
Abstract
Background
The improved prevention of migraine, driven by the introduction of calcitonin gene-related peptide (CGRP)-targeted therapies, has prompted the International Headache Society (IHS) to propose new goals in migraine management. This study aimed to evaluate the safety and effectiveness of atogepant for migraine prevention over 24 weeks, in accordance with the IHS-defined goals.
Methods
This is a multicenter, prospective, observational, real-world study on patients starting atogepant 60 mg for migraine prevention. We describe efficacy and safety outcomes at weeks 9–12 (T3) and 21–24 (T6) relative to baseline (T0) from the initiation of atogepant treatment. We also report the proportion of patients achieving migraine freedom, optimal, modest or insufficient control according to the IHS position paper categories.
Results
One hundred twenty-eight (n = 128) patients (63 (49.2%) with chronic migraine, 115 (89.9%) female, aged 48.3 ± 13.3 years) from 17 centers were analyzed. Monthly migraine days decreased from 16.5 ± 8.5 at T0 to 8.0 ± 8.4 at T3 (p < 0.001 vs. T0) and 8.6 ± 9.2 at T6 (p < 0001 vs. T0, p = 0.265 compared to T3). Overall, 64.8% of patients were responders (at least 50% reduction in monthly migraine days from T0) at T3 and 61.7% at T6; 81.9% of responders at T3 maintained the responder status at T6, while 24.4% of patients among non-responders at T3 became responders at T6. At T3 and T6, 39.1% of the entire cohort achieved at least optimal migraine control (20.6% in chronic migraine and 56.9% in episodic migraine).
Conclusions
The STAR study demonstrates, in a real-world setting, the safety and the sustained effectiveness of atogepant 60 mg over 24 weeks and indicates that more than one-third of treated patients can achieve at least optimal disease control, with markedly better outcomes in episodic than in chronic migraine.
Trial Registration
The study was preregistered on clinicaltrial.gov, NCT06414044.
This is a visual representation of the abstract.
Introduction
Migraine is among the most prevalent disorders worldwide and among the leading causes of health loss. 1 It is associated with clinically relevant comorbidities and thus significantly affects patients’ personal, emotional and professional spheres.2,3 In the last decades, pivotal randomized clinical trials (RCTs) demonstrated the efficacy of onabotulinumtoxinA (BoNTa) 4 and calcitonin gene-related peptide (CGRP) targeting therapies.5,6 in migraine prevention, reporting excellent tolerability profiles as well.
The extensive experience with BoNTa and monoclonal antibodies against CGRP or its receptor (anti-CGRP mAbs), derived from clinical practice and real-world studies, confirms their effectiveness, rapidity of action and tolerability, with up to two-thirds of patients reporting a reduction in headache frequency by half.5–7 Nevertheless, a percentage of patients do not respond properly to these therapies, and even those who respond can be impacted by a significant residual burden of migraine. 8
In this scenario, the more recent introduction of gepants, small molecules antagonizing the CGRP receptor, opened new perspectives in the prevention and acute management of migraine. 6
Atogepant 60 mg daily was approved by the Food and Drug Administration and European Medicines Agency for episodic migraine (EM) and chronic migraine (CM) prevention, based on the evidence from the randomized ADVANCE 9 and PROGRESS 10 trials. The efficacy of atogepant over placebo was also demonstrated in patients with EM who previously had an inadequate response to two to four classes of conventional oral preventive treatments. 11 Based on these and other results, gepants are recommended for preventive treatment by the recent guidelines12,13 and practice recommendations 14 of the International Headache Society (IHS).
Evidence from real-world studies showed that atogepant is effective in preventing migraine also in patients who did not previously benefit from anti-CGRP monoclonal antibodies (mAbs), although to a lesser extent than participants who were naïve to anti-CGRP mAbs.15–18 Indeed, up to 7% of the non-naïve patients achieved a 100% response rate (RR) after 12 weeks of therapy. 15
With the higher treatment response made real by novel, specific drugs, the headache scientific community has grown increasingly aware that minimizing the burden of migraine can become possible. In this context, the IHS has recently advocated raising the standards for migraine prevention by setting ambitious yet pragmatic treatment goals. 19 More specifically, the IHS suggests categorizing the outcomes of migraine treatment into 4 tiers: migraine freedom, optimal control (< 4 monthly days with migraine or moderate-to-severe headache), modest control (4–6 days with migraine or moderate-to-severe headache), and insufficient control (> 6 days with migraine or moderate-to-severe headache). To date, no data are available on the level of disease control, as defined by these standards, that can be achieved with treatment using gepants.
