
Abstract
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder primarily affecting male children and resulting in progressive degeneration and attempted regeneration of muscle with replacement of myofibers by adipose and fibrotic tissue. DMD patients have mutations in the DMD gene encoding for the large cytoskeletal protein dystrophin, which plays an essential role in the dystrophin-glycoprotein complex (DGC) as a structural connector within cardiac and skeletal muscle. The absence of dystrophin and DGC proteins leads to increased membrane fragility, dysregulation of calcium homeostasis, oxidative damage in muscle cells, and finally, premature death of the patients. Rhabdomyosarcoma (RMS), a soft tissue sarcoma arising from subpopulations of muscle cells and their precursors, has previously been reported in DMD patients and mouse models of DMD. Here, we report a case series of RMS arising from skeletal muscles in a Dmd-mutated (Dmd mdx ) rat model developed using transcription activator-like effector nucleases targeting exon 23 of the Dmd gene on a Sprague Dawley background. Subcutaneous firm masses were noted grossly in 8 male Dmd mdx rats aged 6 to 17 months. The histologic findings were consistent with a sarcoma of skeletal muscle, with a population of small round cells in addition to spindle-shaped cells and occasional large multinucleated cells. The neoplasms were immunoreactive for myogenin, MyoD1, and desmin. Histology and immunohistochemistry supported the diagnosis of RMS. This represents a potentially novel animal model of DMD-associated RMS.
Duchenne muscular dystrophy (DMD) is recognized as one of the most severe forms of inherited muscular dystrophies and the most common congenital neuromuscular disease.8,25 Currently, there are limited treatments for the disease, and patients generally develop an onset of symptoms in early childhood and experience severe progressive muscle wasting, respiratory and cardiac failure, and premature death. 8 Mutations in the DMD gene are responsible for the clinical presentation.
The DMD gene is the largest gene in the human genome, which contains 79 exons, and encodes the dystrophin protein. Dystrophin is primarily expressed in skeletal and cardiac muscles, as well as in the brain and retina. Mutations lead to a deficiency or complete absence of dystrophin, disrupting the DGC, an essential component of muscle cell membranes. Without functional dystrophin, muscle cells become highly fragile and prone to damage, with excessive membrane permeability, disrupted calcium homeostasis, and increased oxidative stress. 3 These factors impair the muscle’s ability to regenerate, leading to the progressive replacement of muscle fibers with connective and fatty tissue.
Animal models play a crucial role in our understanding of DMD. Currently, there are nearly 60 animal models 15 of DMD, with canine and mouse models being most commonly used. Preclinical studies involving mice often use the Dmd-mutated (Dmdmdx) mouse, which has a nonsense mutation in exon 23 of Dmd, leading to a premature stop of protein translation and nonfunctional dystrophin. 1 This mutation results in an acute onset of skeletal muscle necrosis at ~21 days of age, followed by repeated cycles of necrosis and regeneration, and culminating in stabilization of the disease by 3 to 4 months of age in the adult mouse.15,26 While the skeletal muscle of these mice undergoes necrosis and regeneration, as it does in human DMD patients, severe muscle weakness, loss of muscle mass, and infiltration of myofibers by adipocytes and fibrosis does not occur in Dmdmdx mice until approximately 2 years of age. 26 The dog is another important model of DMD, as clinical signs of affected dogs are generally more severe than in Dmdmdx mice, with muscular lesions similar to those observed in humans. 23 In addition to canine and mouse models, rats have been more recently used to model DMD. Larcher et al 13 generated 2 lines of Dmdmdx rats targeting exon 23 of the Dmd gene and observed necrosis and regeneration of skeletal muscle by 3 months of age. By 7 months of age, the limb and diaphragm muscles underwent necrosis with severe fibrotic and fatty tissue infiltration, which was associated with a significant reduction in muscle strength and decrease in motor activity. The rats also had cardiac lesions similar to those of human patients. This new animal model has recently been used to evaluate the efficacy of gene therapy products.4,14
Rhabdomyosarcoma (RMS) is a rare and aggressive soft tissue sarcoma. In humans, it typically affects the skeletal muscle of children and young adults. 27 RMS accounts for approximately 3% of all childhood malignant tumors and is the third most common pediatric extracranial solid tumor. 22 Despite recent advances in diagnostics and treatment modalities, survival rates for high-risk and recurrent disease are poor, and significant morbidity is associated with treatment. 27 Mutations in varying stages in the differentiation of myogenic cells are presumed to result in the development of RMS.22,27 RMSs are classified into distinct categories based on clinicopathologic features and genetic abnormalities. Morphological subtypes include embryonal, alveolar, spindle cell/sclerosing, and pleomorphic,2,9,10,20 while molecular subtypes are based on the presence or absence of DNA translocations that juxtapose PAX3 or PAX7 with FOXO1 (PAX3::FOXO1 or PAX7::FOXO1), as well as other genetic translocations or mutations. 7 Embryonal and alveolar RMSs are typically diagnosed in children, whereas pleomorphic RMS is often seen in adult patients. 27
Rhabdomyosarcomas have been described in a limited number of human DMD patients, with alveolar RMS reported in 3 of 4 case reports.6,17,19 Dmdmdx mice greater than 12 to 16 months of age are prone to spontaneous development of RMS,5,11 and a markedly increased incidence of RMS has been reported in other mouse models of DMD. 12 RMS also occurred in over 50% of Dmdmdx rats crossed with rats lacking the tumor suppressor gene p16. 24 Here, we describe and characterize the presence of spontaneous RMS diagnosed in 8 Dmdmdx rats bred on a Sprague Dawley background.
