
Abstract
Bromodomain and extraterminal proteins have emerged as key epigenetic regulators that coordinate transcriptional programs fundamental to cardiovascular physiology and disease. Although bromodomain and extraterminal protein inhibitors exert multiple cardiovascular effects, they cannot fully explain clinical heterogeneity in outcomes and responses, highlighting the need to clarify the context- and disease-specific roles of individual bromodomain and extraterminal protein family members. This narrative review delineates the distinct molecular roles of bromodomain-containing protein 2, bromodomain-containing protein 3, bromodomain-containing protein 4, and bromodomain testis, summarizing their domain architecture and epigenetic regulatory functions. We then synthesize current evidence linking bromodomain and extraterminal protein activity to cardiovascular disease pathogenesis, emphasizing their contributions to major pathological processes, including inflammatory amplification, fibrotic remodeling, dysregulated energy metabolism, aberrant cell proliferation, endothelial–mesenchymal transition, and ferroptotic cell death. Furthermore, we evaluate advances in bromodomain and extraterminal protein inhibitors across preclinical and clinical research, highlighting agents that demonstrate anti-inflammatory, antifibrotic, and cardioprotective efficacy in animal models of cardiovascular disease. By integrating mechanistic and translational evidence, this review provides a framework for understanding bromodomain and extraterminal proteins in cardiovascular diseases and guides the rational design of targeted therapies.
Keywords
Introduction
Cardiovascular diseases (CVDs) remain the leading cause of mortality and disability worldwide, with absolute case numbers and years lived with disability continuing to rise despite therapeutic advances. 1 This burden is particularly pronounced in China, where an estimated 330 million individuals are currently affected, reflecting an alarming trajectory driven by rapid population aging and escalating exposure to modifiable risk factors. 2 Globally, epidemiologic data demonstrate that a substantial proportion of CVD events is attributable to a limited set of preventable risks, underscoring the urgent need for earlier, system-level intervention strategies. 3 Risk accumulation begins early in life and is influenced by lifestyle, behavioral, and environmental exposures, although the relative impact varies across populations. 4 Collectively, these observations highlight the persistent, preventable burden of CVD and emphasize a critical gap in understanding the molecular determinants of clinical heterogeneity, which continues to impede progress in improving patient outcomes.1,3
CVD is influenced not only by genetic susceptibility but also by modifiable risk factors such as hypertension, obesity, and smoking. Environmental exposures, including air pollution and dietary patterns, are now recognized as powerful modulators of gene expression programs that maintain cardiovascular homeostasis, acting primarily through epigenetic mechanisms that regulate transcription without altering the underlying DNA sequence. 5 Among these epigenetic regulators, the bromodomain (BRD) and extraterminal (BET) protein family—consisting of bromodomain-containing protein 2 (BRD2), bromodomain-containing protein 3 (BRD3), bromodomain-containing protein 4 (BRD4), and bromodomain testis (BRDT)—has emerged as a key integrator of chromatin signaling. BET proteins bind acetylated lysine residues on histones and directly regulate the transcription of numerous genes and pathways implicated in cardiovascular pathology. Preclinical studies demonstrate that pharmacologic inhibition of BET proteins, particularly BRD4, can mitigate inflammation, fibrosis, and hypertrophy in models of heart failure and atherosclerosis.6–9 However, clinical translation has revealed considerable heterogeneity in patient responses to BET inhibition, underscoring the need for a more refined understanding of the context-dependent and disease-specific roles of individual BET family members.
This article is a narrative review that focuses on the molecular characteristics of the BET protein family (BRD2, BRD3, BRD4, and BRDT), their pathological mechanisms in CVD, and the therapeutic progress of BET inhibitors (BETi). The primary objective is to review existing evidence, clarify the roles of BET protein, identify gaps in current research, and provide a theoretical basis for the future development of BET-targeted therapies. The literature search was conducted across databases such as PubMed and Web of Science using core keywords such as “BET proteins,” “cardiovascular disease,” “epigenetic regulation,” and “BET inhibitors.” High-quality basic research studies, clinical studies, and review articles published within the past 10 years, particularly within the past 5 years, were screened. After the exclusion of duplicate studies and low-quality literature, the selected publications were categorized, synthesized, and analyzed to ensure the scientific rigor, comprehensiveness, and timeliness of the review. This review was guided by the Scale for the Assessment of Narrative Review Articles (SANRA). 10
Molecular architecture and functions of the BET family
The BRD is a highly conserved evolutionary protein module that specifically recognizes and binds acetylated lysine residues on histone tails, thereby exerting a central influence on chromatin structure and transcriptional regulation. BRDs are broadly distributed across numerous nuclear proteins involved in essential cellular processes, including chromatin remodeling, transcriptional control, and DNA repair. 11 Structurally, each BRD contains a characteristic hydrophobic “pocket” composed of four α-helices that stabilize the interaction with acetylated lysine residues. This structural configuration enables BRD to function as a key epigenetic reader, translating histone acetylation patterns into gene expression programs. 12 To date, 61 BRDs have been identified within 46 human BRD-containing proteins, which are classified into 8 families based on sequence and structural similarity. Among them, the BET family is one of the most extensively studied families given its central role in regulating transcription and chromatin dynamics.13,14 The BET family consists of four highly conserved members—BRD2, BRD3, BRD4, and the testis-restricted BRDT—each characterized by a domain architecture containing two tandem BRDs (BD1 and BD2) and a C-terminal extraterminal (ET) domain. These tandem BRDs confer the capacity to selectively recognize acetylated histone tails, enabling BET proteins to orchestrate transcriptional regulation, participate in chromatin remodeling, and contribute to the maintenance of genomic stability (Figure 1).15,16
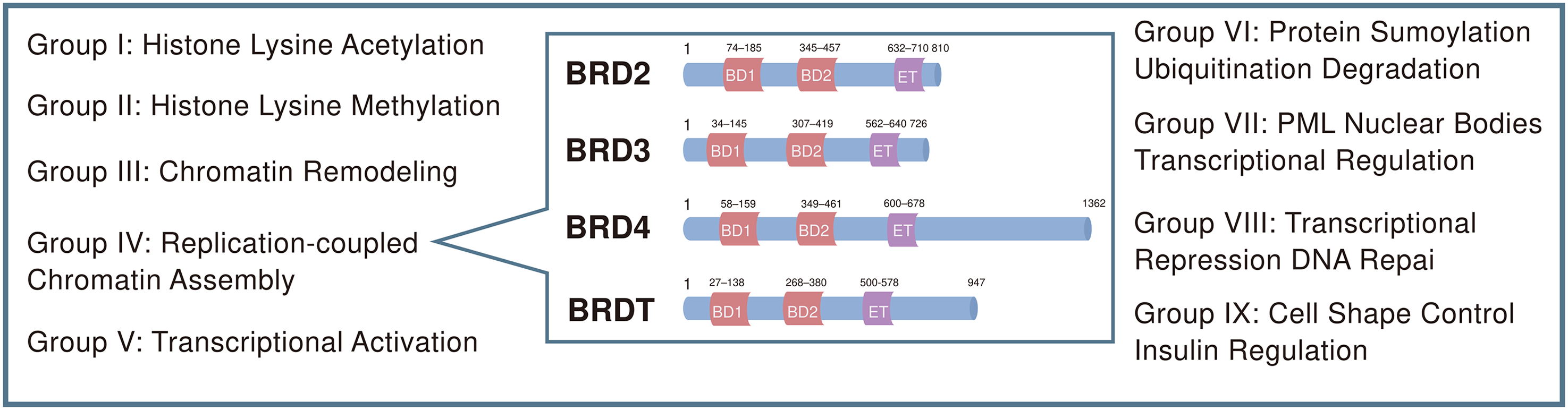
Domain architecture of BET family proteins in humans. The numbers indicate the amino acid boundaries of each domain within a single protein. BET family members BRD2, BRD3, and BRD4 as well as the testis-restricted BRDT each contain two bromodomains (BD-1 and BD-2) and an extraterminal (ET) domain. The unique C-terminal domain (CTD) of BRD4 enables interaction with P-TEFb and RNA polymerase II (RNAPII).
