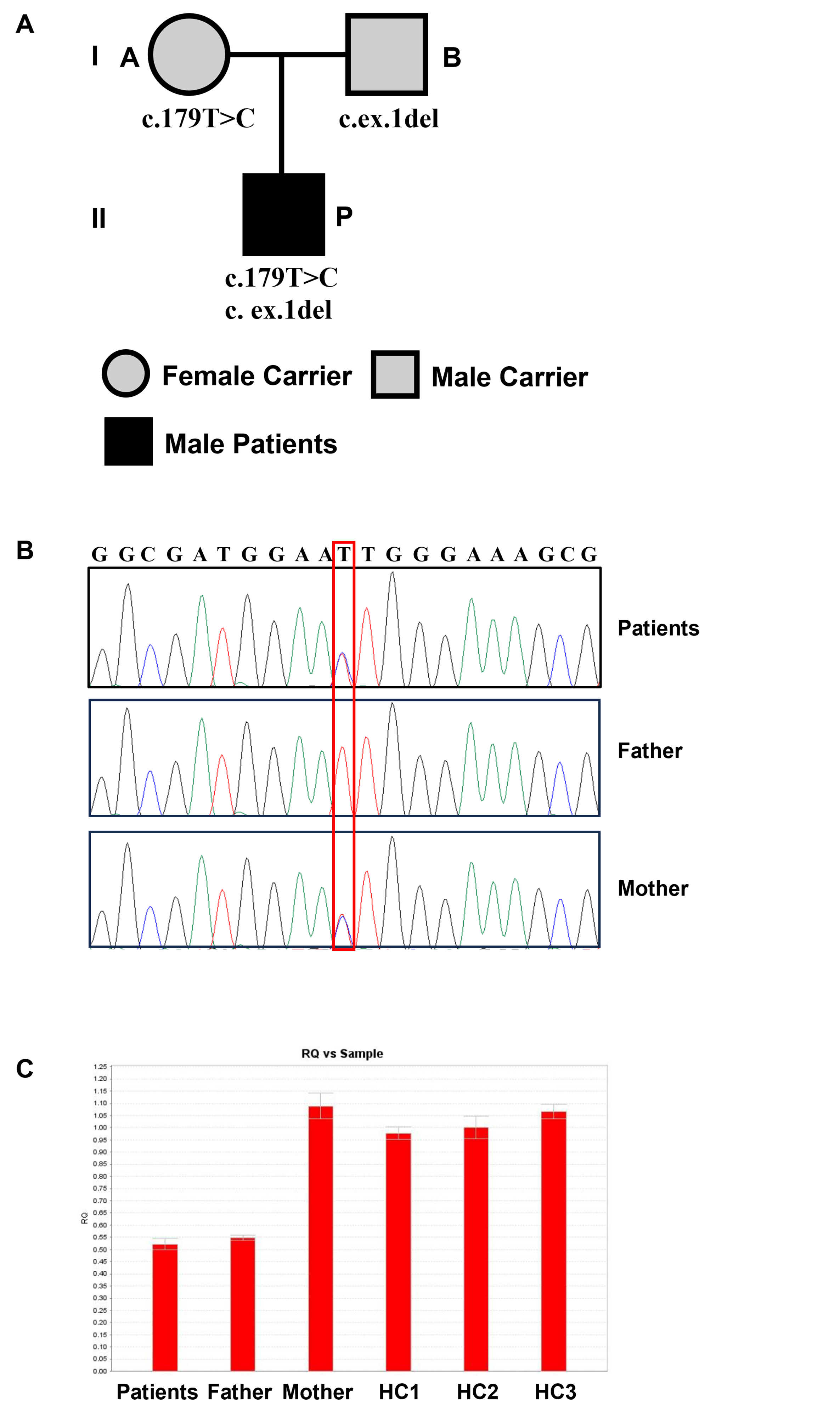
Pedigree and missense mutations of 17β-HSD3 deficiency patients. (a) Family pedigree. (b) HSD17B3 DNA sequencing of the patients; the mutation site in HSD17B3 is marked with a box. (c) Exon 1 expression.
To assess pathogenicity, we analyzed the missense variants in the HSD17B3 genes using SIFT, PolyPhen-2, MutationTaster, and MutPred2. The p.I60 T mutations were predicted to be damaging (Table 2). According to the ACMG criteria, the HSD17B3 variant was classified as likely pathogenic (PM3_Strong + PM2_Supporting + PP3).
Predictions of HSD17B3 mutations.
