
Abstract
Hereditary spastic paraplegia is characterized by progressive motor features, including spastic gait and lower limb weakness, and encompasses numerous genetic subtypes. We report a family of six siblings born to asymptomatic consanguineous parents, in which three siblings exhibited overlapping spastic paraplegia phenotypes with developmental delay. The index patient demonstrated significant lower limb weakness and spasticity despite multiple orthopedic interventions. Next-generation sequencing identified a homozygous SELENOI c.797C > T (p.Pro266Leu) variant of uncertain significance, interpreted cautiously in the context of his SPG81-like phenotype. His older sister reportedly harbored a homozygous LAMA1 c.4579C > T (p.Gln1527Ter) variant consistent with Poretti–Boltshauser syndrome, explaining her cerebellar ataxia and nonprogressive course. His younger brother carried the same homozygous SELENOI variant as the index patient together with a de novo SATB2 c.860C > T (p.Pro287Leu) variant of uncertain significance, which was interpreted as a possible contributor to his mixed neurodevelopmental presentation. This report highlights that siblings presenting with similar early-onset spastic gait phenotypes may have distinct underlying genetic findings, supporting the value of comprehensive genetic evaluation even within a single family.
Introduction
Hereditary spastic paraplegia (HSP) is a clinically and genetically heterogeneous group of disorders characterized by progressive lower limb spasticity and weakness, with more than 80 genetic subtypes identified to date. 1 We encountered a consanguineous family in which three of six siblings presented with spastic gait, initially suggestive of childhood-onset HSP. However, genetic analysis using next-generation sequencing revealed distinct genetic findings among the affected siblings. We describe and compare the clinical features, neuroimaging findings, gene analysis, and rehabilitative courses of these three siblings.
Case presentation
All six siblings were born to healthy consanguineous parents. The index patient, the fourth brother in his early 20s, the second sister in her mid-20s, and the fifth brother in his late teens presented with spastic paraplegia and developmental delay. The eldest sister in her late-20s, the third brother in his mid-20s, and youngest brother in early childhood, were unaffected. The family pedigree is presented in Figure 1.
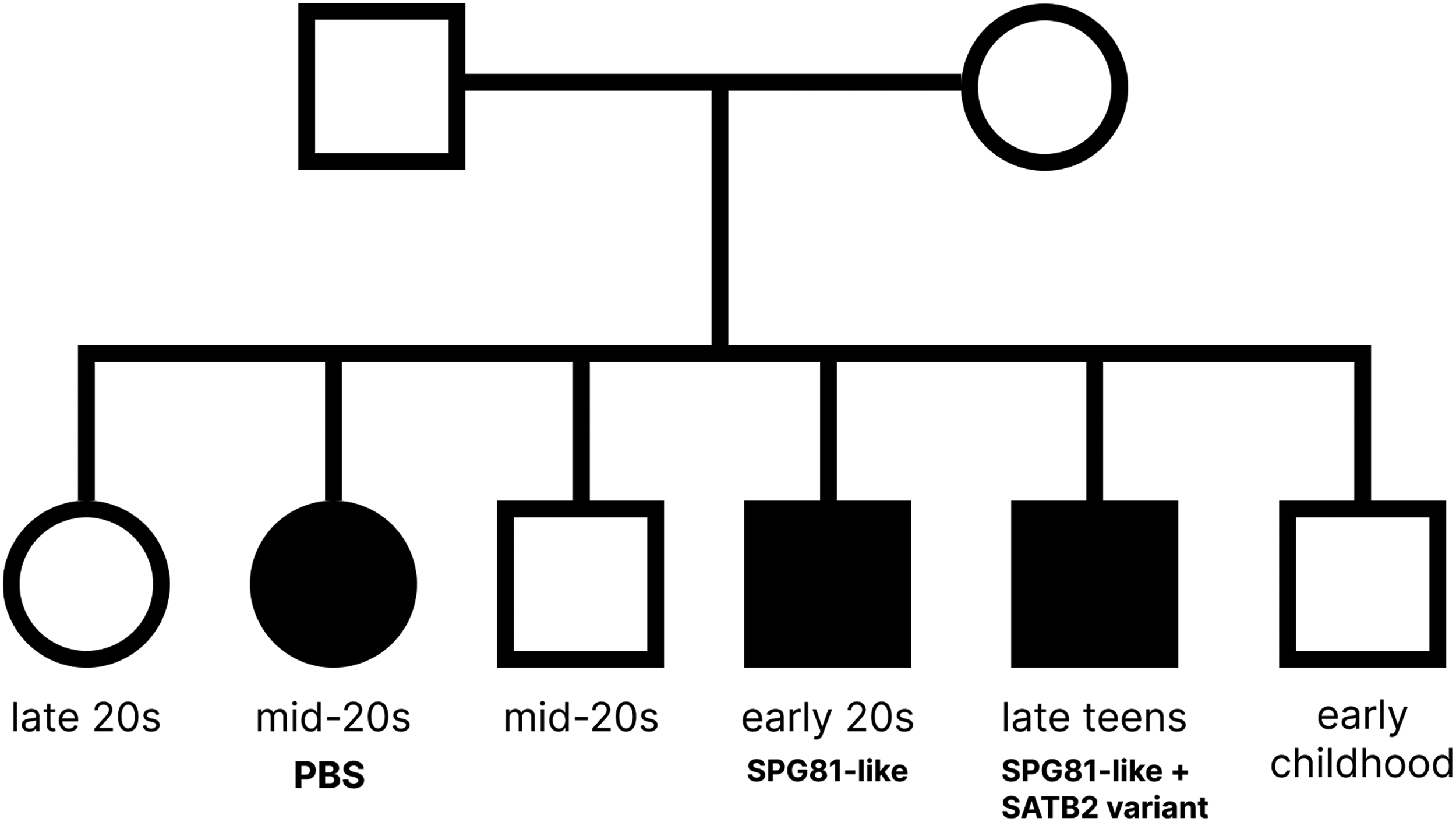
A family pedigree showing three affected siblings. Pedigree showing three affected siblings from consanguineous parents, including one with Poretti–Boltshauser syndrome (PBS), one with an SPG81-like phenotype associated with a SELENOI variant, and one with an SPG81-like phenotype and an additional SATB2 variant.