Molecular structure and function of BRD4
BRD4 is a central member of the BET protein family and functions as an essential epigenetic reader linking histone acetylation to transcriptional activation. Through its two highly conserved BRDs, BD1 and BD2, BRD4 recognizes and binds acetylated lysine residues (KAc) on histone tails, thereby coupling chromatin acetylation states to gene regulatory programs.17,18 BD1 preferentially engages mono- and poly-acetylated H4 peptides, whereas BD2 exhibits broader substrate specificity, binding acetylated residues on both H3 and H4 and interacting with acetylated transcription factors.19,20 In addition to these tandem BRDs, BRD4 possesses a C-terminal ET domain that coordinates transcriptional elongation and mediates the assembly of regulatory protein complexes. 21
BRD4 exerts a dual enzymatic function that positions it as a master regulator of transcription. First, its intrinsic histone acetyltransferase (HAT) activity acetylates histone H3, promoting nucleosome displacement and chromatin accessibility. 22 Second, acting as an atypical kinase, BRD4 phosphorylates the Ser2 site of the carboxy-terminal domain (CTD) of RNA polymerase II, thereby releasing promoter-proximal pausing and enabling productive transcriptional elongation.23,24 These enzymatic activities operate synergistically to regulate the continuum of transcriptional processes, from chromatin remodeling to transcription completion (Figure 2). 25 This central regulatory role also underlies the therapeutic relevance of BRD4, as BETi competitively bind its BRDs and disrupt both HAT- and kinase-dependent functions, attenuating aberrant gene activation in pathological states such as cardiovascular stress.
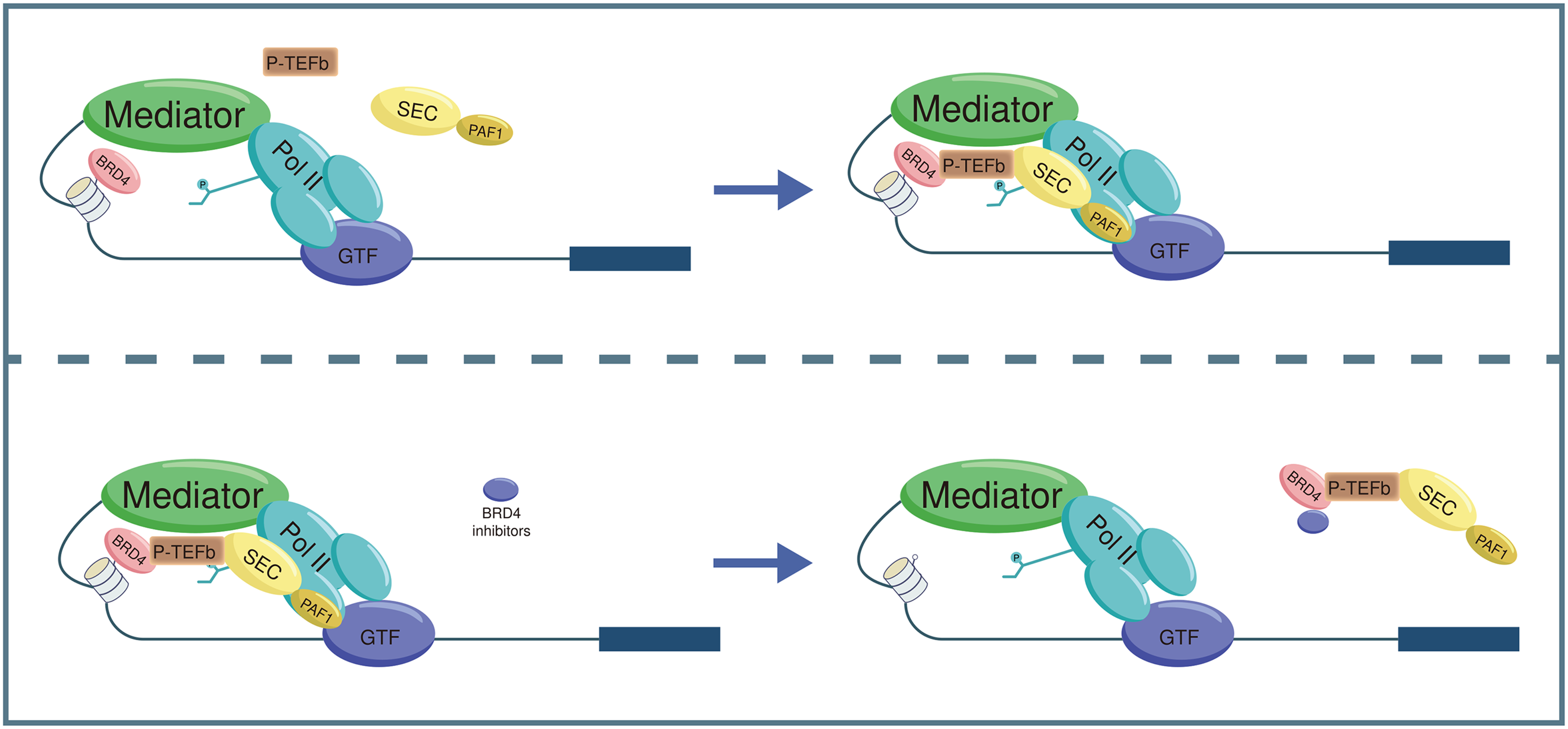
Schematic diagram of BRD4-mediated transcriptional activation and the mechanism of action of BET inhibitors. BRD4 recognizes and binds acetylated lysine residues on histone tails via its bromodomains, functioning as a scaffold that recruits transcriptional regulators such as P-TEFb to chromatin. BET inhibitors (BETi) competitively bind the BRD4 bromodomain, thereby disrupting its chromatin-binding and scaffolding functions and ultimately suppressing transcriptional activation.
Although best known for its transcriptional activation functions, BRD4 also contributes to transcriptional repression. BRD4 interacts with polycomb repressive complex 2 (PRC2) to promote deposition of the repressive histone mark H3K27me3, thereby silencing specific gene subsets. 26 Within the cardiovascular system, such repressive mechanisms may participate in the regulation of cell proliferation, differentiation, and other signaling pathways. BRD4 additionally interacts with histone-modifying enzymes such as G9a, modulating the expression of genes involved in cellular homeostasis processes including autophagy. 27 Dysregulated autophagy has been implicated in the pathogenesis of myocardial hypertrophy and heart failure, highlighting the broader physiological implications of BRD4 activity.
BRD4's pathological relevance in CVD has been extensively documented. It is recognized as a major driver of inflammatory responses across immune cells, cardiomyocytes, and fibroblasts. Through its HAT and kinase functions, BRD4 regulates the transcription of inflammatory mediators such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), thereby amplifying and sustaining inflammatory signaling.21,28 Furthermore, BRD4 serves as a key coactivator of the NF-κB signaling pathway and markedly enhances the transcription of NF-κB-dependent inflammatory genes. 29 These molecular actions provide critical mechanistic insights into the persistent inflammatory activity characteristic of CVDs such as atherosclerosis and myocarditis.
