
Abstract
Background:
Synovial sarcoma (SS) is molecularly defined by the pathognomonic SS18::SSX fusion, a master epigenetic driver. While NTRK gene fusions are high-priority tissue-agnostic targets for TRK inhibitors like larotrectinib, their co-occurrence with established lineage-defining drivers in sarcomas is exceptionally rare and presents a therapeutic paradox.
Case presentation:
A 78-year-old female was diagnosed with metastatic biphasic SS. Molecular profiling via targeted RNA sequencing identified a pathogenic PDE3A-NTRK2 fusion (Tier 1A) alongside the canonical SS18 rearrangement. Despite the presence of this usually actionable target, second-line treatment with larotrectinib resulted in rapid clinical deterioration and "explosive" tumor growth, signifying absolute primary resistance to TRK inhibition. The disease followed an aggressive course typical of chemorefractory SS, leading to the patient's death ten months after initial diagnosis.
Conclusion:
This case provides critical clinical evidence of the "driver versus passenger" phenomenon in precision oncology. It demonstrates that the epigenetic dominance of the SS18::SSX fusion can override the therapeutic relevance of a concurrent NTRK fusion. Our findings serve as a cautionary note: actionable targets detected via next-generation sequencing must be interpreted within the tumor’s canonical biological context, as lineage-defining translocations may render secondary kinase alterations therapeutically inert.
Introduction
Synovial sarcoma (SS) is an aggressive mesenchymal malignancy accounting for approximately 5–10% of all soft tissue sarcomas. While it predominantly affects adolescents and young adults, the disease can present at any age. 1 SS is molecularly defined by the pathognomonic t(X;18) chromosomal translocation, which generates the SS18::SSX fusion oncogene. This fusion protein acts as the primary oncogenic driver by hijacking the SWI/SNF chromatin-remodeling complex, leading to aberrant transcriptional reprogramming. 2 Standard management for advanced disease relies heavily on cytotoxic chemotherapy, specifically anthracycline and ifosfamide regimens, but the prognosis for metastatic patients remains poor. 3
In the era of precision oncology, neurotrophic tyrosine receptor kinase (NTRK) gene fusions have emerged as distinct, actionable targets. Larotrectinib, a highly selective tropomyosin receptor kinase (TRK) inhibitor, has demonstrated marked and durable antitumor activity in NTRK fusion-positive cancers. In a landmark integrated analysis of 55 patients, larotrectinib achieved an overall response rate of 75% regardless of tumor histology or patient age. 4 Consequently, the identification of an NTRK fusion typically provides a strong rationale for targeted therapy, particularly in chemorefractory settings.
Historically, SS18 rearrangements and NTRK fusions are considered mutually exclusive driver events in sarcoma pathology. The co-occurrence of both alterations creates a significant therapeutic dilemma regarding pathway dominance. Herein, we report a rare case of an elderly patient with biphasic SS harboring concurrent SS18 rearrangement and a PDE3A-NTRK2 fusion. We document the clinical course of rapid disease progression on larotrectinib, illustrating a "driver versus passenger" phenomenon where the epigenetic dominance of the SS18 fusion likely mediated primary resistance to targeted TRK inhibition.
Case presentation
A 78-year-old female presented with a rapidly enlarging, hard, and painful mass in her left leg. Magnetic resonance imaging of the lower extremity revealed a lobulated and heterogeneous mass in the proximal anterolateral compartment, measuring approximately 15 x 7.5 x 6 cm. The lesion demonstrated intense contrast enhancement with areas of internal hemorrhage and necrosis. An excisional biopsy confirmed the diagnosis of a malignant mesenchymal tumor with morphological and immunohistochemical features consistent with biphasic SS (Figure 1). Molecular characterization was performed to guide therapeutic management. Fluorescence in situ hybridization analysis was positive for the pathognomonic SS18 rearrangement (19q11.2), with a break-apart pattern observed in 80% of cells. Concurrently, a targeted RNA next-generation sequencing (NGS) panel identified a PDE3A-NTRK2 gene fusion, classified as a Tier 1A pathogenic variant. Pan-TRK immunohistochemistry showed diffuse, moderate-to-strong cytoplasmic staining, confirming TRK fusion protein expression.