Materials and Methods
Animal Model
Dmdmdx rats were generated by creating a pair of transcription activator-like effector nucleases (TALENS) targeting exon 23 of the rat Dmd gene on a Sprague Dawley background. 13 The generated mutations were in a similar region to that of the Dmdmdx mouse model. Rats included in this report came from 2 colonies. In the first colony (Nantes, France), heterozygous females carrying either a frameshift 7 bp deletion of or an 11 bp frameshift deletion in the Dmd gene 13 were bred to CD IGS (Sprague Dawley) rats (Charles River, L’Arbresle, France) to generate hemizygous Dmdmdx males and littermate wild-type controls. In the second colony (Seattle, Washington), founder heterozygous females carrying the 11 bp frameshift deletion were obtained from the first colony and were bred to CD IGS (Sprague Dawley) rats (Charles River, Hollister, California) to generate hemizygous Dmdmdx males and littermate wild-type controls. Colony 1 rats were pair housed in a non-specific pathogen-free facility but with a controlled environment (temperature 21 ± 1°C, 12 h light/dark cycle). Special Diets Services and tap water (human consumption; quality controlled) were given ad libitum. Colony 2 rats were pair housed on a 12:12 light-dark cycle in an individually ventilated caging system with acidified and autoclaved water and fed a commercial rodent diet (Lab Diet, PicoLab Rodent Diet 20, 5053) ad libitum. Excluded pathogens in the facility of colony 2 included rat coronavirus (rat coronavirus/sialodacryoadenitis virus), rat parvoviruses, Mycoplasma pulmonis, Sendai virus, pneumonia virus of mice, pinworms, and fur mites. All rats were on a longitudinal study, with no experimental manipulation except for 1 rat (colony 2, animal 6) that underwent noninvasive antemortem echocardiography and muscle force testing procedures. Animal use was approved by the Ethics Committee on Animal Experimentation of the Pays de la Loire Region (Permit Numbers: CEEA-PdL-2011-45 and CEEA-PdL-01579.01, France), in accordance with the guidelines from the French National Research Council for the Care and Use of Laboratory Animals or by the Institutional Animal Care and Use Committee at the University of Washington and following recommendations set forth by the Guide for the Care and Use of Laboratory Animals. 16
Necropsy and Histology
Rats with masses noted clinically were subjected to complete gross necropsy. One rat from colony 2 had only the head submitted for histology. One additional rat from colony 2 had the heart collected by the investigative group, and the lungs were examined grossly but not submitted for histology. For all other cases, in addition to the mass, routine tissues involving major organs (including lung, liver, spleen, lymph node, kidney, and heart) were examined histologically. Neoplasms and other tissues harvested at gross necropsy were fixed in 10% neutral-buffered formalin. Fixed tissues were routinely processed, paraffin-embedded, sectioned at 4 to 5 μm, and stained with hematoxylin and eosin or with hematoxylin, eosin, and saffron. Subtype classification for each neoplasm was assigned using histologic and immunohistochemical criteria (Supplemental Table S1). All slides were evaluated by one or more board-certified veterinary pathologists.
Immunohistochemistry
Formalin-fixed, paraffin-embedded neoplastic tissues were immunolabeled with antibodies for some combination of the following: desmin, MEOX2, myosin heavy chain (MHC) fast twitch, MHC slow twitch, MHC neonatal, pancytokeratin, MYOD1, and myogenin (Tables 1 and 3). Distribution and intensity of immunolabeling were scored semi-quantitatively on a scale of 0 to 4, where 0 indicated no positive labeling of neoplastic cells, 1 indicated immunoreactivity of <10% of neoplastic cells and/or faint labeling, 2 indicated mild immunoreactivity of 10% to 33% of neoplastic cells, 3 indicated moderate immunoreactivity of 34% to 66% of neoplastic cells, and 4 indicated intense immunoreactivity of >67% of neoplastic cells. Intermediate scores were assigned if an increased percentage of cells displayed less intense immunoreactivity or a lower percentage of cells displayed more intense immunoreactivity.
Primary antibody type, concentration, supplier, and technical factors.