Molecular structure and function of BRD3
BRD3 is a key member of the BET protein family and functions as an epigenetic reader that regulates gene transcription by recognizing acetylated lysine residues on histone tails. 14 Structurally, BRD3 contains two highly conserved BRDs, BD1 and BD2, as well as a C-terminal ET domain. Each BRD consists of approximately 110 amino acids organized into 4 α-helices (αZ, αA, αB, αC) and 2 flexible loop regions (the ZA and BC loops), which together constitute the structural framework that enables selective binding to acetylated histones. 18
Functionally, BRD3 participates in both transcriptional activation and repression. Studies have demonstrated that BRD3 modulates cell proliferation and differentiation by regulating the expression of cell cycle–related genes such as p21, 30 a mechanism that may be particularly relevant to phenotypic switching in cardiovascular cells. Additionally, the colocalization of BRD3 with histone H3K18ac suggests that it plays an important role in shaping dynamic chromatin states and controlling the expression of genes implicated in pathological processes. 31
The activity of BRD3 is further refined through post-translational modifications, among which phosphorylation is especially critical. The nuclear tyrosine kinase TYRO3 has been shown to phosphorylate BRD3, modulating its chromatin-binding affinity and downstream transcriptional regulatory functions. 32 This phosphorylation not only strengthens BRD3–histone interactions but may also influence BRD3-mediated gene reprogramming during cardiovascular stress responses.
Pathologically, BRD3 has been implicated in a range of disease conditions. It is closely associated with β-cell dysfunction in metabolic diseases such as type 2 diabetes, 33 indicating a broader role in metabolic regulatory pathways. These mechanisms may also contribute to the development of CVDs, including atherosclerosis and myocardial reperfusion injury, suggesting that BRD3 warrants further investigation as a potential mediator of cardiovascular pathology.
Molecular structure and function of BRD2
BRD2 is a crucial member of the BET protein family and shares a high degree of structural similarity with BRD3 and BRD4. Structural studies have shown that folding of the second BRD, BRD2(2), follows a hierarchical mechanism in which the C-terminal region forms the initial structural core, while the N-terminal region stabilizes at later stages. 34 Additionally, the ZA loop of BRD2 demonstrates notable conformational flexibility in its interaction with acetylated lysine residues, a property thought to underpin its functional versatility in chromatin binding. 35
BRD2 recognizes acetylated histones through its tandem BRDs and mediates both intra- and internucleosomal interactions, thereby contributing uniquely to chromatin organization and transcriptional regulation. In genome organization, BRD2 participates in the dynamic balance between loop extrusion and chromatin compartmentalization by binding chromatin via its BRDs and low-complexity domain (LCD), promoting spatial mixing of active chromatin regions.36,37 BRD2 also plays a critical role in DNA damage repair and adaptive immune responses. During B-cell antibody class switch recombination (CSR), BRD2 stabilizes the cohesin-loading protein NIPBL, preventing aberrant recombination events and ensuring proper DNA repair pathway selection. 38 BRD2 frequently acts in concert with BRD3, with the two proteins demonstrating synergistic or antagonistic regulatory effects on specific promoters, thereby fine-tuning gene expression. For example, during embryonic stem cell differentiation, BRD2 facilitates promoter “pre-priming” to regulate gene expression, whereas BRD3 contributes to the subsequent state transitions. 36
At the pathological level, dysregulation of BRD2 is associated with multiple disease processes. In immune-inflammatory responses, BRD2 modulates the expression of inflammation-related transcription factors in macrophages through recognition of chromatin regions enriched with H3K18ac. 17 In B cells, BRD2 enhances DNA double-strand break repair by stabilizing NIPBL, a mechanism essential for maintaining genomic stability.28,39 These functions not only support immune system integrity but may also contribute to cellular homeostasis within the cardiovascular system, highlighting the need to further explore BRD2's involvement in CVD pathogenesis.
Molecular structure and function of BRDT
BRDT is a BET family member distinguished by its highly tissue-specific expression profile. Structurally, BRDT contains two tandem BRDs, BD1 and BD2, which recognize acetylated histone residues with high specificity. BRDT is predominantly and robustly expressed in testicular tissue, where it plays an essential role in spermatogenesis. 40 Functional studies indicate that the two BRDs of BRDT exhibit distinct regulatory properties, with selective inhibition of the BD1 domain markedly impairing spermatogenic progression. 15 This domain specificity has positioned BRDT as a promising candidate for the development of non-hormonal contraceptive strategies.
In summary, BRD2, BRD3, and BRD4 collectively regulate chromatin architecture, transcriptional activation, and gene expression programs governing cell proliferation, apoptosis, and metabolic homeostasis. The functional diversity and mechanistic complexity of these BET proteins underscore their importance in maintaining normal cellular physiology and provide a strong conceptual foundation for their exploration as therapeutic targets in CVD.27,41
BET family in CVD
Epidemiological and genetic association of BET proteins in CVDs
Over the past decade, genome-wide association studies (GWAS) have identified a broad spectrum of genetic variants linked to CVDs and related traits. Notably, most of these variants reside in non-coding genomic regions, often far from protein-coding loci, complicating efforts to identify causal genes and decipher functional mechanisms. Many cardiovascular GWAS signals are enriched within regulatory elements marked by histone modification marks or embedded within distinct chromatin states.42,43 Approximately 60% of long-range chromatin interactions exhibit partial overlap across cell types, yet more than 99% display clear lineage- or cell type–specific signatures. 44 These findings indicate that during cellular differentiation, higher-order chromatin architecture undergoes coordinated remodeling, dynamically shaping transcriptional programs essential for cardiovascular development and CVD.
Although advances in cardiovascular genetics have uncovered new pathways and therapeutic targets, genomic inheritance represents only one component of an individual's overall disease risk. Epigenetic modifications—heritable alterations in gene expression that occur without changes to the DNA sequence—constitute a parallel regulatory layer with substantial relevance to cardiovascular pathobiology. 45 Integrating genetic and epigenetic information is expected to enhance individual risk prediction and enable the development of more precise therapeutic strategies. BET proteins play pleiotropic roles across mechanistic and clinical domains of CVD. For example, in vascular calcification, the LIM-domain protein FHL5 cooperates with cAMP response element-binding protein (CREB) to activate pro-calcification genes such as RUNX2, whereas BETi have the capacity to disrupt CREB–FHL5–BET complexes and attenuate calcification. 46 BET family members also contribute to the maintenance of endothelial heterogeneity. FRW peptide motifs regulated by BET proteins can selectively target endothelial junctions within cerebral vessels, offering potential molecular tools for high-precision imaging and treatment of blood–brain barrier–related cardiovascular complications. 47
Genetic studies further support an association between BET family variation and cardiovascular risk. In a cohort of 666 hypertensive patients and 232 normotensive controls, a significant association was observed between the BRD4 single nucleotide polymorphism (SNP) rs4808278 and pulse pressure (PP). Individuals harboring the TT genotype exhibited a markedly lower risk of elevated PP than AA/AT carriers. Moreover, a significant interaction with diabetes status was identified, suggesting that diabetes may potentiate the influence of BRD4 genetic variation on PP. 48 Taken together, these findings suggest that BRD4 polymorphisms may contribute to genetic susceptibility to CVD.