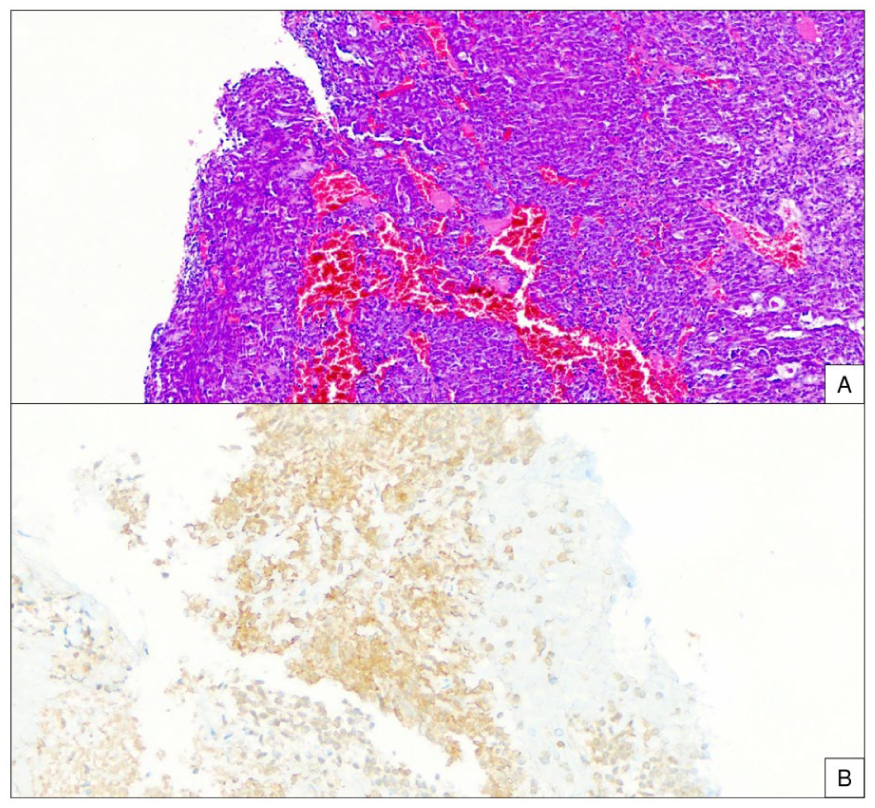
Histopathological and immunohistochemical features of the synovial sarcoma. (A) The tumor is composed of monotonous spindle cells with a high rate of mitotic activity (H&E stain, x200). (B) Corresponding immunohistochemistry shows strong and diffuse cytoplasmic staining for Pan-TRK, confirming protein expression secondary to the NTRK fusion (Ventana anti-TRK [EPR17341] antibody, x200).
Initial staging with 18F-fluorodeoxyglucose positron emission tomography–computed tomography (18F-FDG PET-CT) demonstrated a hypermetabolic primary tumor (SUVmax: 15.9) with invasion of adjacent musculature and fibular destruction. Additionally, the scan revealed mildly hypermetabolic inguinal and parailiac lymph nodes and multiple non-FDG-avid bilateral pulmonary nodules, consistent with metastatic disease. The patient commenced first-line systemic chemotherapy with doxorubicin (every 21 days). Following the third cycle, an interim PET-CT indicated a mixed response; while the primary tumor showed partial metabolic regression, the patient exhibited systemic disease progression, evidenced by dimensional and metabolic enlargement of nodal metastases and the appearance of new pulmonary nodules.
Given the disease progression and the identification of a potentially actionable NTRK fusion, the patient was switched to second-line targeted therapy with larotrectinib (100 mg twice daily). Over a period of two months, the primary tumor exhibited rapid, explosive growth, evolving into a large exophytic, fungating mass with extensive necrosis and ulceration (Figure 2). Restaging 18F-FDG PET-CT confirmed unequivocal disease progression, demonstrating marked metabolic and dimensional increase in both the primary lesion and systemic metastases. This progression was complicated by severe malodorous discharge and intractable hemorrhage, resulting in grade 4 anemia (hemoglobin nadir: 6 g/dL) necessitating multiple red blood cell transfusions. Palliative amputation was recommended but declined by the patient. She subsequently underwent a short course of palliative radiotherapy (5 fractions) to the left leg, achieving only temporary hemostasis. Systemic therapy was switched to third-line ifosfamide; however, the patient withdrew after two cycles due to rapid clinical deterioration and poor performance status. The patient died approximately ten months after the initial diagnosis.
