
Abstract
Objective
We aimed to characterize the clinical, magnetic resonance imaging, and genetic features of Chinese patients with dentatorubral–pallidoluysian atrophy.
Methods
In this retrospective case-series and literature-review study, we analyzed three affected members belonging to the same family diagnosed at our institution. We also reviewed genetically confirmed Chinese dentatorubral–pallidoluysian atrophy cases reported in the China National Knowledge Infrastructure, Wanfang, and PubMed databases up to 31 May 2025 and included additional 42 patients from separate families. Cases with available age at onset, initial symptoms, and CAG repeat size were included; cases without magnetic resonance imaging data were excluded only from imaging-specific analyses. Thus, the final cohort included 45 Chinese dentatorubral–pallidoluysian atrophy patients.
Results
Forty-five patients were analyzed. The median ATN1 CAG repeat count was 62 (range, 53–79), and mean age at onset was 28.22 ± 17.48 (range, 2–69) years. Seizures (46.7%) and gait instability (42.2%) were the most common initial manifestations. Multiple clinical manifestations frequently co-occurred, including gait instability (88.9%), cognitive impairment (73.3%), speech impairment (66.7%), seizures (64.4%), and involuntary movements (48.9%). Patients with seizure onset had larger CAG expansions and younger age at onset than those with gait-instability onset. Age at onset negatively correlated with CAG repeat count in the overall cohort and seizure-onset subgroup, but not in the gait-instability subgroup.
Conclusions
Larger ATN1 CAG repeat expansions may be associated with earlier onset, especially in seizure-predominant dentatorubral–pallidoluysian atrophy. Magnetic resonance imaging commonly shows cerebellar atrophy and white-matter or brainstem involvement. After excluding common causes, dentatorubral–pallidoluysian atrophy should be considered in patients presenting with unexplained combinations of seizures, ataxia, cognitive impairment, and a compatible family history.
Keywords
Introduction
Dentatorubral–pallidoluysian atrophy (DRPLA) is a rare autosomal dominant polyglutamine disorder caused by CAG repeat expansion in ATN1. 1 Clinical manifestations include epilepsy, ataxia, choreoathetosis, and cognitive decline, with larger expansions associated with juvenile onset and seizures (SZs).2–4 Epilepsy in juvenile-onset DRPLA may be difficult to control and can resemble progressive myoclonic epilepsy. 2 Although most frequently reported in Japan, DRPLA occurs worldwide and may be under-recognized in Chinese populations, where evidence remains limited to isolated reports.5,6 A pooled analysis may clarify the phenotypic and genotypic spectrum in Chinese patients.
Methods
Study design and participants
This was a retrospective case series combined with a literature review. The reporting of this study conforms to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guideline. 7 Three DRPLA patients from one family were diagnosed at the Neurology Department of the Fourth Medical Center of PLA General Hospital. The family members were identified through evaluation of the proband and cascade family assessment. Additionally, we searched the China National Knowledge Infrastructure, Wanfang, and PubMed for eligible articles published up to 31 May 2025 using the terms: (“DRPLA” OR “dentatorubral–pallidoluysian atrophy” OR “dentatorubropallidoluysian atrophy”) AND (“China” OR “Chinese”).
Inclusion criteria were as follows (a) genetically confirmed DRPLA (ATN1 gene CAG repeats ≥48); (b) available data on age at onset, initial symptoms, and CAG repeat count; and (c) Chinese ethnicity. Magnetic resonance imaging (MRI) data were not required for inclusion in the clinical-genetic analyses. Patients without MRI data were retained for demographic, clinical, and genetic analyses but were excluded from MRI-specific summaries. Duplicate reports and articles with insufficient clinical information were excluded. Newly published studies that reported only aggregate cohort-level data without extractable individual-patient variables were reviewed qualitatively but were not merged with the individual-patient pooled dataset to avoid ecological bias and possible duplicate counting.