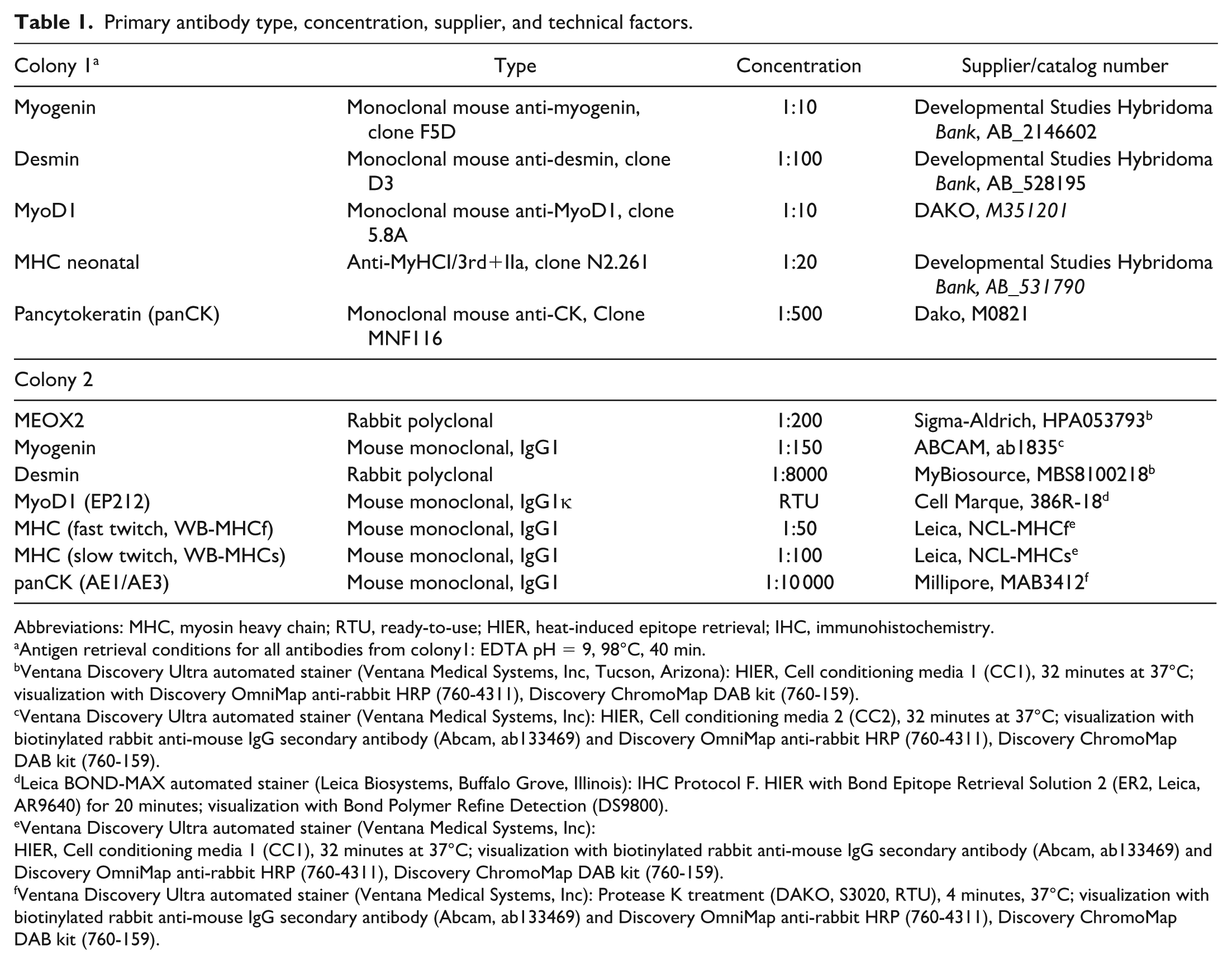
Abbreviations: MHC, myosin heavy chain; RTU, ready-to-use; HIER, heat-induced epitope retrieval; IHC, immunohistochemistry.
Antigen retrieval conditions for all antibodies from colony1: EDTA pH = 9, 98°C, 40 min.
Ventana Discovery Ultra automated stainer (Ventana Medical Systems, Inc, Tucson, Arizona): HIER, Cell conditioning media 1 (CC1), 32 minutes at 37°C; visualization with Discovery OmniMap anti-rabbit HRP (760-4311), Discovery ChromoMap DAB kit (760-159).
Ventana Discovery Ultra automated stainer (Ventana Medical Systems, Inc): HIER, Cell conditioning media 2 (CC2), 32 minutes at 37°C; visualization with biotinylated rabbit anti-mouse IgG secondary antibody (Abcam, ab133469) and Discovery OmniMap anti-rabbit HRP (760-4311), Discovery ChromoMap DAB kit (760-159).
Leica BOND-MAX automated stainer (Leica Biosystems, Buffalo Grove, Illinois): IHC Protocol F. HIER with Bond Epitope Retrieval Solution 2 (ER2, Leica, AR9640) for 20 minutes; visualization with Bond Polymer Refine Detection (DS9800).
Ventana Discovery Ultra automated stainer (Ventana Medical Systems, Inc):
HIER, Cell conditioning media 1 (CC1), 32 minutes at 37°C; visualization with biotinylated rabbit anti-mouse IgG secondary antibody (Abcam, ab133469) and Discovery OmniMap anti-rabbit HRP (760-4311), Discovery ChromoMap DAB kit (760-159).
Ventana Discovery Ultra automated stainer (Ventana Medical Systems, Inc): Protease K treatment (DAKO, S3020, RTU), 4 minutes, 37°C; visualization with biotinylated rabbit anti-mouse IgG secondary antibody (Abcam, ab133469) and Discovery OmniMap anti-rabbit HRP (760-4311), Discovery ChromoMap DAB kit (760-159).