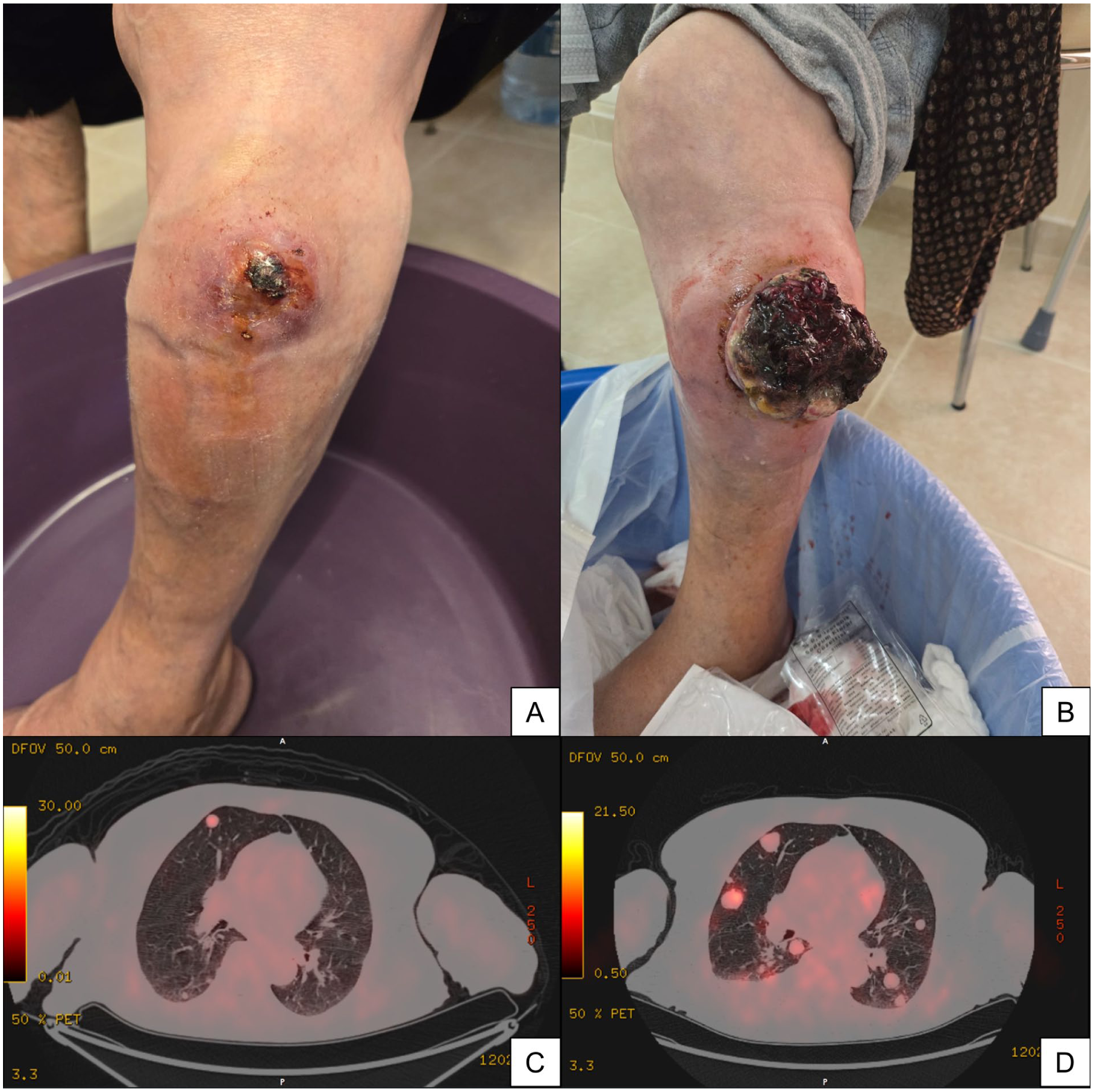
Clinical appearance of the primary tumor on the left leg at the initiation (A) and after two months of larotrectinib treatment (B), demonstrating local progression characterized by a large, exophytic, and fungating mass with extensive necrosis. Pre-treatment axial 18F-FDG PET-CT (C) versus restaging scan (D) confirming systemic progression, evidenced by a marked increase in the size, number, and metabolic activity of pulmonary metastases.
Discussion
This case represents a unique report of a biphasic SS harboring the pathognomonic SS18 rearrangement concurrently with a PDE3A-NTRK2 fusion. The rapid clinical deterioration observed upon administration of larotrectinib strongly suggests that the SS18::SSX fusion acted as the dominant, obligate driver, while the NTRK2 fusion functioned merely as a passenger alteration or a non-essential secondary event. This phenomenon of "driver versus passenger" dominance provides clinical evidence that the presence of a druggable target does not guarantee therapeutic vulnerability when a potent epigenetic driver is simultaneously present.