The index patient was born at term without perinatal complications. Motor developmental delay became evident after 1 year of age when he developed progressive lower limb weakness and delayed developmental milestones. Early language development was markedly delayed as he spoke his first words at approximately 4 years of age and his first simple sentences at approximately 5 years of age. At 4 years of age, he underwent orthopedic surgery such as tenotomy. Despite these interventions, he did not achieve independent ambulation and was only able to take a few assisted steps for the first time at 10 years of age. He was not receiving any medication for spasticity or seizures.
The index patient was first admitted to Severance Hospital, Seoul, Republic of Korea, in July 2023 for inpatient rehabilitation for gait disturbance. At the initial evaluation at our hospital, he demonstrated poor grade strength at both lower limbs on manual muscle testing (MMT) and bilateral lower limb spasticity graded 2 on the modified Ashworth scale (MAS). 2 Deep tendon reflexes were hyperactive at the knees and ankles, with sustained ankle clonus. He exhibited impaired motor coordination with ataxia, dysmetria, hyperreflexia, and a spastic gait pattern. Light touch, pinprick, and proprioception were intact in all four limbs. Bladder and bowel control were normal. The Berg balance scale (BBS) 3 score was 15 with impaired balance function. The functional independence measure (FIM) 4 was 101 and the modified Barthel index (MBI) 5 was 84. He could walk only a few steps indoors with a walker. Gait analysis revealed a severe anterior pelvic tilt. His lordosis angle was 98.63° on standing whole-spine X-ray. Electrodiagnostic studies revealed no evidence of peripheral neuropathy, radiculopathy, or motor neuron disease. Language assessment identified mild dysarthria; however, articulation and intelligibility remained functional. Cognitive evaluation demonstrated global intellectual impairment, with a full-scale intelligence quotient (IQ) of 58 (below the 1st percentile). He exhibited bilateral myopia without other visual abnormalities and he had no history of seizure.
The second sister, the first affected sibling, was born at term by normal spontaneous delivery with no perinatal complications. Spasticity and weakness in both lower limbs became apparent at approximately 1 year of age, when she showed delayed motor milestones with difficulty in walking. She underwent surgical reduction for bilateral hip dislocation at 8 years of age followed by tendon lengthening at 9 years of age. Following these procedures, she achieved independent indoor ambulation but continued to require a wheelchair for longer outdoor distances. She had an approximately 20-year history of left-sided hearing loss. Her ophthalmologic history included left esotropia surgery at 7 years of age and laser barrier procedures in both eyes for retinal pathology. She also had myopia and astigmatism. There was no history of seizures and bladder or bowel dysfunction. She was not receiving any medication for spasticity or seizures.