Negative controls (omission of primary antibody or IgG isotype control) were performed for MyoD1, MHC, pancytokeratin, and myogenin. Negative and positive tissue controls were performed for MEOX2 and desmin. Internal positive controls included vessel wall and skeletal muscle (desmin), hair follicle (pancytokeratin), and skeletal muscle (MHC fast twitch). Other controls were not performed.
Results
Clinical information and results of immunohistochemistry are presented in Tables 2 and 3.
Clinical and histologic findings in 8 cases of rhabdomyosarcoma in Dmdmdx rats.
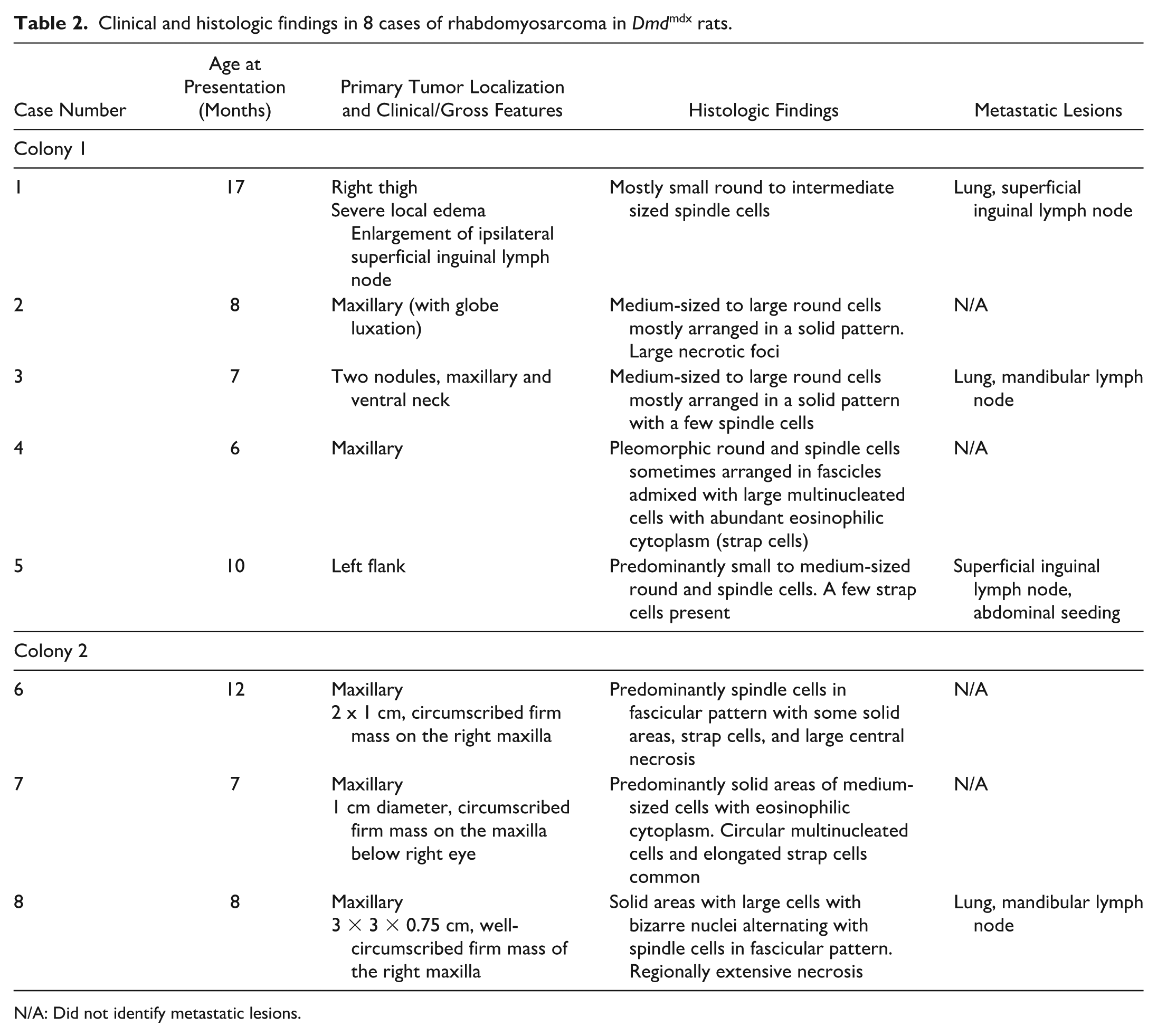
N/A: Did not identify metastatic lesions.
Immunohistochemical findings in 8 cases of rhabdomyosarcoma in Dmdmdx rats.