Data extraction
From each eligible report, we extracted data on sex, age at onset, initial symptoms, CAG repeat count, presence of SZs, MRI findings (cerebellar atrophy, brainstem atrophy, cerebral cortical atrophy, and leukoencephalopathy), and family history. We additionally extracted data regarding multiple clinical manifestations, including gait instability (GI)/ataxia, involuntary movements (IMs), SZs/epilepsy, cognitive impairment (CI), psychiatric symptoms (PSs), and speech impairment (SI). Data from 42 previously reported Chinese patients and three new patients from our center were combined for a total sample size of 45.
Genetic testing
Genomic DNA was analyzed from peripheral blood samples using polymerase chain reaction and capillary electrophoresis to determine ATN1 CAG repeat length.
Statistical analysis. Continuous variables were assessed for normality using the Shapiro–Wilk test. Normally distributed variables are presented as mean ± SD (range), and non-normally distributed variables (including CAG repeat lengths) as median (range or interquartile range) values. Categorical variables are presented as frequencies (%). For normally distributed variables, group comparisons were performing using unpaired independent-samples t-tests; for non-normally distributed variables, the Mann–Whitney U test was used. Correlations were assessed using Pearson's r (or Spearman's rank correlation for non-normal data). A two-sided p-value <0.05 was considered statistically significant. Analyses were performed using the Statistical Package for Social Sciences software, version 26.0. This was an observational retrospective study with a modest sample size; therefore, the findings should be considered exploratory.