The older sister was first evaluated at Severance Hospital, Seoul, Republic of Korea, in 2023 and later underwent next-generation sequencing in February 2024. At her initial evaluation at our hospital, she showed good-grade strength in hip and knee flexion and fair grade in ankle dorsiflexion and plantar flexion on MMT. Muscle tone was increased and spasticity was graded 1 in both lower limbs on MAS. Finger-to-nose testing revealed intentional tremor. Eye movement examination demonstrated catch-up saccades consistent with cerebellar and oculomotor involvement. BBS was 35 and MBI was 96. Gait analysis demonstrated a spastic gait pattern with genu valgus and marked internal rotation of both feet. She was able to ambulate independently indoors; however, bilateral calf pain and increasing spasticity with longer ambulation necessitated wheelchair use for longer distances. Electrodiagnostic studies revealed no evidence of peripheral neuropathy, radiculopathy, or motor neuron disease. Cognitive evaluation demonstrated global intellectual impairment with a full-scale IQ of 55.
The fifth brother, the youngest affected sibling, was born at 40 weeks by normal spontaneous delivery with no perinatal complications. Global developmental delay and spastic paraplegia became evident at approximately 1 year of age. He never achieved independent ambulation and could only take a few assisted steps while holding onto support. Expressive language was markedly delayed, with first words at approximately 4 years of age and simple sentences at approximately 5 years. At 9 years of age, he underwent left hip derotation varus osteotomy with bilateral gastrocnemius–soleus recession. At 15 years of age, he experienced a first generalized tonic seizure event. At presentation, he was receiving baclofen for lower limb spasticity.
The younger brother was first evaluated at Severance Hospital, Seoul, Republic of Korea, in June 2023 and was later admitted for orthopedic surgery and inpatient rehabilitation. At his first admission to our hospital, he exhibited spastic and ataxic motor deficits and left-side predominant involvement. He demonstrated good grade in the hip flexion, fair grade in knee flexion and extension, and poor grade in ankle dorsiflexion and plantarflexion on MMT. He exhibited lower limb spasticity on MAS, indicating G2/G2 in hip adductor, G1/G2 in hamstring, and G1/G1 in heel cord. Deep tendon reflexes at the knees and ankles were hyperactive, with sustained ankle clonus. Functionally he could stand while holding a support bar and walk approximately 3–5 meters with an anterior walker with maximal assistance. His speech showed mild dysarthria. He exhibited difficulty swallowing tablets, and video fluoroscopic swallowing study revealed delayed swallowing reflex without aspiration. Electrodiagnostic studies showed low amplitude of sensory nerve action potential in left sural nerve. Cognitive assessment indicated intellectual disability, with a full-scale IQ of 56. He exhibited no visual or hearing impairment and no bladder or bowel dysfunction. During follow-up at our hospital, baclofen was adjusted and later discontinued. After his first seizure event, antiseizure treatment was initiated with levetiracetam and subsequently transitioned to lamotrigine with gradual dose escalation.
Neuroimaging findings
Brain magnetic resonance imaging (MRI) of the index patient revealed thinning of the posterior body and splenium of the corpus callosum, accompanied by delayed white matter myelination. Follow-up MRI findings showed persistent peritrigonal white matter signal change and mild syringomyelia (Figure 2(a)). Brain MRI of the second sister demonstrated markedly abnormal cerebellar foliation with multiple retrocerebellar cysts and a molar tooth–like configuration of the superior cerebellar peduncles. Mild ventriculomegaly was present with accompanying periventricular T2 hyperintensities (Figure 2(b)). Brain MRI of the fifth brother demonstrated delayed white matter myelination with subtle, symmetric peritrigonal hyperintensities on T2-weighted images, consistent with maturational delay rather than structural injury. Follow-up MRI findings revealed progression to diffuse white matter volume loss with thinning of the corpus callosum (Figure 2(c)).