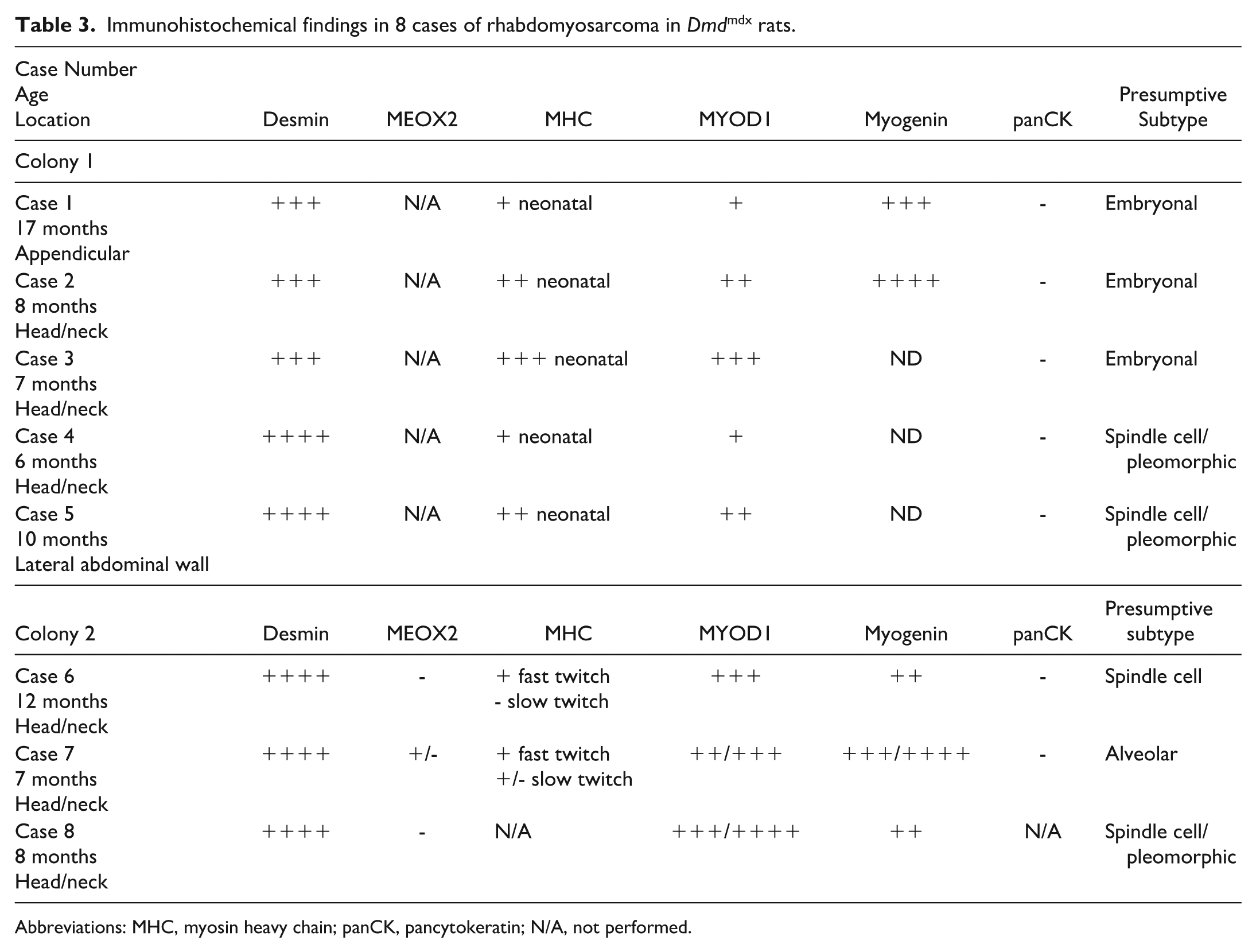
Abbreviations: MHC, myosin heavy chain; panCK, pancytokeratin; N/A, not performed.
From a group of 43 Dmdmdx male rats from colony 1 that were followed for a minimum of 6 months for a longitudinal study, 2 rats between 7 and 8 months of age presented for masses affecting the head or neck (Fig. 1a), 2 others between 6 and 17 months of age presented for masses affecting the thigh, and one 10-month-old rat presented for a mass affecting the flank (5 of 43 rats total presenting with masses). The masses in these 5 rats ranged from 1 to 4 cm in diameter and were firm on palpation. One rat had an associated malocclusion with displacement of the lower incisors approximately 3 mm from midline and involvement of the right suborbital region with ipsilateral globe luxation, epiphora, and porphyrin discharge. The 2 rats with masses on the thigh had severe local edema, and 1 rat had marked enlargement of the ipsilateral superficial inguinal lymph node.