Cardiovascular risk factors and the BET family
In addition to inherited susceptibility, modifiable risk factors such as hypertension, obesity, and smoking substantially contribute to the development of CVD. Environmental exposures—including air pollution, dietary patterns, and lifestyle factors—further modulate cardiovascular risk by shaping gene expression programs through epigenetic mechanisms that regulate chromatin states and transcriptional activity.
Aging is characterized by a progressive decline in physiological function and an increased predisposition to age-related diseases, including CVDs. BRD4 has been implicated in cellular aging processes and is closely linked to age-associated cardiovascular pathophysiology.49–51 Inhibition of BRD4 appears to exert anti-apoptotic effects in specific contexts. In ischemia–reperfusion (I/R) injury models, treatment with the BETi JQ1 or BRD4 knockdown reduces the expression of endoplasmic reticulum stress markers and pro-apoptotic proteins. Similar protective effects have been reported in cellular hypoxia–reoxygenation (H/R) models. 52 Furthermore, BRD4 expression is upregulated in chronic obstructive pulmonary disease (COPD), and its knockdown decreases cigarette smoke extract-induced apoptosis in BEAS-2B cells, 43 suggesting a broader role for BET proteins in stress responses spanning the cardiovascular and respiratory systems.
Obesity, a major contributor to cardiovascular risk, is also closely associated with BET protein activity. In diet-induced obesity mouse models that recapitulate features of human metabolic syndrome, treatment with the BRD4 inhibitor JQ1 reduces adipose tissue mass and preserves skeletal muscle mass and function. 53 Mechanistically, BRD4 in adipose tissue macrophages promotes expression of the antilipolytic factor Gdf3 in a peroxisome proliferator-activated receptor gamma (PPARγ)–dependent manner, suppressing lipase activity (e.g. adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL)), reducing lipid breakdown, and facilitating lipid accumulation. BRD4-deficient mice demonstrate enhanced fat oxidation, improved insulin sensitivity, and lower circulating triglyceride (TG) and low-density lipoprotein cholesterol levels. In addition, BRD4 augments adipose inflammation and systemic insulin resistance by regulating inflammatory cytokines such as TNF-α and IL-6, thereby contributing to hyperlipidemia. 54 In high-fat diet models, BETi such as apabetalone and JQ1 ameliorate obesity-related metabolic abnormalities and attenuate endothelial pro-inflammatory phenotypes, indicating their potential to reduce cardiovascular risk in obese individuals. 55
Hypertension represents another major cardiovascular risk factor in which BET proteins play a mechanistic role. Studies using spontaneously hypertensive rats (SHRs) demonstrate that JQ1 treatment downregulates key mediators associated with vascular dysfunction, including angiotensin II (Ang II), endothelin-1 (ET-1), malondialdehyde (MDA), IL-6, and TNF-α, and increases levels of nitric oxide (NO), nitric oxide synthase (NOS), and superoxide dismutase (SOD). JQ1 also enhances endothelial nitric oxide synthase (eNOS) activity in aortic tissue and decreases superoxide and nitrite accumulation. 56 These findings support a central role for BRD4 in blood pressure regulation and hypertensive vascular pathology.
Collectively, current evidence suggests that BET proteins integrate genetic, environmental, and metabolic cues, positioning them as critical regulators of cardiovascular risk and promising targets for refined cardiovascular risk stratification.
The mechanism of BET protein family involvement in CVD
The BET protein family acts as a central epigenetic regulatory system that governs multiple molecular processes fundamental to the initiation and progression of CVD. BET family members function as integrative feedback hubs across diverse signaling pathways, including inflammatory amplification, fibrotic remodeling, dysregulated energy metabolism, aberrant cell proliferation, epithelial–mesenchymal transition (EndoMT), and ferroptosis, thereby amplifying or attenuating pathological gene expression programs.57,58 The following sections systematically examine these core pathways to delineate how BET proteins act in a coordinated and network-level manner to drive cardiovascular pathophysiology (Figure 3).
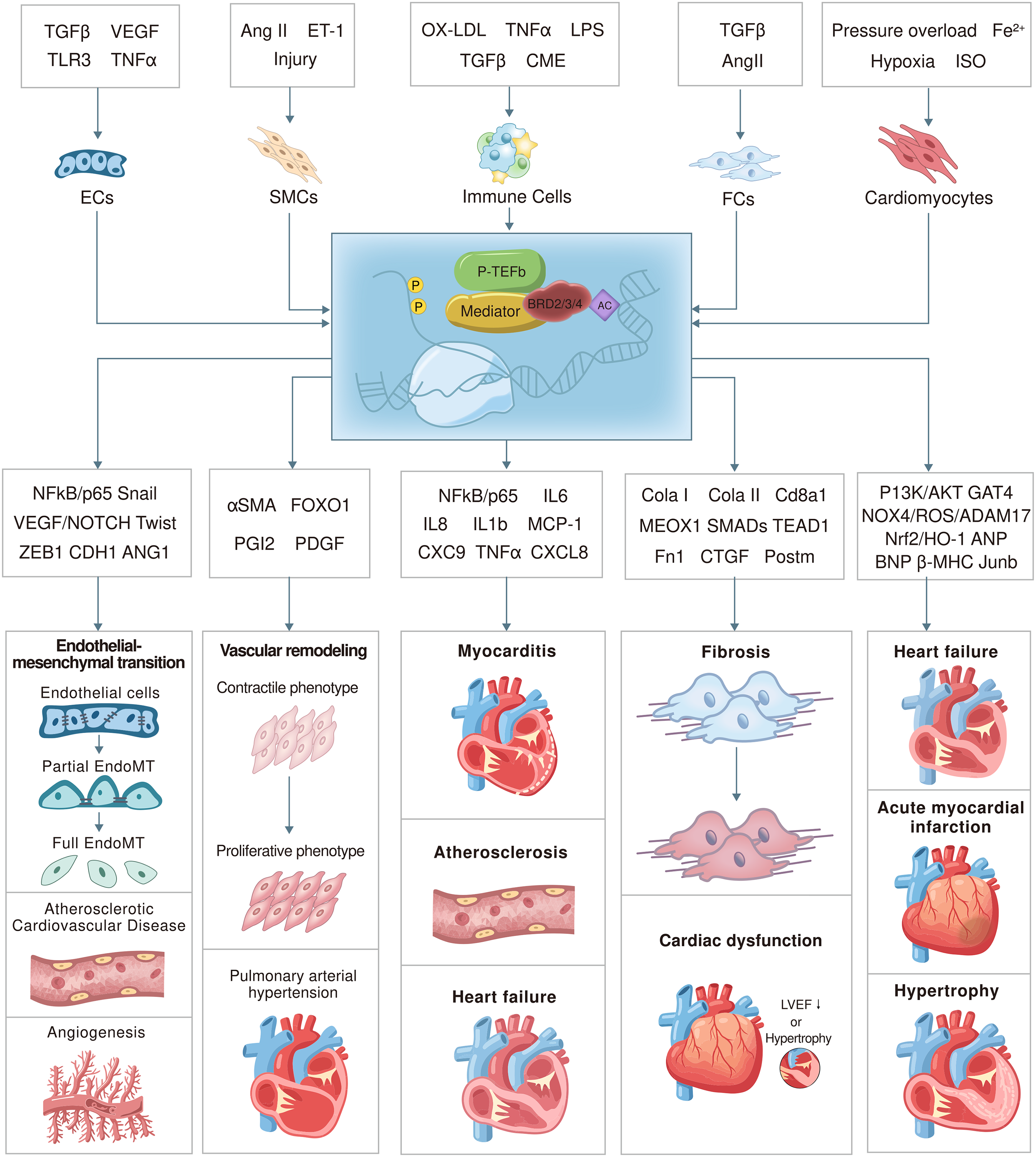
Cell-specific regulatory mechanisms of BET proteins in the cardiovascular system. This figure illustrates the regulatory roles of BET proteins in endothelial cells (EC), smooth muscle cells (SMCs), immune cells (such as monocytes/macrophages; Mo/MP), fibroblasts (FCs), and cardiomyocytes. It highlights their responses to pathological stimuli, key target genes, and associated cardiovascular disease–related phenotypes.