Brain MRI findings of the index patient, the second sister, and the fifth brother. (a) Brain MRI of the index patient demonstrating thinning of the corpus callosum and delayed peritrigonal white matter myelination, with mild progression to diffuse white matter loss. (b) Brain MRI of the second sister showing cerebellar dysplasia with abnormal foliation, multiple retrocerebellar cysts, molar tooth sign, and mild ventriculomegaly. (c) Brain MRI of the fifth brother revealing delayed white matter myelination and diffuse white matter atrophy with corpus callosum thinning.
Genetic analysis by next-generation sequencing
Genetic evaluation was performed using different next-generation sequencing approaches across the affected siblings, including targeted HSP panel sequencing, trio-based whole-exome sequencing, and family-based exome sequencing with parental and sibling samples. Variant detection and interpretation were based on the original clinical diagnostic reports, and variant classification followed the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP)-based classifications provided in those reports. Both parents were included in the family-based exome analyses, and segregation and inheritance were assessed on the basis of family-based exome data and follow-up familial Sanger sequencing reports. However, not all reported variants were independently confirmed by Sanger sequencing, and follow-up familial Sanger sequencing was available only for selected segregation analyses.
The index patient carried homozygous SELENOI c.797C > T (p.Pro266Leu), a novel variant of uncertain significance that was interpreted cautiously in the context of his SPG81-like phenotype, characterized by early-onset lower limb spasticity and brain atrophy. The second sister had a homozygous LAMA1 c.4579C > T (p.Gln1527Ter), consistent with Poretti–Boltshauser syndrome. The fifth brother harbored the same homozygous SELENOI c.797C > T (p.Pro266Leu) together with a heterozygous SATB2 c.860C > T (p.Pro287Leu) variant that was identified as de novo, as it was detected only in the proband and was absent from both parents and siblings. This novel variant of uncertain significance was interpreted cautiously as a possible contributor to his additional neurodevelopmental features. He also carried a homozygous SPAST (Spastin) c.131C > T (p.Ser44Leu) modifier variant. Family-based analyses further showed heterozygous carrier status for the LAMA1 c.4579C > T (p.Gln1527Ter) variant in both parents, whereas the SELENOI c.797C > T (p.Pro266Leu) and SPAST c.131C > T (p.Ser44Leu) variants in the younger brother were reportedly inherited from both parents in the original family-based exome report. The detailed genetic findings, including available mean sequencing coverage, population frequency data, ACMG/AMP evidence codes, and supporting in silico or conservation information from the original diagnostic reports, are summarized in Table 1.
Summary of the genetic findings in three affected siblings.

SELENOI c.797C > T (p.Pro266Leu) and SATB2 c.860C > T (p.Pro287Leu) were novel variants absent from gnomAD and the Greater Middle East Variome database in the original diagnostic reports. Computational prediction and conservation analyses suggested a possible impact, but the evidence was insufficient for pathogenic classification.
LAMA1 c.4579C > T (p.Gln1527Ter) was classified as likely pathogenic with ACMG/AMP evidence codes PVS1 + PM2.
SPAST c.131C > T (p.Ser44Leu) was reported as benign on its own but a possible disease modifier.
Mean exome coverage in the diagnostic reports was 119× for the older sister, 124× for the index patient, and 115× for the younger brother. Both parents were included in the family-based exome analysis. Variant detection and interpretation were based on the original clinical diagnostic reports. Not all reported variants were independently confirmed by Sanger sequencing; follow-up familial Sanger sequencing was available only for selected segregation analyses.
HGVS: Human Genome Variation Society; VUS: variant of uncertain significance; WES: whole-exome sequencing; PVS1: pathogenic very strong criterion for a predicted loss-of-function variant; PM2: pathogenic moderate criterion based on absence or rarity in population databases; ACMG/AMP: American College of Medical Genetics and Genomics/Association for Molecular Pathology.
Clinical comparisons and rehabilitation course
All three siblings exhibited early-onset lower-extremity spasticity and gait impairment, requiring prolonged rehabilitation with gait training, orthotic management, and orthopedic interventions. Despite these shared motor features, their clinical trajectories diverged. Index patient followed a progressive clinical course characterized by worsening spasticity, white matter atrophy, and greater dependence on gait aids over time. His inpatient rehabilitation focused on spasticity management and compensatory gait strategies. Lower limb spasticity improved from grade 2 to grade 1 on MAS. The BBS score increased from 15 to 21 points, FIM from 101 to 117, and MBI from 84 to 88, thereby indicating better balance and independence in daily activities. His lumbar lordosis angle also improved from 98.63 to 66.93, consistent with strengthened abdominal and hip muscles and improved pelvic control.