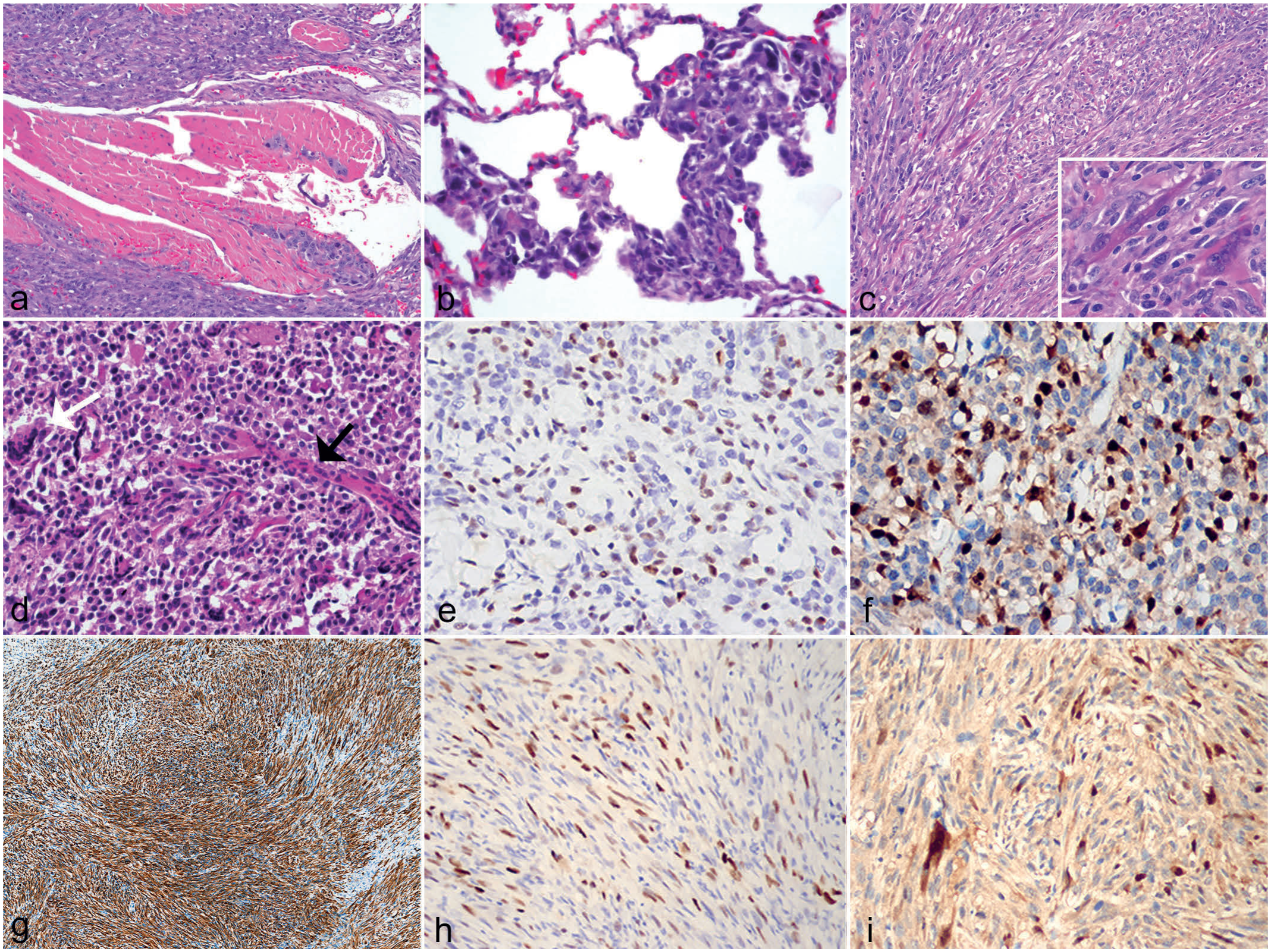
Macroscopic, histologic, and immunohistochemical findings of rhabdomyosarcomas affecting Dmdmdx rats from colony 1. (a) Sub-orbital maxillary neoplasm with globe luxation. Case 2. (b) The neoplasm is mainly composed of small, monomorphic round cells arranged in a solid pattern with some multinucleated cells (arrows). Case 3. Hematoxylin and eosin saffron (HES). (c) The neoplasm is composed of solid areas of atypical, large polygonal to plasmacytoid cells with abundant eosinophilic cytoplasm and prominent, often multiple nucleoli growing in a haphazard fashion. Case 4. HES. (d) Tumor infiltrating the transverse abdominis muscle. Case 5. HES. (e) Pulmonary metastases. Case 3. HES. (f) Metastasis to the mandibular lymph node. Case 3. HES. (g) Anti-desmin immunohistochemistry (IHC) shows strong, cytoplasmic immunolabeling of most neoplastic cells. Case 4. (h) Anti-MyoD1 IHC shows nuclear immunolabeling of many neoplastic cells. Case 2. (i) Anti-myogenin IHC shows nuclear immunolabeling of some neoplastic cells. Case 2.
From colony 2, a total of 3 Dmdmdx male rats between 7 and 12 months of age from a colony generating approximately 40 Dmdmdx male rats per year were presented with masses involving the maxilla over a 1-year period. Two years prior to these 3 cases, a similar mass had been documented in a 10-month-old rat, but histology was not performed. Masses in these 3 rats ranged from 1 to 3 cm in diameter and were firm on palpation. One rat had associated malocclusion with the displacement of the lower incisors approximately 5 mm from midline. In 1 rat, the mass also involved the right suborbital region and was associated with epiphora and porphyrin discharge from the ipsilateral eye. The rat with the largest mass had a 2.5 × 0.5 × 1 cm heterogeneous lobulated mass at the level of the cervical lymph node that was semi-firm and dark red to pale yellow-tan; histologically, this was confirmed to be ventral extension of the neoplasm.