Ethical approval
Written informed consent was obtained from both patients and their relatives. Ethical approval for this study was granted by the ethics committee of the Fourth Medical Center of PLA General Hospital (approval No. 2025KY001-KS001). This study was conducted in accordance with the Declaration of Helsinki of 1975, as revised in 2024. All patient details were de-identified before analysis and reporting. No artificial intelligence tools were used for research methods, including data collection, data extraction, statistical analysis, or scientific interpretation.
Results
Clinical characteristics of the new family
To better illustrate the family relationships, we constructed a family pedigree chart (presented in Figure 1).
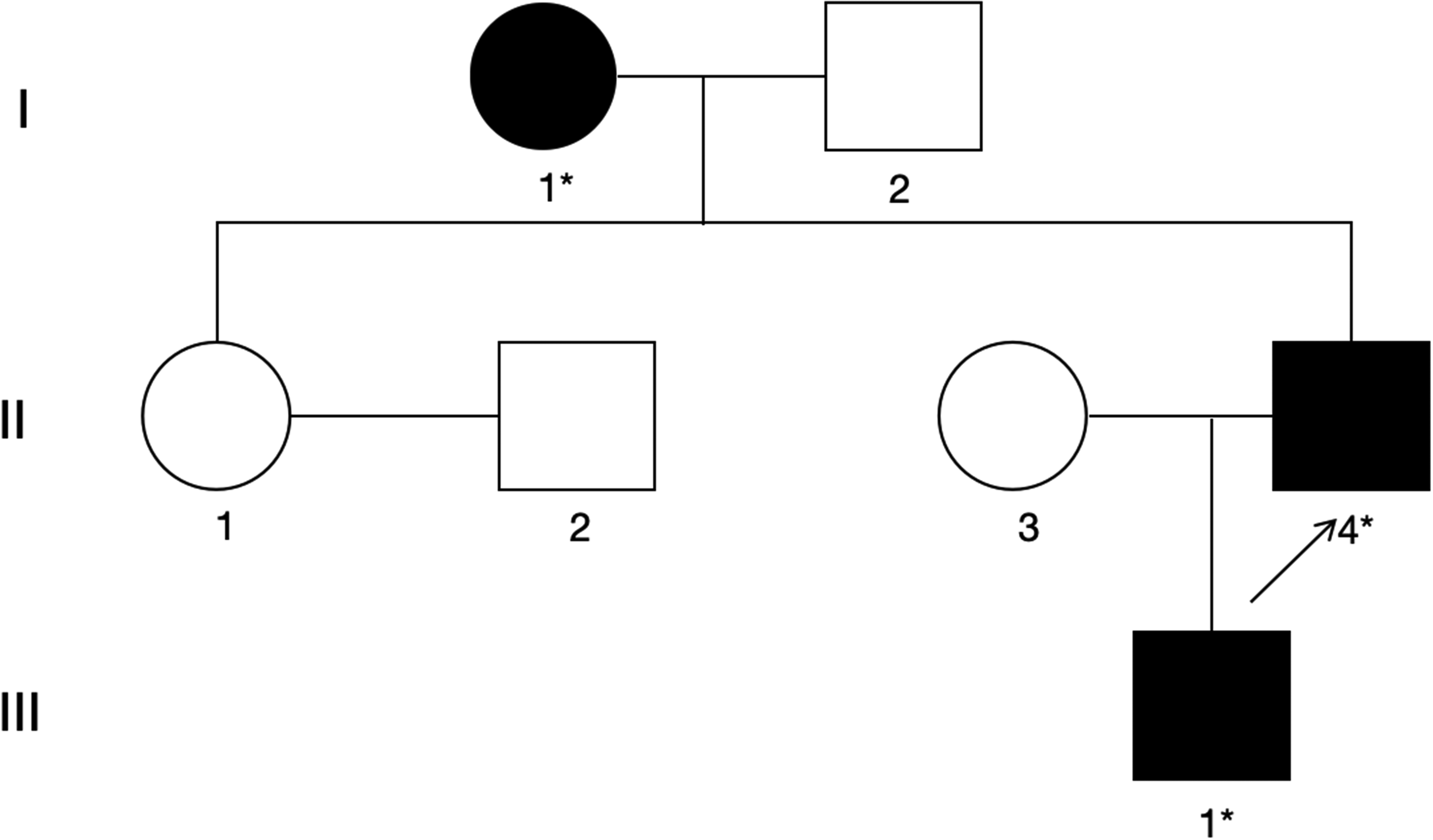
Pedigree analysis of the patient's family.
Proband. A 56-year-old man presented with clinical symptoms such as walking instability, IMs, and speech impediment. According to the available retrospective records, the speech difficulty was documented as dysarthria rather than aphasia or dysphonia. The available retrospective record did not contain sufficient detail to classify the IMs reliably as chorea, dystonia, myoclonus, or another movement phenomenology.
Proband's mother. She was diagnosed with cerebellar atrophy at approximately 50 years of age, was unable to walk independently, and struggled to communicate normally. According to her family history, she developed progressive GI and cognitive decline at approximately 50 years of age. The original MRI reports were not available for review, and she was therefore not included in MRI-specific analyses.