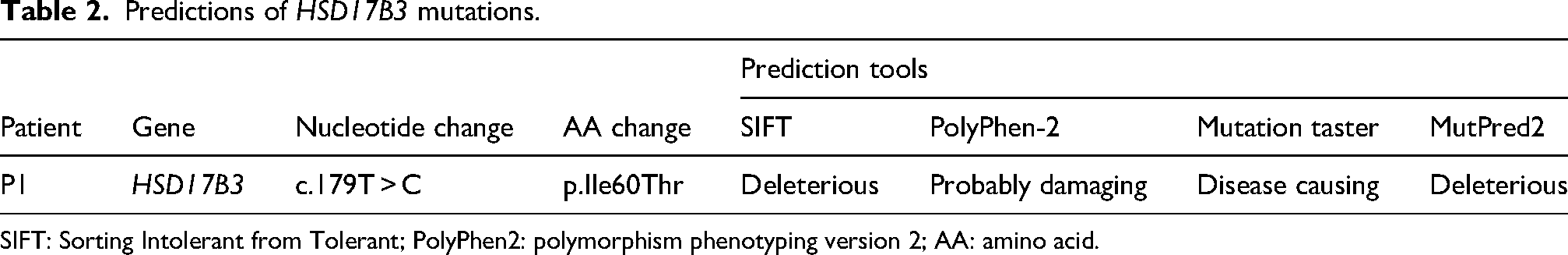
SIFT: Sorting Intolerant from Tolerant; PolyPhen2: polymorphism phenotyping version 2; AA: amino acid.
Protein structure change
To elucidate the potential functional consequences of the identified variants, we performed in silico conformational analyses. After the p.Ile60Thr mutation in the HSD17B3 gene, the structure of the 60th amino acid changed; however, the hydrogen bonds formed with adjacent amino acids remain unchanged, suggesting that this mutation did not affect the three-dimensional structure of the protein. The predicted three-dimensional protein structures are presented in Figure 3.
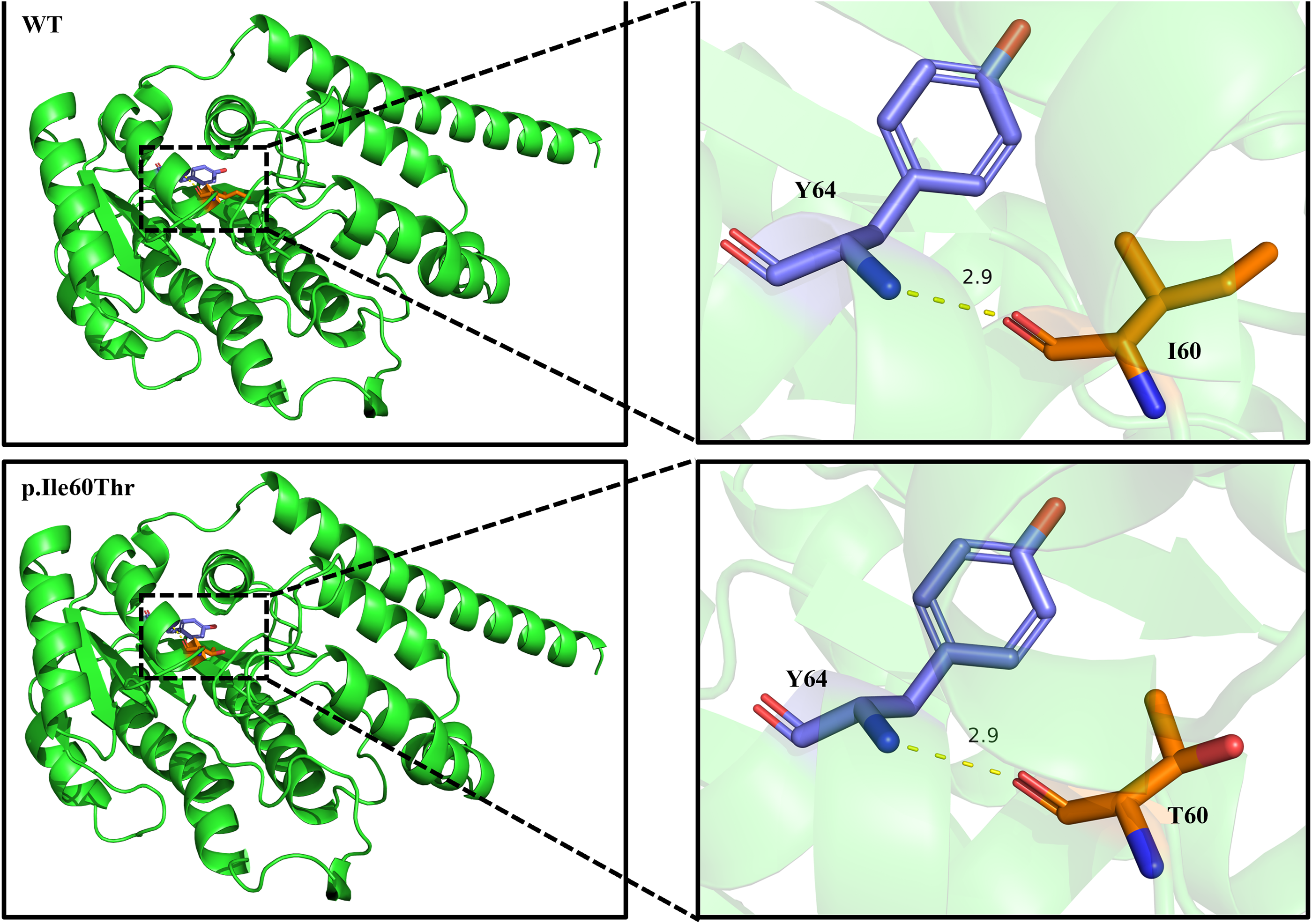
In silico three-dimensional protein structural prediction of HSD17B3.