Histology of all 8 neoplasms (Figs. 1b–f and 2a–d) revealed soft tissue sarcoma involving skeletal muscle, with central necrosis in the 4 largest tumors and mineralization in 1 case. Neoplasms were mostly nonencapsulated, locally invasive, reasonably well demarcated, and composed of round and elongated cells with moderate cytoplasm arranged in a solid pattern, streams, and/or bundles supported by thin to moderately thick fibrovascular stroma. Multinucleated cells with abundant hypereosinophilic cytoplasm were frequent in some cases, occasionally with >20 nuclei. The contralateral and adjacent skeletal muscle had changes including myofiber size variation, central nuclei, individual fiber necrosis, hyaline fibers, and mild multifocal chronic inflammatory cell infiltration, as well as fibrosis and adipose tissue infiltration in older animals, consistent with the DMD phenotype. Four rats (both colonies considered) had metastatic lesions in the lymph node and/or lung (Figs. 1e and f; 2b). Notably, 1 rat with a tumor in the flank displayed multiple peritoneal metastases infiltrating the diaphragm, mesentery, and mesenteric lymph nodes. Results of histology and immunohistochemistry are summarized in Tables 2 and 3, respectively. In short, all of the neoplasms strongly expressed desmin and were variably immunoreactive for MyoD1 and, when performed, for myogenin. Neoplasms also did not express pancytokeratin, in 7 of 8 cases tested. Based on the results of histology and immunohistochemistry (Tables 2 and 3; Figs. 1g–i and 2e–i), RMS was diagnosed in all cases.
Histology and immunohistochemistry (IHC) of rhabdomyosarcomas affecting the head and neck of Dmdmdx rats from colony 2. (a–c) Eight-month-old rat with a maxillary mass. Case 8. (a) The neoplasm infiltrates the skeletal muscle. Hematoxylin and eosin (HE). (b) Pulmonary metastasis with polygonal to round cells expanding alveolar capillaries and the interstitium. HE. (c) Cells in the central portion of the maxillary mass have a spindle cell morphology with interspersed multinucleated cells consistent with strap or tadpole-shaped rhabdomyoblasts (inset). HE. (d–f) Seven-month-old rat with a maxillary mass. Case 7. (d) There are solid regions of smaller round to polygonal cells with larger elongated (black arrow) or ovoid (white arrow) multinucleated cells. HE. (e) Anti-MyoD1 IHC shows moderate nuclear immunolabeling of many neoplastic cells (semi-quantitative score of 2–3). (f) Anti-myogenin IHC shows mild cytoplasmic but stronger nuclear immunolabeling of neoplastic cells (semi-quantitative score of 3–4). (g–i) Twelve-month-old rat with a maxillary mass. Case 6. (g) Anti-desmin IHC shows strong, diffuse immunolabeling of predominantly spindle-shaped neoplastic cells arranged in streams and bundles. (h) Anti-MyoD1 shows moderate positive immunolabeling of many neoplastic cells (semi-quantitative score of 3). (i) Anti-myogenin IHC shows mild cytoplasmic but stronger nuclear immunolabeling of neoplastic cells (semi-quantitative score of 2).
Discussion
In this case series, we describe 8 Dmdmdx rats presenting between 6 and 17 months of age with rapidly growing masses localized predominantly to the muscles of mastication (n = 5) but also of the limb (n = 2) and flank (n = 1) that were diagnosed as RMS on the basis of histomorphology and desmin, myogenin, and MyoD1 immunoreactivity. RMS was not identified in littermate control rats. Importantly, RMSs were diagnosed in Dmdmdx rats with 2 different mutations in Dmd exon 23. There is growing evidence that animal models of DMD are prone to developing RMSs. Chamberlain et al 5 reported that 6 out of 94 Dmdmdx mice (and no wild-type littermates) developed alveolar RMS (subtype diagnosis based on histology and immunohistochemistry) affecting the limbs, with nearly all tumor cells demonstrating positive labeling for myogenin and PAX7 expression, and approximately 50% exhibiting positive MyoD1 expression. It was postulated that the increased susceptibility of Dmdmdx mice to this neoplasm may be related to the continuous myofiber degeneration and regeneration occurring in DMD and associated with activation and proliferation of muscle progenitor cells, promoting an environment prone to mutations affecting the progeny of satellite cells. A 2010 study of mice lacking dystrophin or alpha sarcoglycan showed the absence of members of the dystrophin-associated glycoprotein complex may establish a permissive environment for spontaneous development of embryonal RMS (subtype diagnosis based on histology, immunohistochemistry, and molecular testing) with mutation of p53 and mutations or altered splicing of Mdm2, a negative regulator of p53. 11 Of these Dmdmdx mice, 9% over the age of 12 months developed spontaneous tumors consistent with embryonal RMS affecting the limbs and paraspinal muscles, whereas similarly aged wild-type mice did not develop such tumors. In a Dmd and Dysf (dysferlin) double mutant mouse model, more than 90% of mice developed RMS at an average age of 12 months. 12 In addition, Schmidt et al. 21 have shown that mutations in muscular dystrophy genes (Dmd, Dysf, Capn3, Large) led to age-related spontaneous formation of skeletal muscle-derived tumors in the mouse, presenting as mixed rhabdo-, fibro-, and liposarcomas. These studies suggest that the proliferation of and genetic mutations in skeletal muscle progenitor cells and other mesenchymal cells (eg, adipocytes, fibroblasts) associated with DMD may facilitate a molecular context permitting sarcoma development.