Proband's son. He underwent an intelligence test at the age of 10 years and scored 70, which is below the normal range. An intelligence quotient evaluation was performed because of concerns about developmental delay and academic difficulties; however, the detailed neuropsychological report was not available in the retrospective records. He began experiencing SZs at approximately 20 years of age. At that time, electroencephalography (EEG) revealed irregular spike-slow and multiple spike-slow complex waves repeatedly erupting throughout the brain (Figure 2). Detailed anti-SZ medication response and long-term follow-up information were not available in the retrospective records.
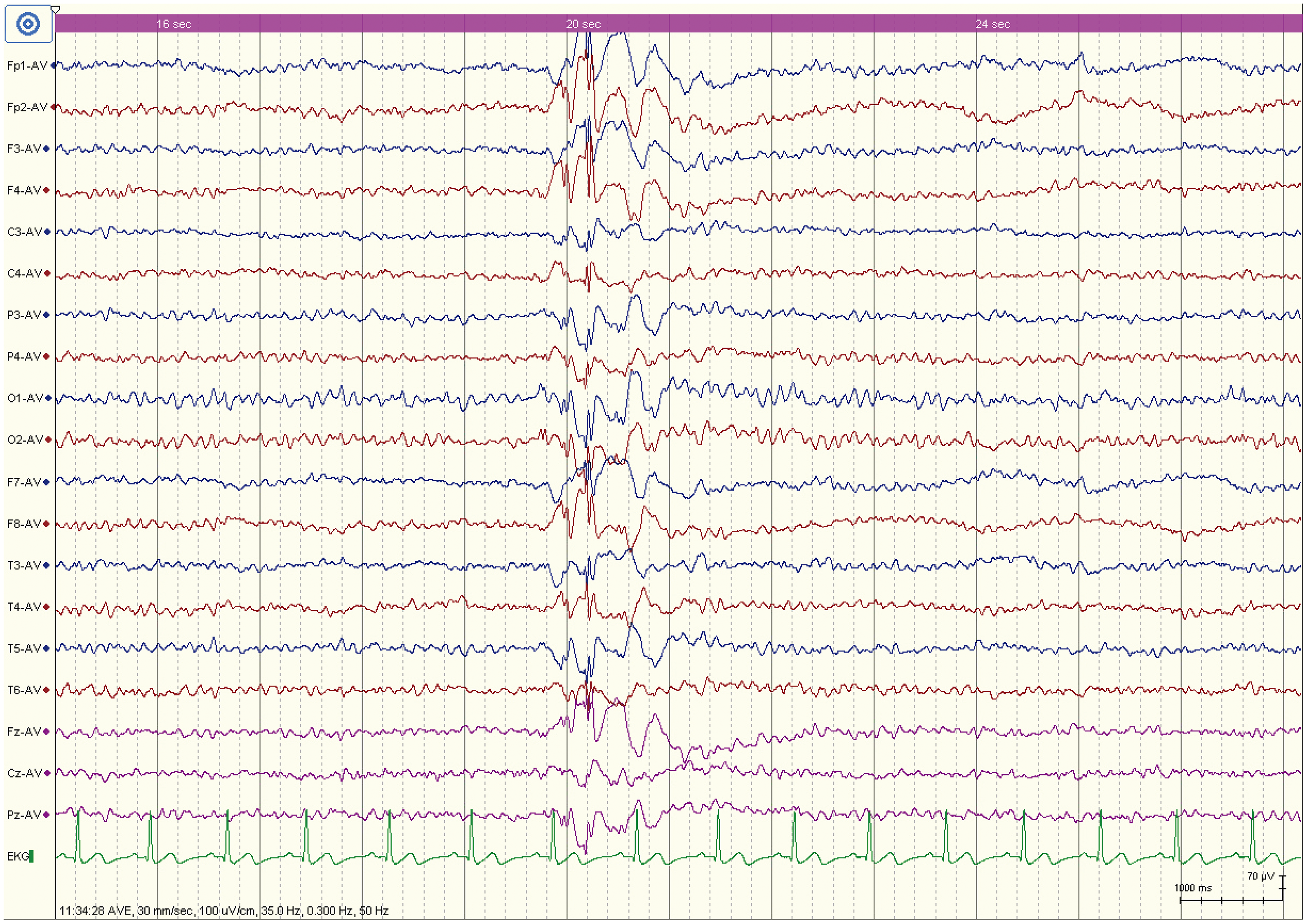
EEG of the proband's son showing irregular spike-slow and multiple spike-slow complex waves.
MRI findings
Figure 3(a) and 3(b) show white matter lesions in the brainstem and periventricular region of the proband. Figure 3(c) presents a fluid-attenuated inversion recovery (FLAIR) image from the proband's son and shows prominence of cortical sulci. A pontine T2 hyperintensity with a mountain-like appearance was observed in the proband (Figure 3(b)).

Fluid-attenuated inversion recovery (FLAIR) sequences: (a, b) white matter lesions in the brainstem and periventricular region of the proband; (c) FLAIR image from the proband's son showing prominence of cortical sulci.
Genetic testing results
According to the ATN1 gene CAG trinucleotide repeat test, the proband exhibited 18/58 CAG repeats (Figure 4(a)); the proband's mother showed 15/57 repeats (Figure 4(b)); and the proband's son displayed 16/65 repeats (Figure 4(c)).