His older sister exhibited a nonprogressive course with stable coordination deficits, consistent with the static cerebellar malformation of Poretti–Boltshauser syndrome. Her rehabilitation program focused on improving standing balance, postural control, and gait stability. Although lower limb motor strength and spasticity remained unchanged, her balance function improved, with BBS score increasing from 35 to 42, and gait stability enhanced.
The fifth brother, who harbored both the homozygous SELENOI variant and the de novo SATB2 variant, exhibited a developmental and mixed phenotype. Lower limb spasticity was comparable to the index patient; however, additional cognitive, speech, and oromotor impairments were considered possibly associated with the SATB2 variant, although its contribution remains uncertain. Rehabilitation was focused on both lower-extremity strengthening and gait training as well as fine motor and oromotor function. Over the course of treatment, he progressed from being unable to walk independently to achieving a few meters of independent indoor ambulation, although remaining wheelchair-dependent for longer distances.
Discussion
HSPs are monogenic disorders that primarily affect the corticospinal tracts and dorsal columns, causing progressive lower limb spasticity, hyperreflexia, and gait impairment with more than 80 genetic subtypes identified. 1 Among the autosomal recessive forms, SELENOI-related SPG81 is a complex subtype characterized by early developmental delay, intellectual disability, seizures, hypomyelination, and progressive spastic paraplegia.6–11 To date, only a small number of families with SELENOI-related SPG81 have been reported, and functional validation has been performed using different approaches across studies, including fibroblast-based enzymatic analysis, yeast complementation assays, and RNA splicing analysis.6,7,9 SELENOI encodes ethanolamine phosphotransferase, the final enzyme of the cytidine diphosphate (CDP)–ethanolamine (Kennedy) pathway, which is required for synthesis of phosphatidylethanolamine and lipids essential for myelin integrity and neuronal membrane stability. Disruption of this pathway leads to impaired plasmalogen production, oxidative vulnerability, defective oligodendrocyte maturation, and progressive white matter degeneration.8,10,11 Although additional functional validation was not feasible in our patients, the strong phenotypic and neuroimaging concordance between our patients and previously reported SPG81 cases supports the clinical relevance of the identified variant to the SPG81-like phenotype. The variant was classified as a variant of uncertain significance in the original diagnostic report; however, its absence from population databases, supportive computational and conservation analyses, recessive segregation in the two affected brothers, and phenotypic concordance with previously reported SPG81 provide supportive evidence for its clinical relevance.
Although both brothers had clinically similar SPG81-like features, their clinical severity differed markedly. A possible contributing factor was the fifth brother's additional de novo SATB2 c.860C > T (p.Pro287Leu) variant, also classified as of uncertain significance in the original report. SATB2-associated syndrome is characterized by global developmental delay, speech and oromotor impairment, behavioral abnormalities, and sometimes seizures.12,13 However, this syndrome shows considerable phenotypic variability. In the largest published cohort, neurodevelopmental impairment was universal and absent or near-absent speech was reported in 16 of 19 affected individuals, whereas craniofacial abnormalities were less consistently present. 14 The younger brother's progressive spastic paraplegia and white matter abnormalities were consistent with the established phenotype of SELENOI-related SPG81, whereas his additional neurodevelopmental features, including dysarthria, dysphagia, and seizure onset, overlapped with SATB2-associated syndrome. Although the relative contribution of SELENOI and SATB2 to his overall phenotype remains uncertain, the coexistence of these variants may provide a plausible, hypothesis-generating explanation for his greater clinical complexity. The siblings also differed in their SPAST genotype. The fifth brother carried the homozygous p.Ser44Leu variant that was absent in the index patient. The SPAST p.Ser44Leu variant has been reported as a disease modifier rather than an independent pathogenic cause. Its relatively high population frequency and prior literature suggest that it is unlikely to explain the phenotype on its own, although a limited contributory effect in the presence of coexisting variants cannot be completely excluded.15,16
Poretti–Boltshauser syndrome is an autosomal recessive cerebellar malformation caused by biallelic LAMA1 variants and is characterized by nonprogressive cerebellar ataxia, ocular motor abnormalities, and variable cognitive impairment.17–19 Neuroimaging typically demonstrates cerebellar dysplasia with cortical or subcortical cysts and vermian hypoplasia, with relative preservation of supratentorial structures. The second sister's presentation was consistent with this profile. She had early-onset but stable coordination deficits, longstanding ocular abnormalities, and mild cognitive issues, and her MRI scan revealed abnormal cerebellar foliation with retrocerebellar cysts without supratentorial involvement. Together with her homozygous LAMA1 variant, these features support a diagnosis of Poretti–Boltshauser syndrome and explain her nonprogressive clinical course compared with that of her siblings. In the index patient, the same LAMA1 c.4579C > T (p.Gln1527Ter) variant was present only in the heterozygous state. Because LAMA1-related Poretti–Boltshauser syndrome is an autosomal recessive disorder caused by biallelic pathogenic variants, this finding was not considered sufficient to explain his phenotype as a primary diagnosis. His clinical and neuroimaging features were also less consistent with Poretti–Boltshauser syndrome than those of his older sister. Nevertheless, we cannot completely exclude the possibility that the heterozygous LAMA1 variant may have represented a coincidental carrier finding or had a limited modifying effect.