Neoplasms in this case series were variably consistent with pleomorphic, spindle cell, embryonal, or alveolar RMS (solid type) subtypes, given the range of their histologic appearance and pattern of immunohistochemistry immunoreactivity. However, molecular diagnostics would be required to more definitively diagnose the subtype. Embryonal RMS, the most common subtype in humans (70–80%), is usually diagnosed in children aged 0 to 5 years and recapitulates the phenotypic and biologic features of embryonic skeletal muscle. 2 This subtype is observed in the head and neck region or the genitourinary tract of patients. Histologically, this neoplasm often has an alternating loose and dense appearance and is composed of primitive oval to spindle, small blue round cells, and/or stellate cells with occasional rhabdomyoblasts in a myxoid background.2,10 Immunohistochemical markers of skeletal muscle differentiation are positive in embryonal RMS, including diffuse labeling for desmin, in addition to varied positivity of MyoD1 and myogenin. 2 Embryonal RMSs may also be more immunoreactive for MEOX2, given that this is a marker of more immature cell populations expressing transcription factors characteristic of paraxial mesoderm. 18
Alveolar rhabdomyosarcoma (ARMS) constitutes around 20% of all RMS diagnoses in humans. 20 ARMS is more frequently diagnosed in adolescents and young adults and tends to develop with a preference for certain anatomical locations, such as the extremities and head and neck regions. Alveolar RMS has been previously diagnosed in a mouse model of DMD. 5 Histologically, ARMSs are composed mostly of monomorphic round cells, arranged in a solid pattern or along a thin fibrovascular stroma (alveolar pattern).2,10 Multinucleated cells are common. ARMS is associated with a poor prognosis; 25% to 30% of patients with ARMS present with metastatic disease. 20 ARMS tends to have strong diffuse (>90% of neoplastic cells) labeling for myogenin, in contrast to embryonal RMS, but has variable expression of desmin and MyoD1. Nevertheless, due to overlap in immunohistochemical findings, molecular testing in addition to immunohistochemistry is preferred in human patients for RMS subclassification.2,10 One important consideration in classification is PAX-FOXO1 gene fusion status, as ARMSs are generally PAX-FOXO1 fusion positive, whereas embryonal RMSs tend to be PAX-FOXO1 fusion negative. 10
In contrast to embryonal and alveolar RMSs, pleomorphic RMS most often involves the distal extremities in human patients and is characterized histologically by enlarged and bizarre pleomorphic neoplastic cells. Immunohistochemically, there is strong desmin immunoreactivity with more limited immunoreactivity for myogenin or MyoD1.9,10
In our case series, 4 of 8 rats had metastatic disease. RMS has been reported in a limited number of human DMD patients, and 3 of 4 case reports describe ARMS developing in children from 4 to 9 years of age, with embryonal RMS reported in the remaining case. Metastatic RMS is reported in 1 patient. 19 In previous reports of RMS in Dmdmdx mouse models, RMS developed at a mean age of 18 to 20 months (range = 12–27 months).5,11 In a previous report of RMS in a rat model of DMD with loss of p16, pleomorphic RMS was diagnosed in rats >9 months of age and predominantly involved the limbs, although one third of the neoplasms involved the cheek and neck. 24 The presence or absence of metastatic disease was not discussed in these previous reports of RMS.5,11,24
This case series adds to the current evidence that links muscular dystrophies in rodent models to a marked increased incidence of RMS, supporting the hypothesis that the microenvironmental context of dystrophic muscle may promote tumorigenesis. Although the lack of molecular data, including gene fusion status, does limit comparison to RMS subtypes in human patients, mouse models of DMD, and that rat model of DMD crossed with rats lacking the tumor suppressor gene p16, this discovery in Dmdmdx rats nonetheless represents a novel animal model of spontaneous RMS that may be of note for future studies to assess the genetic, molecular, and phenotypic characteristics, ultimately paving the way for therapeutics in a rare childhood disease.
Supplemental Material
sj-pdf-1-vet-10.1177_03009858261457961 – Supplemental material for Spontaneous rhabdomyosarcomas in Dmdmdx rats
Supplemental material, sj-pdf-1-vet-10.1177_03009858261457961 for Spontaneous rhabdomyosarcomas in Dmdmdx rats by Rachel H. Moore, SiWei Luo, Aude Lafoux, Caroline Le Guiner, Corinne Huchet, Shanny H. Kuo, Olivia Terceve, David L. Mack, Heather Sheppard, Thibaut Larcher and Jessica M. Snyder in Veterinary Pathology
Footnotes
Acknowledgements
We would like to acknowledge the Histology and Imaging Core at the University of Washington for assistance with the preparation of HE slides from cases from colony 2. We would also like to acknowledge Emily Spaulding, LVT, for expert veterinary care of the rats in colony 2.
Supplemental material for this article is available online.
Author Contributions
JMS, SHK, HS, and TL coordinated and interpreted histology and immunohistochemistry. RHM, TL, and JMS wrote the manuscript. SL, AL, CLG, CH, OT, and DLM managed the rat colonies and assisted with data collection; all authors read and approved the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.