BET and cardiovascular inflammation
The BET protein family plays a central role in the inflammatory processes underlying a wide range of CVDs, and extensive experimental evidence has validated its involvement in immune activation and inflammatory signaling. BET proteins promote cardiovascular pathology largely by enhancing the expression of inflammatory mediators and facilitating immune cell recruitment to diseased tissues. A key regulatory circuit is the TFEB–P300–BRD4 molecular axis, which is critically involved in the progression of atherosclerosis. Oxidative low-density lipoprotein (oxLDL) activates this pathway, impairing macrophage autophagy and intensifying inflammatory responses. 59 Mechanistically, BRD4 augments nuclear factor kappa B (NF-κB) transcriptional activity by recruiting cyclin-dependent kinase 9 (CDK9), thereby increasing the expression of leukocyte adhesion molecules and multiple pro-inflammatory cytokines. 60 In pulmonary vascular endothelial cells and smooth muscle cells from patients with pulmonary arterial hypertension (PAH), BRD2 and BRD4 synergistically enhance NF-κB p65 enrichment at IL-6 and CXCL8/IL-8 promoter regions, promoting robust transcription of these inflammatory genes. 61 Importantly, BET proteins exhibit context-dependent and cell type–specific functions. In a hypoxia model using rat H9C2 cardiomyocytes, BRD2 has a protective role, activating the Nrf2/HO-1 signaling axis and attenuating H/R-induced myocardial injury. 62
Macrophage phenotype regulation represents another critical mechanism through which BET proteins modulate cardiovascular inflammation. Under physiological conditions, BRD4 activity promotes differentiation into pro-inflammatory macrophages (MHC-IIhigh). Inhibition of BRD4 instead shifts macrophages toward a reparative phenotype (MHC-IIlow), characterized by suppressed NF-κB signaling and reduced MHC-II expression, resulting in diminished inflammation and improved cardiac remodeling. 63 In the coronary microembolism (CME), BRD4 protein abundance is markedly elevated despite unchanged mRNA levels, indicating regulation at the post-translational level. Serine/arginine-rich splicing factor 1 (SRSF1) reduces BRD4 O-GlcNAcylation by modulating ENPP3 splicing and lowering uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) availability. Mutation of the S484 site (S484R) prevents BRD4 glycosylation, leading to protein accumulation and amplified inflammatory signaling. 64 BRD3 also contributes to innate immune regulation, promoting recruitment of the IRF3/p300 complex to the Ifnb1 promoter and increasing histone H3/H4 acetylation, which enhances type I interferon production. In BRD3-deficient RAW 246.7 macrophages, virus-induced interferon-gamma (IFN-γ) expression is markedly suppressed. 65
Beyond macrophages, BET proteins influence inflammation-related pathological processes in several additional cell types. In atherosclerosis, BRD4 forms a transcriptional complex with NF-κB, remodels chromatin through its histone acetyltransferase activity, and drives vascular smooth muscle cell phenotypic switching and vascular remodeling. 66 In cardiomyocytes, BRD4 activates the Nox4/ROS/ADAM17 signaling cascade, intensifying oxidative stress, promoting inflammatory responses, and contributing to cardiac hypertrophy. 63 Conversely, BRD4 inhibition significantly upregulates Nrf2 and HO-1 expression, suggesting that BRD4 may impair cellular antioxidant defenses by suppressing the Nrf2/HO-1 axis. 67 BET proteins also modulate the TGF-β1/SMAD signaling pathway, influencing fibrosis-related gene expression and playing an important role in oxidative stress-induced fibrotic remodeling.
BET and fibrosis
BET proteins play a critical regulatory role in myocardial fibrosis, influencing fibroblast activation, extracellular matrix deposition, and maladaptive cardiac remodeling. Inhibition of BRD4 has been shown to markedly suppress the activity of fibrosis-associated super-enhancers, thereby attenuating fibroblast activation and reducing myofibroblast differentiation through modulation of key transcription factors such as MEOX1. 68 This epigenetic reprogramming contributes to substantial improvement in myocardial fibrotic remodeling. In pressure overload (TAC)- or Ang II-induced cardiac fibrosis models, TEAD1 expression is significantly elevated in cardiac fibroblasts. TEAD1 directly interacts with BRD4 and binds the promoter of the Wnt4 gene, synergistically enhancing Wnt4 transcriptional activity. Upregulated Wnt4 activates canonical Wnt/β-catenin signaling, driving the expression of fibrosis-related genes including ACTA2 (α-SMA), collagen I, and collagen III. This signaling cascade promotes myofibroblast proliferation, collagen deposition, and expansion of the myocardial interstitial matrix, ultimately contributing to progressive cardiac fibrosis. 69
During heart failure, activation of the TGF-β signaling pathway increases BRD4 chromatin occupancy through p38 mitogen-activated protein kinase (MAPK)–mediated regulation. This enhances BRD4 binding to the Sertad4 gene regulatory region, upregulating Sertad4 expression and promoting fibroblast activation and fibrotic progression. 70 Evidence from isoproterenol-induced cardiac hypertrophy models further demonstrates that BRD4 contributes to maladaptive remodeling by promoting expression of hypertrophic marker genes such as atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP), and beta-myosin heavy chain (β-MHC) through regulation of H3K122 acetylation and RNA polymerase II phosphorylation, leading to structural and functional cardiac abnormalities. 71 Additionally, BRD4 enhances TGF-β1/Smad2/3 pathway activity, driving excessive proliferation and collagen synthesis in valvular fibroblasts. This mechanism intensifies valvular extracellular matrix remodeling and exacerbates cardiac valve fibrosis. 19
BET and metabolic abnormalities