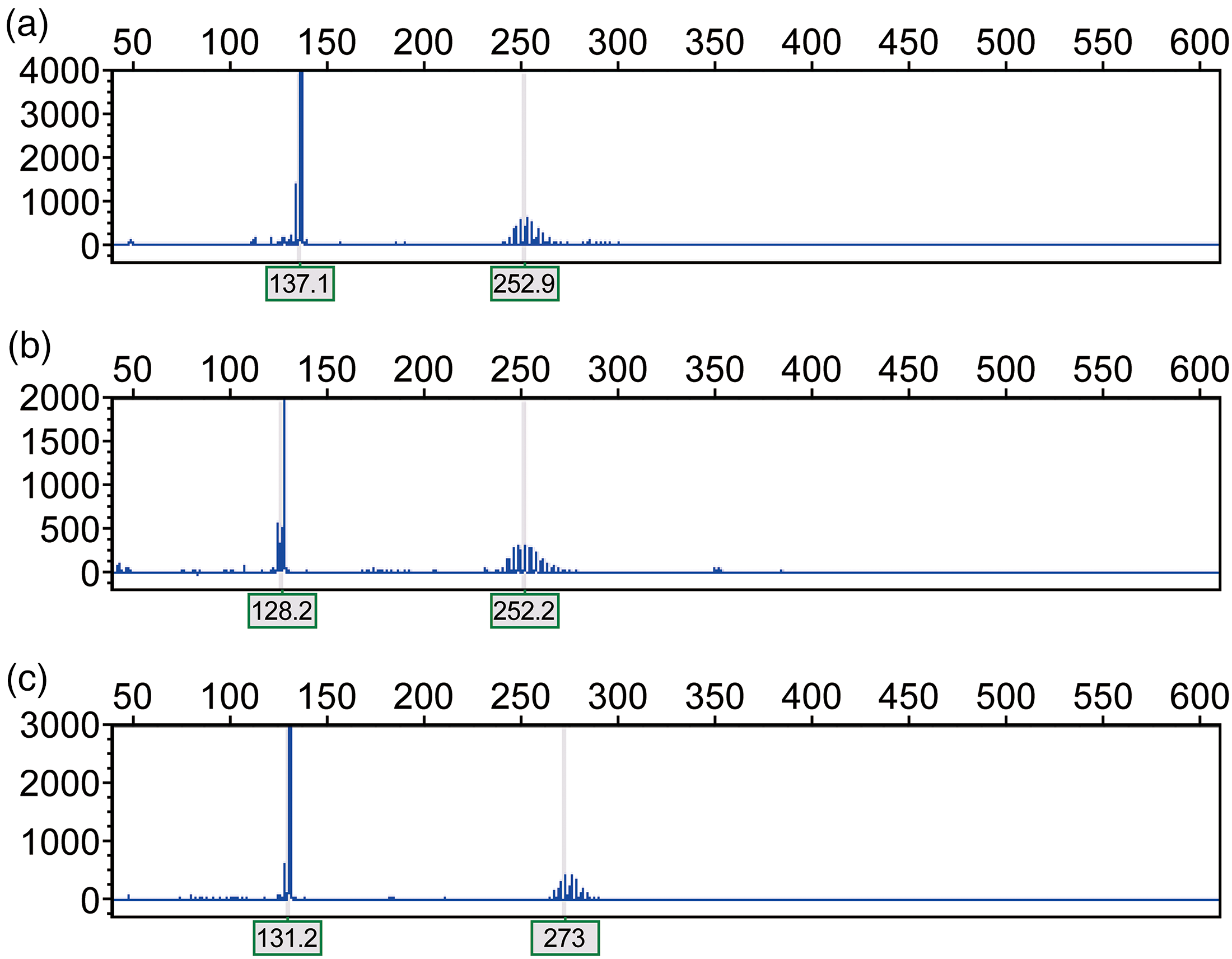
ATN1 gene CAG repeat analysis: (a) proband (18/58), (b) proband's mother (15/57), and (c) proband's son (16/65).
Literature review and demographic and clinical summary
In total, 45 genetically confirmed DRPLA patients were analyzed. Demographic and clinical characteristics are summarized in Table 1.3,5,8–27. Table 1 shows multiple co-occurring clinical manifestations for each patient in a compact single column. Based on age at onset, patients were classified as having juvenile type (≤ 20 years, n = 17), early-onset adult type (21–39 years, n = 18), or late-onset adult type (≥ 40 years, n = 10) DRPLA. 2 The overall median CAG repeat length was 62 (range, 53–79).
Clinical characteristics of the 45 DRPLA patients.