The present study aimed to evaluate the effectiveness and safety of atogepant for migraine prevention over 24 weeks and to assess treatment performance in terms of migraine control, as defined by the IHS Position Statement on setting new standards for migraine prevention.
Methods
Study design and patients
The STAR study design and patients’ enrollment have been previously described. 15 The STAR is a real-world, prospective, multicenter, investigator-initiated and independent study, considering all consecutive outpatients treated with atogepant 60 mg orally for EM or CM. The study has a pre-planned 2-year follow-up.
In the present study, we included all patients enrolled in the STAR study who had potentially at least 24-week follow-up since the first intake and who took at least one 60 mg tablet of atogepant, regardless of discontinuation for any reason, from June 2024 to April 2025.
The monthly migraine days (MMDs) were fully available at baseline, T3 and T6 for the entire cohort. The study was pre-registered on clinicaltrials.gov (NCT06414044). This study is reported in accordance with the STROBE guidelines for observational study.
The local Ethics committee approved the study as part of the Registro Italiano Cefalee (RICe) study (Studio RICe, 14591_oss CEAVC Studio RICe, 14591_oss and subsequent amendments). Detailed information on the RICe study is reported elsewhere.20,21 All patients provided their written informed consent before starting treatment with atogepant. The open online database Research Electronic Data Capture (REDCap) and the Empedocle electronic platform (developed for the RICe study) were used for data collection.
Inclusion and exclusion criteria
Inclusion criteria were: (i) individuals aged 18 years or older; (ii) a diagnosis of migraine without aura, migraine with aura, or CM according to the International Classification of Headache Disorders, 3rd edition 22 ; (iii) at least four MMDs in the 3 months before enrollment; (iv) good compliance with study procedures; (v) availability of headache diaries for at least 1 month before enrollment; and (vi) clinical indication for the prescription of atogepant 60 mg for migraine prevention.
Exclusion criteria were: (i) subjects with any contraindications to gepants; (ii) concomitant diagnosis of medical diseases and/or comorbidities that could undermine the study according to clinicians (e.g. medical conditions with a life expectancy shorter than 2 years, or mental disabilities/psychiatric conditions, that may interfere with the questionnaire fill-in); (iii) pregnancy and breastfeeding; and (iv) introduction or change in concomitant therapies in the 12 weeks prior to enrollment.
Participants were enrolled regardless of the number of prior ineffective or non-tolerated preventive treatments, if any, in accordance with clinical practice. We have defined “lack of efficacy” as no meaningful clinical migraine improvement after the administration of drugs for ≥ 6 weeks at the appropriate dose for oral standard of care and two cycles (24 weeks) for BoNTa and at least 3 months for CGRP mAbs according to the European Headache Federation criteria. 23 We also considered patients with “lack of efficacy” who presented with a loss of effectiveness for ≥ 6 weeks at the appropriate dose for oral standard of care and one cycle (12 weeks) for BoNTa, and at least 3 months for CGRP mAbs.
According to the recent IHS recommendations for the design of real-world studies in migraine, 24 our study provides Data Quality Level 1 evidence. The checklist is reported in the Supplementary material (Table S1).
Collected variables
Clinical and demographic features, as well as concomitant and prior preventive treatments, were recorded after obtaining an accurate medical history. We recorded clinically relevant conditions, defined as active or recent medical conditions or symptoms that have a meaningful impact on a patient's health or require treatment or diagnostic follow-up.
MMDs and the number of monthly acute medications (MAMs) were collected from paper or electronic headache diaries prior to the first intake (i.e. baseline).
We defined patients with medication overuse (MO), regardless of CM diagnosis, as those with overuse of acute or symptomatic headache medication (≥ 10 days/month for triptans and combination analgesics, or ≥ 15 days/month for non-opioid analgesics) for more than 3 months. 25
Finally, a panel of questionnaires was administered at baseline (T0) and after 12 weeks (T3) and 24 weeks (T6) weeks of therapy: the Headache Impact Test (HIT-6, 36–78 scale), 26 the Migraine Disability Assessment (MIDAS, 0–270 scale) questionnaire, 27 the Migraine Treatment Optimization Questionnaire-6 (mTOQ-6, 6–24 scale), 28 the Migraine-Specific Quality-of-Life Questionnaire (MSQ, 0–100 scale), 29 the 12-item Allodynia Symptom Checklist (ASC-12, 0–24 scale) 30 and the Migraine Interictal Burden Scale (MIBS-4, 0–12 scale). 31 The Patient's Global Impression of Change (PGIC) 32 was administered only at T3 and T6.