Although LAMA1-related Poretti–Boltshauser syndrome and SELENOI-related SPG81 have each been reported in consanguineous families, there are no published cases in which both disorders occurred independently within the same family.20,21 Similarly, although SATB2-associated syndrome and the SPAST p.Ser44Leu modifier variant have been described individually, their coexistence with SPG81 and Poretti–Boltshauser syndrome in a single sibling has not been previously documented. Although the coexistence of multiple recessive conditions may be biologically plausible in consanguineous families, this case illustrates an unusual intrafamilial combination of distinct genetic findings associated with overlapping early-onset spastic gait phenotypes. These findings emphasize the importance of considering multiple concurrent genetic diagnoses when siblings exhibit superficially similar motor phenotypes but differ in associated features, progression, or neuroimaging patterns.
This report has several limitations. First, the SELENOI and SATB2 variants remain classified as variants of uncertain significance, and their pathogenicity cannot be established definitively on the basis of current evidence. Second, additional functional validation, including fibroblast-based assays for the SELENOI variant, was not feasible in this retrospective study. Third, although family-based exome sequencing supported the reported segregation patterns, the SELENOI variant may not fully explain the phenotype. The possibility of additional undetected variants or a more complex multilocus contribution cannot be excluded. Fourth, the original clinical diagnostic reports provided selected variant-level evidence, including population frequency data, ACMG/AMP classification, and qualitative in silico or conservation information. However, complete raw annotation data, such as full ACMG/AMP evidence codes, numerical in silico prediction scores, and conservation metrics, were not available for all variants. Finally, disease-specific severity measures for HSP, such as the Spastic Paraplegia Rating Scale, were not available because this was a retrospective study based on routine clinical assessments.
Conclusion
Three siblings presented with overlapping spastic paraplegia phenotypes; however, genetic evaluation revealed distinct genetic findings in each affected individual. This case highlights the potential value of comprehensive genetic evaluation when similar motor phenotypes occur within the same family.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605261463615 - Supplemental material for Hereditary spastic paraplegia in three siblings with distinct genetic mutations
Supplemental material, sj-pdf-1-imr-10.1177_03000605261463615 for Hereditary spastic paraplegia in three siblings with distinct genetic mutations by Tae Kwon Lee, Kyung Min Kim, Su Min Lee and Sung-Rae Cho in Journal of International Medical Research
Footnotes
Acknowledgments
The authors have no acknowledgments to declare.
Ethics approval and informed consent
Written informed consent for publication of this case report and accompanying images was obtained from all patients or their legally authorized representatives. Signed consent to treatment was obtained from the patients or their legally authorized representatives. Ethics approval was not sought, as this report describes three patients and is based on anonymized retrospective clinical data obtained during routine clinical care. All patient details were deidentified by removing all personal details to ensure the patient's privacy. The reporting of this study conforms to CARE guidelines. 22
Author contributions
TKL contributed to data curation, investigation, and drafting of the original manuscript. KMK and SML contributed to data curation and manuscript review and editing. SRC contributed to conceptualization and supervision of the study and revised the manuscript. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (RS-2022-KH129545, RS-2022-KH129919, RS-2025-02215487).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.