F: female; M: male; GI: gait instability/ataxia; IM: involuntary movement; SZ: seizures/epilepsy; CI: cognitive impairment; PS: psychiatric symptom; SI: speech impairment; DRPLA: dentatorubral–pallidoluysian atrophy
In the clinical manifestations column, the listed abbreviations indicate manifestations reported as present; absence of an abbreviation indicates that the manifestation was not reported/present in the source data. In the leukoencephalopathy column, N indicates no leukoencephalopathy on available MRI, and - indicates MRI not done or not available.
Across all 45 patients, initial symptoms were SZs in 21 patients (46.7%), GI in 19 (42.2%), CI in 3 (6.7%), SI in 1 (2.2%), and developmental delay in 1 (2.2%). Multiple clinical manifestations were common. GI was present in 40 patients (88.9%), CI in 33 (73.3%), SI in 30 (66.7%), SZs in 29 (64.4%), IMs in 22 (48.9%), and PSs in 8 (17.8%).
Initial symptoms by subtype
SZs predominated in juvenile-onset cases (82.4%), whereas adult-onset cases were characterized by GI (50%–100%). GI was the initial symptom in nine out of 18 early-onset adult cases (50.0%) and all 10 late-onset adult cases (100.0%). As a clinical manifestation, GI occurred in 12 out of 17 juvenile-onset cases (70.6%), 18 out of 18 early-onset adult cases (100.0%), and all 10 late-onset adult cases (100.0%). Median CAG repeat lengths were 65 (range, 63–79) in the juvenile group, 59 (range, 54–65) in the early-onset adult group, and 57 (range, 53–63) in the late-onset adult group.
MRI characteristics
Among the 45 patients, MRI findings were available for 37 and are summarized in Table 2. The remaining eight patients were excluded from MRI-specific analyses only. Cerebellar atrophy was the most frequent abnormality (86.5%). After rechecking the original case-level MRI data, brainstem atrophy, leukoencephalopathy, and cerebral cortical atrophy were confirmed in 25/37 (67.6%), 20/37 (54.1%), and 19/37 (51.4%) patients, respectively. White matter involvement progressed from periventricular regions to brainstem and supratentorial areas.
MRI characteristics of three subtypes of DRPLA patients.
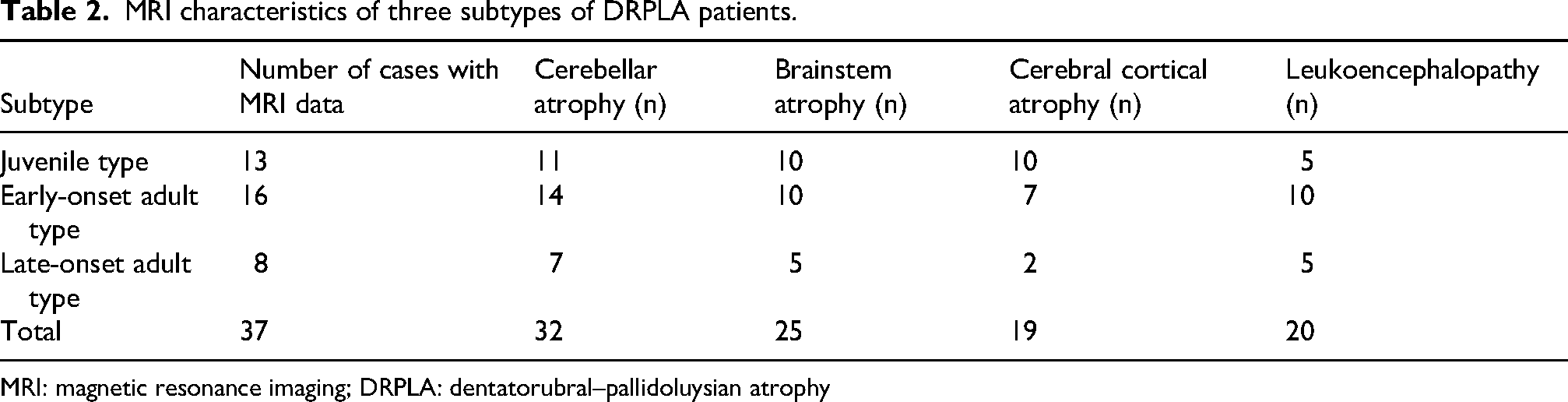
MRI: magnetic resonance imaging; DRPLA: dentatorubral–pallidoluysian atrophy
Correlation analyses
The age at onset in the 45 DRPLA patients showed a strong negative correlation with the CAG repeat count of the ATN1 gene (r = –0.749, p < 0.001) (Figure 5(a)). For the 21 patients with SZs as the primary symptom, age at onset was negatively correlated with the CAG repeat count (r = –0.771, p < 0.001) (Figure 5(b)). In contrast, for the 19 patients with GI as the initial symptom, no significant correlation was observed (r = –0.097, p = 0.692) (Figure 5(c)).