BET proteins contribute to the onset and progression of CVDs by regulating myocardial energy metabolism and coordinating transcriptional networks essential for mitochondrial function. BRD4 deficiency disrupts metabolic homeostasis by inducing dissociation of the nuclear receptor ESRRα from the promoter region of its target genes, leading to impaired activity of mitochondrial electron transport chain complexes I, III, and IV as well as key enzymes of the tricarboxylic acid (TCA) cycle. These metabolic defects culminate in reduced adenosine triphosphate (ATP) production and contribute to the development of dilated cardiomyopathy. 57 As a central cardiac transcription factor, GATA4 co-regulates mitochondrial function-related genes such as PPARGC1A (PGC-1α) and PPARGC1B (PGC-1β) in coordination with BRD4. Loss of BRD4 results in significant downregulation of these genes, impairing mitochondrial biogenesis, oxidative phosphorylation, and ultimately myocardial contractile performance. 58
Genome-wide analyses indicate that BRD2 plays a broad regulatory role in metabolic gene networks, particularly those governing the TCA cycle. In cardiomyocytes stimulated with the β-adrenergic agonist isoproterenol, BRD2 overexpression markedly increases the expression of TCA cycle genes, enhances cardiac metabolic activity, and drives the expression of hypertrophic markers alongside increases in cell surface area.6,72 Additionally, BRD4 regulates key genes involved in fatty acid metabolism, including HADH, ACSL1, and ACAA2, thereby modulating mitochondrial fatty acid oxidation, influencing lipid peroxide accumulation, and altering myocardial sensitivity to ischemia–reperfusion injury. 73 BRD4 further promotes H3K122 acetylation and RNA polymerase II phosphorylation, enhancing transcription of hypertrophic marker genes and Nox4, a critical mediator of oxidative stress. These actions drive pathological metabolic remodeling and accelerate the progression of myocardial hypertrophy. 74
BET and cell proliferation
The BET protein family exerts multilayered regulatory control over cell proliferation within the cardiovascular system, influencing vascular remodeling, endothelial function, and myocardial survival. In PAH, aberrant histone acetylation leads to dysregulated expression of genes that drive vascular structural remodeling. BRD4 directly promotes transcription of key pro-proliferative mediators such as ET-1 and platelet-derived growth factor (PDGF) by recognizing specific histone acetylation marks, recruiting CDK9, and assembling transcriptionally active complexes. Concurrently, BRD4 suppresses anti-proliferative genes such as eNOS and prostacyclin synthase (PGIS) through epigenetic repression, thereby shifting the balance toward smooth muscle cell proliferation and resistance to apoptosis. From a cell cycle perspective, BRD4 facilitates transition from quiescence into active proliferation by upregulating G1-phase regulators, including cyclin D1 and CDK4. Additionally, through modulation of Bcl-2 family protein ratios, BRD4 inhibits apoptosis, jointly driving pathological vascular remodeling. 75
In endothelial cells, the FAM222A (C12orf34) gene has emerged as an essential regulator of proliferation. FAM222A controls endothelial cell growth, migration, and tube formation by finely modulating the balance between vascular endothelial growth factor (VEGF) and Notch signaling. BET inhibition with JQ1 significantly induces FAM222A expression, indicating that BRD4 normally represses FAM222A transcription by occupying its promoter under basal conditions. When BRD4 is inhibited, FAM222A expression increases, promoting endothelial proliferation and angiogenic capacity through activation of VEGF signaling and coordinated modulation of the Notch pathway.76,77
In myocardial protection, the PI3K/AKT axis serves as a central survival pathway during reperfusion, with its activation critically determining cardiomyocyte fate. BRD4 influences this pathway by modulating the phosphorylation state of the PI3K p85 regulatory subunit. Through recruitment of histone deacetylases (HDACs) and specific phosphatases, BRD4 reduces PI3K activity and suppresses downstream AKT activation, a mechanism that may exacerbate apoptosis during myocardial ischemia–reperfusion injury. 78
During vascular injury repair, mechanical injury establishes a local inflammatory and growth factor–rich microenvironment that markedly upregulates BRD4 expression in vascular smooth muscle cells. Elevated BRD4 binds directly to the transcription factor FOXO1 via its bromodomain, suppressing FOXO1 transcriptional activity and downregulating the cyclin-dependent kinase inhibitor p21 (CDKN1A). Reduced p21 expression releases the inhibitory constraint on the G1/S transition, thereby enhancing smooth muscle cell proliferation. Simultaneously, BRD4 promotes smooth muscle cell migration by upregulating matrix metalloproteinases MMP2 and MMP9. These combined actions facilitate smooth muscle migration toward the vascular lumen and drive abnormal proliferative expansion of the neointima, ultimately resulting in pathological luminal stenosis. 79
BET and EndoMT
EndoMT is a critical biological process in which endothelial cells acquire mesenchymal characteristics, contributing substantially to vascular remodeling across multiple CVD states. Emerging evidence demonstrates that BRD4 is a key regulator of EndoMT in both human and rodent endothelial cells. 80 Specifically, BRD4 controls the expression of the transcription factor ZEB1 through its second BRD (BD2). ZEB1 is a master regulator of EndoMT whose expression level determines the extent of endothelial–mesenchymal phenotypic transition. In TGF-β1-induced EndoMT models, BRD4 knockdown completely abolishes the transition process, underscoring its essential role in EndoMT signaling. 81
In pathological vascular remodeling, BET proteins shape vascular architecture through dual mechanisms: they directly regulate smooth muscle cell proliferation and migration, and they modulate endothelial cell function by governing the EndoMT process. BRD4-specific inhibitors have been shown to markedly suppress EndoMT and improve vascular function and structural integrity in models of vein graft failure and atherosclerosis. Notably, during the adaptive remodeling process of transplanted veins, BRD4—particularly through its BD2 domain—serves as a central driver of pathological remodeling via EndoMT-dependent mechanisms. 81
BET and ferroptosis