Histological and IHC staining of the testis tissue samples
The seminiferous tubules were structurally intact with regular lumen, and the germ cells at all levels were arranged neatly and in an orderly manner. It is important to emphasize that no cellular tumors were identified in the patient, indicating an absence of malignancy risk. This conclusion was further corroborated by negative staining results using specific markers for these cells, as illustrated in Figure 4.
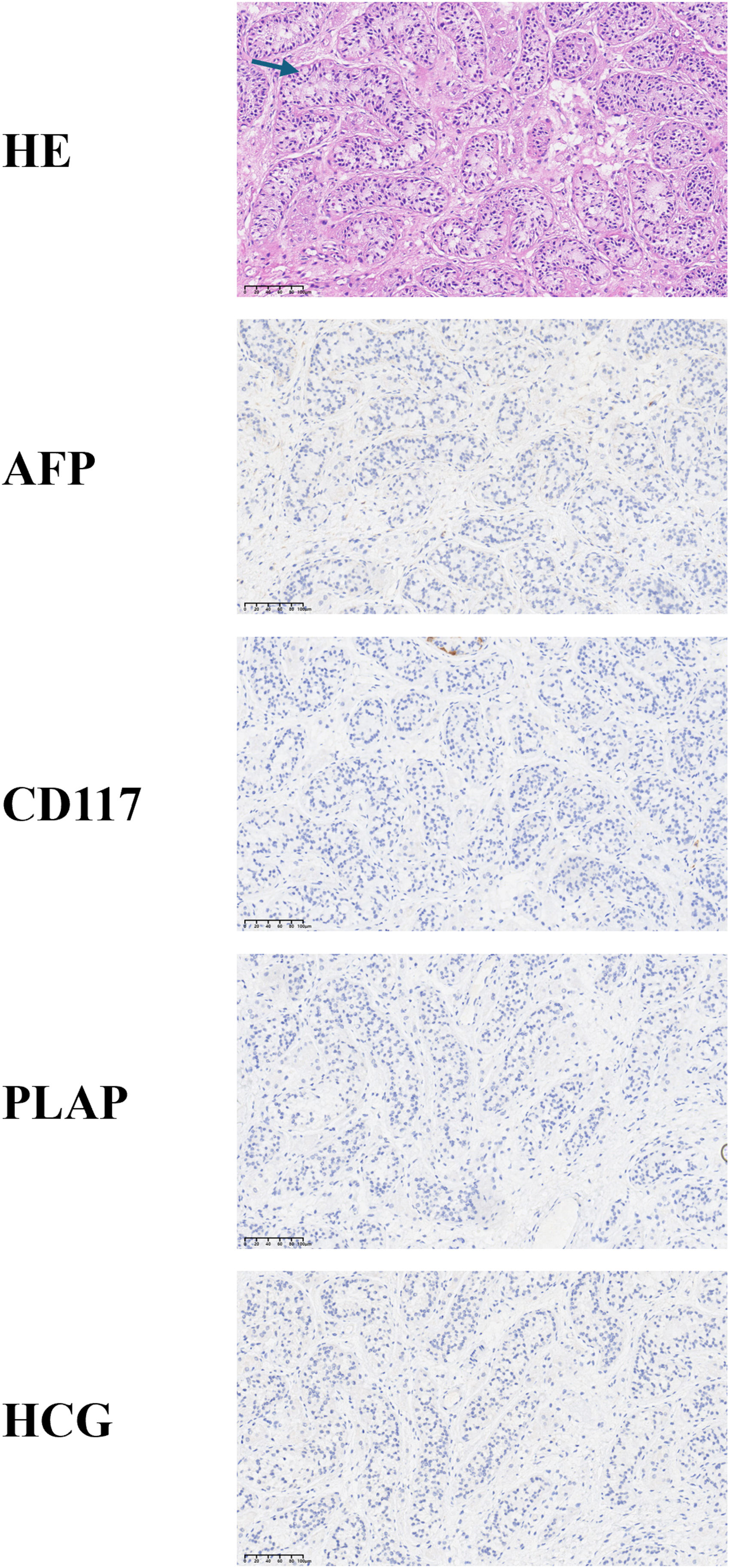
HE staining and representative images of immunohistochemistry were used to assess testicular development and tumor risk. Arrows show structurally intact and neatly arranged seminiferous tubules. AFP, CD117, hCG, and PLAP staining confirming the absence of malignancy in the testes.