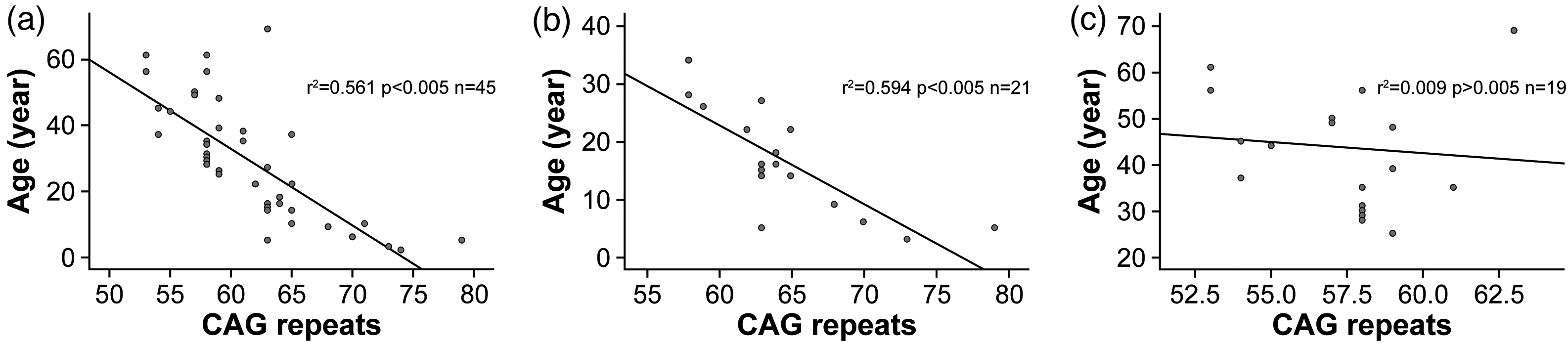
Correlation between age at onset and CAG repeat length: (a) all 45 patients, (b) 21 patients with seizures as the initial symptom, and (c) 19 patients with gait instability as the initial symptom.
Comparison between SZ-onset and GI-onset groups
Patients with SZ onset had significantly higher CAG repeat lengths (median 63, range 58–79) than those with GI onset (median 58, range 53–63; Mann–Whitney U test, p < 0.001). Patients with SZ onset also had a significantly younger age at onset (mean, 16.43 ± 8.68 vs. 43.58 ± 12.93 years; independent t-test, p < 0.001).
Discussion
Our findings confirm the inverse correlation between ATN1 CAG repeat length and age at onset.3,4,28 Patients with SZ onset had larger expansions and earlier onset than those with GI onset, underscoring repeat length as an indicator of clinical severity. However, because this was an observational retrospective analysis, these associations should be interpreted as supportive rather than definitive.