The efficacy of larotrectinib in NTRK-positive cancers is well-established. In the landmark integrated analysis, the agent demonstrated a durable overall response rate of 75% across diverse histologies, leading to its approval as a tissue-agnostic therapy. 4 The identification of a PDE3A-NTRK2 fusion in our patient provided a compelling rationale for targeted inhibition, particularly following the failure of first-line anthracycline-based chemotherapy. However, the majority of patients in the pivotal trials possessed tumors where NTRK fusions were the singular, mutually exclusive driver events (e.g., infantile fibrosarcoma or salivary gland carcinomas). Our case diverges significantly from this cohort because SS is fundamentally defined by the SS18::SSX translocation, a genetic event present in over 95% of cases that is essential for tumorigenesis. 5
The lack of response to larotrectinib in this patient can be mechanistically understood through the biology of the SS18::SSX fusion protein. As detailed by El Beaino et al., 2 SS18::SSX is not a classic kinase driver but a master epigenetic regulator. It integrates into the BAF (SWI/SNF) chromatin-remodeling complex, ejecting the tumor suppressor SMARCB1 and recruiting polycomb repressive complexes to aberrant loci. 6 This results in a profound, global transcriptional reprogramming that maintains the stem-cell-like state of SS. We hypothesize that because the tumor’s survival was dependent on this epigenetic catastrophe rather than downstream kinase signaling, inhibiting the TRK pathway was biologically futile. The PDE3A-NTRK2 fusion likely arose as a stochastic event during the genomic evolution of the tumor but did not supplant the SS18::SSX complex as the central engine of tumor viability.
Beyond the epigenetic dominance of SS18::SSX, the primary resistance to larotrectinib in this context can be further elucidated by a well-established mechanistic bypass pathway. SS18::SSX directly induces the expression of insulin-like growth factor 2 (IGF2), a process critical for synovial sarcoma tumorigenesis. 7 Consequently, the activation of the IGF-1 receptor (IGF-1R) axis is a hallmark of this disease, with nuclear IGF-1R expression serving as an independent adverse prognostic factor. 8 Crucially, the activation of the IGF-1R signaling cascade is a recognized class-wide, off-target resistance mechanism to TRK inhibitors, explicitly including larotrectinib. 9 Therefore, in our patient, the resistance to TRK inhibition was not an acquired, adaptive response, but rather a constitutive state. The continuous, upstream activation of the IGF-1R pathway by the lineage-defining SS18::SSX oncoprotein effectively bypassed any therapeutic blockade of the passenger NTRK2 fusion, making the clinical failure of larotrectinib biologically predictable.
Furthermore, the explosive progression observed during larotrectinib therapy may mirror the aggressive natural history of chemorefractory SS rather than a hyper-progressive response to targeted therapy. While standard intensive chemotherapy with doxorubicin and ifosfamide can achieve long-term survival in localized disease, 10 metastatic SS remains largely incurable with cytotoxic agents. 1 The failure of chemotherapy in this case aligns with findings by Chakiba et al., 11 who noted that mechanisms of metastatic relapse and resistance in SS are intrinsic to the tumor's biology and often independent of genomic complexity.
The emerging landscape of sarcoma therapeutics is rapidly evolving beyond cytotoxic chemotherapy. Recent approvals of agents like tazemetostat for epithelioid sarcoma and afami-cel for MAGE-A4-expressing SS represent significant strides in targeting histology-specific mechanisms. 12 However, the widespread use of broad-panel NGS has increased the frequency of detecting "incidental" fusions. In soft tissue sarcomas, which are frequently driven by specific translocations or complex karyotypes, 13 distinguishing between a true "addiction" to a kinase fusion and a bystander event is crucial. While NTRK inhibitors are transformative for patients with NTRK-driven malignancies, our case cautions against their indiscriminate use in tumors with established, potent alternative drivers, even when an NTRK fusion is detected.
In conclusion, this case illustrates primary resistance to larotrectinib in a patient with SS18-rearranged synovial sarcoma and a concurrent NTRK fusion. It underscores the dominance of the SS18::SSX epigenetic driver over secondary kinase alterations. Clinicians must exercise caution when interpreting NGS results in translocation-associated sarcomas; the detection of a "targetable" fusion should be contextualized within the tumor's canonical molecular pathology. Future research should focus on elucidating the functional interplay between concurrent drivers to better stratify patients for targeted therapies versus evolving cellular or epigenetic therapies.
Footnotes
Author contributions
Conceptualization, R.I.; Data curation and investigation (clinical), R.I., A.O., and O.A.; Investigation (pathology and molecular analysis), B.O.; Writing—original draft, R.I.; Writing—review and editing, R.I., B.O., A.O., and O.A.; Supervision, O.A. All authors have read and agreed to the published version of the manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Patient consent
Written informed consent was obtained from the patient's next of kin for the publication of this case report and accompanying images.
AI use statement
AI tools were used exclusively for language editing. ChatGPT-5.2 (OpenAI) assisted with clarity and fluency, and Grammarly with grammatical and stylistic accuracy. All scientific content was generated by the authors, who assume full responsibility for its accuracy and integrity.