Discussion
17β-HSD3 is one of the enzymes necessary for androgen synthesis, as it converts Δ4-A to the active androgen T in the testes. Defects in T synthesis can lead to insufficient differentiation of male external genitalia and defective virilization. However, a deficiency of the 17β-HSD3 enzyme is a rare cause of 46, XY DSD. The phenotypes of the patients varied from typical female external genitalia to predominantly male genitalia with micropenis or hypospadias, depending on the residual activity of enzymes. 10 In certain nations, most pediatric patients with 17β-HSD3 deficiency and predominantly female external genitalia are raised as girls and undergo gonadal surgery. 11 Similar to our patient, other patients mainly seek medical attention during puberty when symptoms of masculinization such as deep voice, hirsutism, and clitoral enlargement appear. Upon examination, palpation may uncover bilateral gonads situated in the inguinal region. These patients are initially considered to have other, more common DSDs, such as partial androgen insensitivity syndrome and 5-α-reductase type 2 deficiency because of analogous clinical manifestations. 12 Therefore, biochemical evaluations are crucial as they can help differentiate these diseases. In 17β-HSD3 deficiency, the conversion of A to T is impaired, leading to a decreased T/A ratio, even after hCG stimulation. Research has shown that a T/A ratio <0.8 after hCG stimulation is a laboratory diagnostic criterion for patients with 17β-HSD3 deficiency.8,11 However, in our patient, the baseline T/A ratio was 1.77, which decreased to 1.32 after hCG stimulation, but remained above 0.8. Other studies have reported similar results, which may be attributable to the activity of other 17β-HSD isoenzymes (17β-HSD5 or 17β-HSD2) and residual enzyme activity.13–15 Thus, hormone levels may mislead our diagnosis, and genetic testing is particularly crucial for confirmation.
17β-HSD3 deficiency is caused by compound heterozygous or homozygous mutations of the HSD17B3 gene, which consists of 11 exons and is located on chromosome 9q22. To date, relevant literature has recorded 70 different mutations of HSD17B3. 4 These mutations are mostly identified in the Arab population residing in the Gaza strip. The p.Arg80Gln mutation located on exon 3 stands out as the most prevalent mutation among this population. 16 The compound heterozygous mutation p.I60 T(c.179T > C) and exon 1 deletion that were identified in our patient have been documented as pathogenic in the existing literature;17–19 however, this is the first time that both were simultaneously detected in a single patient. Existing literature has indicated that there is no correlation between genotype and phenotype in 17β-HSD3 deficiency. 20 Furthermore, p.I60 T(c.179T > C) is a unique mutation site that has only been reported in the Chinese population. We speculate that the p.I60 T mutation represents a founder mutation in the Chinese population. The founder effect refers to a genetic variation that arises when a new population is established from a limited number of individuals drawn from a larger gene pool. 21 Further research is necessary to confirm this finding.