The age-dependent phenotypic distribution observed in our pooled cohort is also in line with previous reports. Juvenile-onset DRPLA is more commonly associated with SZs, myoclonus, and CI, whereas adult-onset disease more often begins with gait ataxia, SI, IMs, or psychiatric and cognitive symptoms. 6 Epilepsy in juvenile-onset DRPLA may be drug refractory, and this issue should be considered during clinical follow-up and counseling. 2 However, these boundaries are not absolute. Accordingly, ATN1 testing should be considered in patients with unexplained combinations of SZs, ataxia, cognitive decline, and a compatible family history.4,6
Neuroimaging is an important diagnostic adjunct. Cerebellar atrophy was the most frequent finding (86.5%), with white matter involvement extending from periventricular regions to the brainstem. Advanced MRI reveals widespread gray- and white-matter degeneration in adult-onset DRPLA. 29 MRI findings should be interpreted in conjunction with clinical and molecular data.29–34
A pontine T2 hyperintensity was noted in our proband; similar findings have been reported in DRPLA32–34 but are not disease-specific. 32
The combination of SZs, ataxia, and cognitive decline has a broad differential diagnosis. More common or clinically urgent causes should be considered first, including acquired inflammatory, infectious, metabolic, toxic, and structural disorders; mitochondrial disease; progressive myoclonic epilepsies; other spinocerebellar ataxias; Huntington disease phenocopies; and other repeat-expansion disorders. DRPLA should be considered when these conditions are excluded and when autosomal dominant inheritance, anticipation, suggestive CAG repeat testing, or compatible MRI findings are present.3,4,28
Recently, Yuan et al. assessed the largest Chinese DRPLA cohort to date (n = 116), identified a novel ATN1 haplotype, and described cognitive, imaging, and plasma neurofilament light findings that help define prodromal and manifest disease stages. 35 That cohort was not merged with the present individual-patient pooled analysis because the publicly available article did not provide extractable individual-level variables for all required fields, including age at onset, initial symptom, CAG repeat count, clinical manifestations, and MRI findings. In addition, possible overlap with previously published Chinese case reports could not be excluded. Combining the aggregate data with the present case-level dataset would have therefore introduced a risk of duplicate counting and ecological bias. The present 45-case analysis thus complements the larger cohort by showing that core genotype–phenotype relationships can still be detected using conventional clinical data.
Comparison with other Asian cohorts also supports the age-dependent clinical pattern. Recently, an Indian family with DRPLA showed adult-onset ataxia in the proband and his father and juvenile-onset epilepsy with CI in the son, consistent with anticipation and the SZ-predominant juvenile phenotype observed in our Chinese cohort. 36
Limitations
This study should be interpreted considering certain limitations. The pooled sample size remained modest, the analysis was retrospective, and clinical descriptors and MRI protocols were not uniform across the published reports. MRI data were unavailable for eight patients, and neuropathological data were not available. Some details requested during peer review, including current age, detailed anti-SZ medication response, and precise classification of IMs for the newly reported family, were not consistently documented in the retrospective records. In addition, literature-based aggregation may overrepresent severe or unusual phenotypes because case reports and small pedigrees are more likely to be published. The larger 2025 Chinese cohort could not be incorporated into our individual-patient pooled analysis because extractable case-level data were unavailable and overlap with published case reports could not be excluded. Prospective multicenter registries with standardized neurological assessment, harmonized MRI protocols, and longitudinal follow-up are needed to refine the natural history of DRPLA in Chinese patients. Such efforts will also be important for biomarker development and future therapeutic studies.28,29,37
Conclusion
DRPLA is a rare but likely under-recognized hereditary neurodegenerative disorder in Chinese patients. In this cohort, larger ATN1 CAG repeat expansions were associated with earlier disease onset and were more common among patients presenting with SZs, whereas GI predominated in adult-onset cases. MRI commonly demonstrated cerebellar atrophy and white-matter or brainstem abnormalities, which may provide important supportive diagnostic clues when interpreted together with clinical findings. In patients with unexplained combinations of SZs, ataxia, CI, and a compatible family history, evaluation with cranial MRI and ATN1 testing should be considered after more common causes have been excluded.
Footnotes
Acknowledgments
A generative artificial intelligence tool was used only for language polishing and revision formatting; it was not used for data collection, data extraction, statistical analysis, or scientific interpretation. All scientific content, data, analyses, and interpretations were checked and approved by the authors.
Ethical approval
This study was approved by the Ethics Committee of the Fourth Medical Center of PLA General Hospital (Approval No. 2025KY001-KS001).
Patient consent statement
Written informed consents for publication were obtained from all participating patients and their legal guardians.
Author contributions
The contributions of the authors are detailed in the title page (blinded for review).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The de-identified data underlying the results are summarized in the revised tables. Additional de-identified extracted data are available from the corresponding author on reasonable request, subject to ethical and privacy restrictions.