BET proteins, particularly BRD4, play a central regulatory role in ferroptosis—an iron-dependent, lipid peroxidation–driven form of programmed cell death that contributes substantially to myocardial ischemia–reperfusion injury. Evidence from studies of the natural compound Wedelolactone (Wed) highlights the mechanistic involvement of BRD4 in this process. Wed reduces myocardial infarct size, improves ventricular function, and reverses ferroptosis-related molecular changes in a dose-dependent manner. Mechanistic analyses have revealed that Wed exhibits high affinity for the RNA methyltransferase METTL3 and suppresses both its expression and enzymatic activity. By inhibiting METTL3, Wed prevents m6A methylation of BRD4 mRNA, leading to reduced post-transcriptional stabilization of BRD4 and consequent downregulation of BRD4 protein abundance. This attenuation of BRD4 expression effectively inhibits ferroptosis, establishing the METTL3–m6A–BRD4 regulatory axis as a key determinant of cardiomyocyte survival during myocardial injury. 82
In parallel, BRD4 directly modulates cellular metabolic status to influence susceptibility to ferroptosis. BRD4 binds the promoter region of the NAMPT gene and recruits the HDAC/CoREST transcriptional repression complex, thereby suppressing NAMPT transcription. NAMPT is the rate-limiting enzyme in nicotinamide adenine dinucleotide (NAD)+ biosynthesis, and its downregulation causes a substantial reduction in intracellular NAD+ levels. NAD+ depletion impairs the deacetylase activity of SIRT1, preventing activation and nuclear translocation of the antioxidant transcription factor Nrf2. As a result, antioxidant defense genes, including GPX4, HO-1, and SLC7A11, are broadly downregulated. The diminished antioxidant capacity promotes accumulation of lipid peroxides, collapse of mitochondrial membrane potential, and loss of cristae integrity, ultimately culminating in irreversible ferroptotic death of cardiomyocytes. 83
Therapeutic targeting: Advances in BETi
Overview of BETi in preclinical and clinical research
In recent years, BETi have emerged as a promising class of epigenetic therapeutics with substantial potential for CVD intervention. By selectively preventing BET proteins from binding acetylated lysine residues on histones, these agents disrupt BET-mediated transcriptional programs that drive pathological gene expression. This targeted mechanism offers a novel strategy for modulating key processes involved in cardiovascular pathology, including inflammation, fibrosis, metabolic dysregulation, and adverse remodeling (Table 1).19,74,84–89
Summary of drugs targeting BET proteins in cardiovascular field
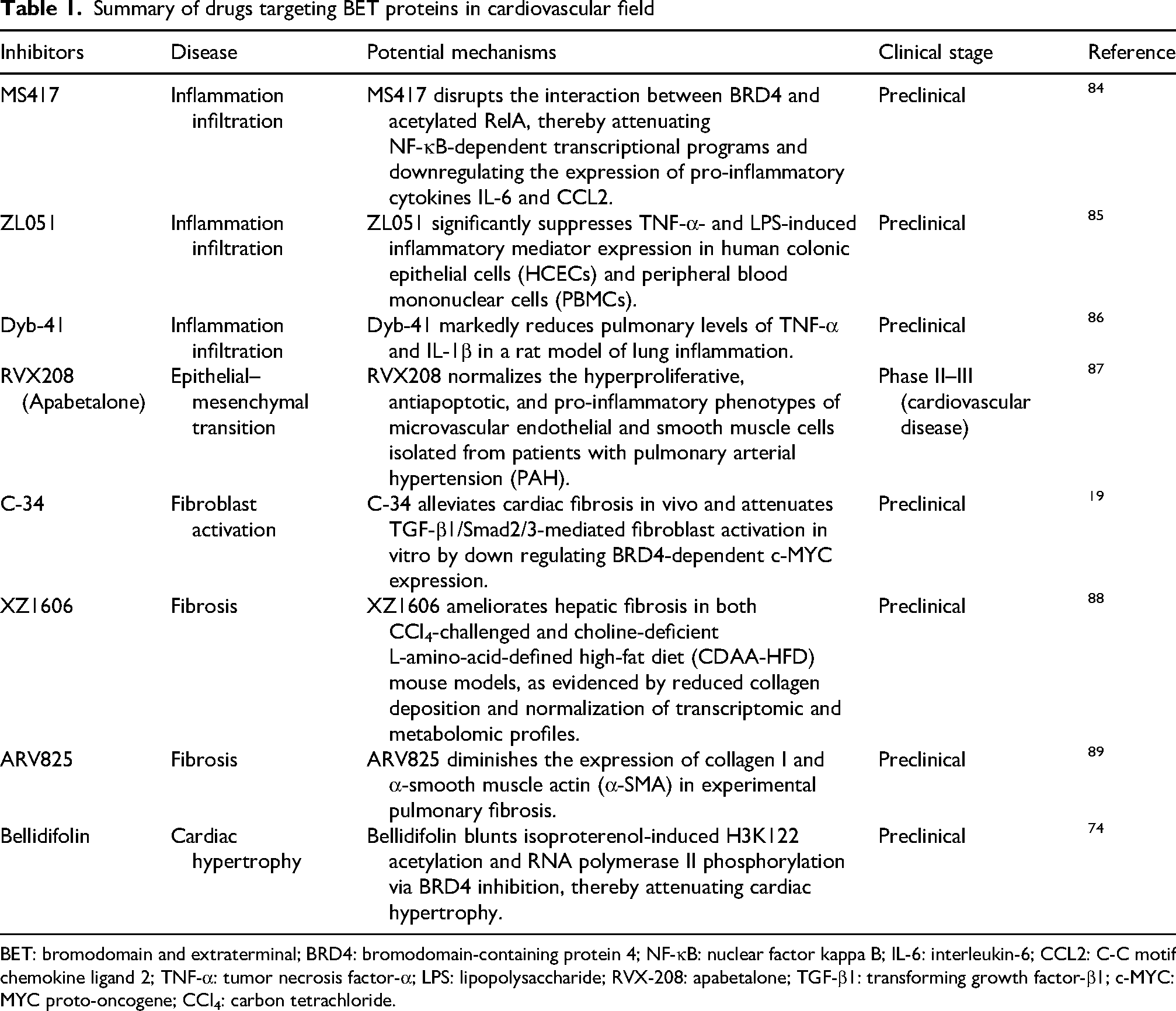
BET: bromodomain and extraterminal; BRD4: bromodomain-containing protein 4; NF-κB: nuclear factor kappa B; IL-6: interleukin-6; CCL2: C-C motif chemokine ligand 2; TNF-α: tumor necrosis factor-α; LPS: lipopolysaccharide; RVX-208: apabetalone; TGF-β1: transforming growth factor-β1; c-MYC: MYC proto-oncogene; CCl4: carbon tetrachloride.
Among currently available BET inhibitors, apabetalone (RVX-208) is the most extensively studied in cardiovascular populations. Clinical trials have demonstrated that apabetalone significantly reduces major adverse cardiovascular events and improves liver fibrosis in patients with type 2 diabetes and acute coronary syndrome.90,91 Additionally, in patients with PAH, apabetalone has been shown to improve pulmonary vascular resistance and right ventricular function, with a favorable safety and tolerability profile. 92
However, the phase III clinical trial of apabetalone did not meet its primary endpoint. Consequently, the trial protocol precluded formal statistical testing of secondary endpoints, limiting the interpretation of potential secondary benefits. Furthermore, apabetalone did not demonstrate significant improvements in traditional cardiovascular biomarkers, providing limited biological support for its therapeutic efficacy. Overall, the phase III trial results were not sufficiently robust to support regulatory approval. In addition, the study population was restricted to specific patient groups (recent acute coronary syndrome, type 2 diabetes, low high-density lipoprotein cholesterol) and consisted predominantly of White participants, limiting the generalizability of the findings. Finally, with respect to safety, the incidence of elevated liver enzymes and the rate of adverse event-related treatment discontinuation were higher in the apabetalone group than in the placebo group, indicating potential safety concerns. Therefore, apabetalone has not been approved by regulatory authorities for cardiovascular indications, and the development of safer and more selective BETi with improved efficacy remains an important objective.
Beyond apabetalone, several other BETi have shown potential therapeutic relevance for CVDs. Compounds such as I-BET762 and CPI-0610 have been evaluated primarily in oncology; however, their ability to modulate inflammatory and fibrotic pathways suggests broader applicability to cardiovascular conditions. Notably, pelabresib (CPI-0610) has demonstrated a favorable safety profile in clinical studies of myelofibrosis, 93 supporting the rationale for exploring its use in cardiovascular indications. Collectively, these data highlight the expanding therapeutic landscape of BETi and underscore their potential to transition from cancer therapeutics to clinically valuable agents for CVD management.
Therapeutic effects and next-generation BETi
The therapeutic effects of BETi in CVD models as well as the distinguishing characteristics of next-generation BETi are summarized in Table 1. In general, BETi attenuate inflammation, fibrosis, and myocardial injury through pathways involving NF-κB, TGF-β/Smad, Nrf2/HO-1, and ferroptosis-related signaling cascades (Figure 4). Emerging strategies to improve selectivity and safety include domain-selective agents (e.g. the BD2-selective inhibitor apabetalone), BRD4-selective compounds (e.g. C-34), PROTAC degraders (e.g. XZ1606 and ARV825), biomimetic nanodelivery systems (e.g. PM&EM/JQ1 nanoparticles), and natural product-derived inhibitors (e.g. bellidifolin and Wed). Table 1 provides detailed compound-specific mechanisms, disease contexts, experimental models, clinical stages, and references.