For patients with 46, XY DSD, determining sex assignment is a difficult challenge. Pediatric patients with 17βHSD-3 deficiency are usually raised as girls. However, 39%–64% of individuals reported a subsequent change in sexual role during puberty. Most of these patients were from the Gaza region where there was a notable preference for the male sex. 22 This situation may be related to local social and cultural influences. In some countries, most children with 17-βHSD3 deficiency and predominantly female external genitalia are raised as girls and undergo gonadal surgery.11,23 A recent systematic review has stated that the rate of sexual identity transition from female to male has decreased to 7%–10%. 24 The authors have concluded that there is currently insufficient data to support male sex reassignment surgery for girls with early symptoms. In our case, the pediatric patient was born with fully developed female genitalia and raised as a girl. She underwent medical consultations after clitoromegaly and palpable gonads in the groin were observed after puberty. After undergoing multiple sex-adjustment evaluations and 6 months of discussions with the family members, the sex council recommended male sex assignment. Endoscopic examination revealed no Müllerian structures (uterus or fallopian tubes) in the abdominal cavity and a blind vaginal pouch; ultrasonography revealed testes in the inguinal region. We performed orchidopexy and two-stage urethroplasty sequentially. At the same time, we retained the blind vaginal pouch to preserve the possibility of future correction if the patient did not agree with the assigned sex for nurturing. At the 6-month follow-up, the patient expressed satisfaction with his assigned sex.
There is evidence that both individuals raised as men due to 17-βHSD3 deficiency and those raised as women exhibit satisfactory sexual function. 25 In addition, fertility issues must be taken into consideration. Although spontaneous or assisted paternity has been documented in patients with 5α-reductase 2 deficiency, 26 a fertile 17-βHSD3-deficiency patient has not been previously identified. Some studies have indicated decreasing tendency of germ cells, suggesting that early surgical intervention on the testes may preserve some spermatogenic capacity. 10 Recent pioneering research has demonstrated that the disruption of the canonical pathway in transgenic mice lacking HSD17B3 exerts minimal influence on male development and fertility. 27 The increase in T mainly comes from the peripheral conversion of androgens and other androgen biosynthesis pathway commonly known as the backdoor pathway. 14 The discovery of compensatory mechanisms for androgen synthesis is essential and may yield significant insights for preserving fertility in 17-βHSD3 deficiency patients.
In fact, the overall risk of gonadal neoplasia for patients with 17-βHSD3 deficiency is currently unclear. It is crucial to consider the risk of malignancy, especially in individuals who choose to preserve their testicular tissue while transitioning into a male sex role. Germ cell irregularities may manifest as early as the second decade of life. 28 The preserved gonads should be carefully lowered into the scrotum and routinely examined for any signs of malignancy. 29 Testicular removal as a preventive measure should be carefully considered due to the minimal risk of testicular malignancy. 30 Patients diagnosed early with 17-βHSD3 deficiency should delay gonadectomy until their sexual identity can be evaluated. 31
Conclusions
Early identification of the DSD type is crucial. Although a T/A ratio <0.8 after hCG stimulation is the laboratory diagnostic criterion for 17β-HSD3 deficiency, hormone levels may lead to misdiagnosis due to the conversion of isoenzymes; genetic testing is the gold standard for diagnosis. The decision regarding sex assignment during upbringing must be guided by the most accurate predictions concerning future sexual function, virilization, and contentment with sexual identity. In this context, it has been proposed that the early removal of gonads may lead to the preservation of a female sexual identity during adolescence in children who exhibit significantly low levels of virilization. A more cautious methodology concerning gonads may be adopted, allowing the affected individuals to actively participate in the decision-making process, thereby facilitating a deeper exploration of their sexual identity.
Footnotes
Ethics statement
Ethical approval was granted by the Medical Ethics Committee of the Children’s Hospital of Soochow University (2025CS367).
Author contributions
Li Tao contributed to conceptualization, data curation, surgical procedures, result analysis, and writing the original draft. All authors read and approved the final manuscript. Enfu Huang contributed to patient data collection and curation and assisted in clinical surgeries. Chao Wang contributed to patient data collection and curation and assisted in clinical surgeries. Tianyi Wang contributed to patient data collection and curation and assisted in clinical surgeries. Xueqian Wang contributed to interpretation of genetic data. Rongrong Xie contributed to data validation and interpretation of results. Haiying Wu contributed to methodology development and data validation. Xiangming Yan was responsible for the acquisition of funding. Yun Zhou contributed to supervision and administered the entire research project. Ting Zhang as corresponding authors, conceived and designed the study. She also conducted thorough review, editing, and final approval of the manuscript for submission.
Funding
This work was supported by the General Project of Suzhou Municipal Health Commission (Grant No. MSXM2025016) and the Suzhou Children’s Structural Anomaly Key Laboratory Construction Project (Grant No. SZS2022018).
Declaration of conflicting interests
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All raw data and code are available upon request.