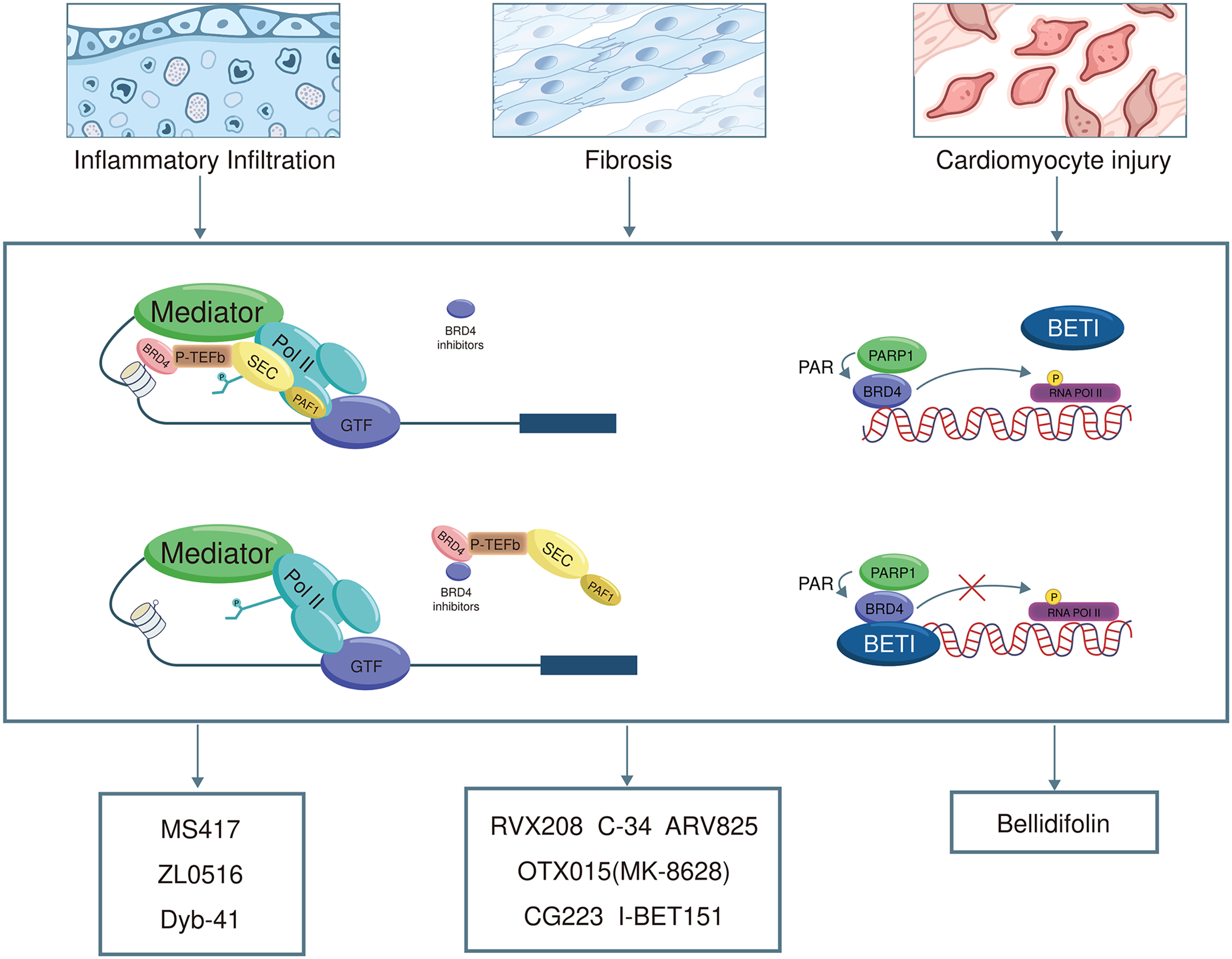
BET inhibitors effective in cardiovascular disease.
Discussion
The development of CVD involves complex epigenetic regulation by BET proteins, yet many mechanistic and translational questions remain unresolved. Previous reviews have primarily focused on BRD4 as a master regulator of NF-κB-driven inflammation and cardiac hypertrophy, 94 whereas the distinct and overlapping roles of BRD2, BRD3, and the testis-restricted BRDT have received less attention. In contrast, the present review systematically compares all four BET family members across multiple pathological processes, including inflammation, fibrosis, metabolic reprogramming, aberrant cell proliferation, EndoMT, and ferroptosis. This broader scope represents a key novelty of this review. Furthermore, whereas previous reviews have summarized the antifibrotic effects of pan-BETi, 95 this review integrates recent advances in domain-selective inhibitors (e.g. the BD2-selective inhibitor apabetalone), PROTAC degraders (e.g. XZ1606 and ARV825), biomimetic nanodelivery systems (e.g. PM&EM/JQ1 nanoparticles), and natural product-derived inhibitors (e.g. bellidifolin and Wed), which have not been comprehensively covered in the cardiovascular literature. By mapping specific pathological mechanisms to individual BET family members and their BRDs, this review provides a conceptual framework that may help explain the heterogeneity in clinical responses to BET inhibition. However, several limitations should be acknowledged. First, most of the evidence discussed is derived from preclinical animal models and in vitro studies, and large-scale clinical validation is lacking, particularly regarding the proposed role of BET proteins as biomarkers. Second, this is a narrative review without formal meta-analysis, and publication bias cannot be excluded. Third, the long-term safety of BETi, especially with respect to testis-specific BRDT inhibition and potential off-target effects on spermatogenesis, requires further investigation. Future research should prioritize cell type–specific epigenetic profiling using single-cell multiomics approaches, the development of next-generation BETi with improved selectivity and reduced toxicity, and refined patient stratification based on BET pathway activation signatures. Addressing these challenges will be essential for translating BET-targeted strategies from experimental findings into precision cardiovascular therapies.
Concluding remarks
BET proteins, particularly BRD2, BRD3, and BRD4, are key epigenetic regulators in CVD, contributing to inflammation, fibrosis, metabolic reprogramming, and other pathological processes. Preclinical studies have demonstrated the anti-inflammatory, antifibrotic, and cardioprotective effects of BETi; however, limited tissue specificity, potential systemic toxicity, and insufficient clinical validation continue to hinder their clinical translation. Future research should focus on elucidating cell type–specific mechanisms, developing next-generation inhibitors with improved selectivity and safety, evaluating combination therapeutic strategies, and refining patient stratification approaches. The potential utility of BET proteins as biomarkers also warrants further clinical investigation.
Footnotes
Acknowledgments
Authors’ contributions
Conceptualization: Yuan Wang, Yangkai Fan, and Kaixuan Zhang; Methodology: Yangkai Fan and Kaixuan Zhang; Formal Analysis: Yangkai Fan and Kaixuan Zhang; Visualization: Kaixuan Zhang and Yangkai Fan; Supervision: Yuan Wang and Yangkai Fan; Writing—Original Draft: Kaixuan Zhang; Writing—Review and revision: Yangkai Fan; Funding Acquisition: Yuan Wang and Yangkai Fan.
All authors read and approved the final version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Noncommunicable Chronic Diseases–National Science and Technology Major Project, Natural Science Foundation of Beijing Municipality, National Natural Science Foundation of China, Beijing Municipal Health Commission, (grant numbers: 2023ZD0503100, 7264239, 91939303, and 11000025T000003321643). This work was supported by the Noncommunicable Chronic Diseases–National Science and Technology Major Project (Grant No. 2023ZD0503100), the National Natural Science Foundation of China (NSFC) Major Program (Grant No. 91939303), the Beijing Municipal Health Commission (Grant No. 11000025T000003321643), and the Beijing Municipal Natural Science Foundation (Grant No. 7264239). The funders had no role in study design, data collection, analysis, interpretation, or the decision to submit for publication. All authors had full access to all data and accept final responsibility for publication.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
Not applicable.