An MMD was defined as any day with a headache with the characteristics of migraine or the use of a triptan. Adverse events (AEs) were collected and reported to the Italian Medicines Agency (i.e. AIFA) in accordance with Italian regulatory requirements.
Outcomes and analysis
According to the IHS guidelines for the design of real-world studies in migraine, 24 the co-primary effectiveness outcomes of the STAR study were: (i) changes in MMDs in weeks 9–12 (T3) and in weeks 21–24 (T6) of treatment compared to baseline and (ii) the percentage of responders (namely patients who presented a reduction of MMDs ≥ 50% (50%RR) compared to baseline) in weeks 9–12 and 21–24. We associated the assessment of the occurrence of treatment-emergent side effects (TEAEs) to evaluate the safety of the drug in a real-world population.
For the present analysis, our key secondary outcome was the assessment of treatment effectiveness at T3 and T6 according to the following IHS position statement categories over the previous three months: migraine freedom (no migraine or moderate-to-severe headache attacks), optimal control (< 4 monthly days with migraine or moderate-to-severe headache), modest control (4–6 days with migraine or moderate-to-severe headache) and insufficient control (> 6 days with migraine or moderate-to-severe headache). 19
Other secondary outcomes assessed at T3 and T6 were: (i) the percentage of patients with MO reverted during treatment; changes from baseline in MAMs; changes from baseline in the following patients’ reported outcomes (PROMs); (ii) MIBS4 scores; (iii) changes from baseline in the MSQ role function restrictive scores; (iv) changes from baseline in the ASC-12 scores; (v) changes from baseline in the mTOQ-6 scores; (vi) changes from baseline in the HIT-6 scores; (vii) changes from baseline in the MIDAS ; (viii) PGIC rating; and (ix) percentage of AEs.
The exploratory outcome was the assessment of the co-primary outcomes in patients who had not previously benefited from anti-CGRP mAbs compared to participants who were naïve to anti-CGRP mAbs.
Statistical analysis
The present study is an a priori analysis for the co-primary outcomes. We included only subjects with complete information regarding the primary studied variables (i.e. MMDs). We declared data availability and performed the analysis only in patients with usable data for the secondary variables.
Based on the results of atogepant clinical trials and real-world data on monoclonal antibodies targeting the CGRP pathway, we calculated a sample size of at least 128 subjects to achieve a power of 80% and a significance level of 5% (two-sided) for detecting an effect size of 0.25 between paired variables (i.e. changes in MMDs).
Between-group differences in interval variables were analyzed using independent t-test (expressed as the mean ± SD) or analysis of variance (ANOVA) when parametric assumptions were met, and Mann–Whitney tests (medians with interquartile range) when they were not. Accordingly, we applied the t-test for repeated measures and the Wilcoxon signed rank test to analyze the variation of interval variables over time. We applied a repeated-measures ANOVA to assess the effects of intersubject factors on variation across evaluation times. To correct the multiple comparisons, we applied Duncan's multiple range test.
Contingency tables (chi-squared tests) and unadjusted odds ratios (OR) with their 95% confidence intervals (CI) were run to compare frequencies between groups. The McNemar test for proportions of paired samples was applied to assess changes in frequency over the evaluation times.
Binary logistic regression (forced entry) was also run to evaluate possible baseline factors (i.e. the presence of clinical comorbidities, CM, previous ineffective mAbs or BoNTa therapies, concomitant preventive therapies, age and sex as independent variables) influencing an OPTIMAL outcome at T6 (dependent variable). The independent variables included in the above-described model were selected based on their association in the univariate model, relevance to the outcome, and the available evidence. 15
All tests were two-tailed. p < 0.05 (two-tailed) was considered statistically significant. Statistical analyses were performed using SPSS, version 27.0 (IBM Corp., Armonk, NY, USA).
Results
Co-primary outcomes
Out of the 398 screened, 128 patients (63; 49.2% with CM, 115; 89.8% female, aged 48.3 ± 13.3 years, with a minimum age of 19 years and a maximum of 79 years) from 17 Italian centers had the 24-week observation period following the first intake of atogepant tablets (Figure 1), regardless of discontinuation. Table 1 summarizes baseline demographic and clinical profiles in EM and CM patients. The overall discontinuation rate was 13.2% (n = 17/128). The time and reasons for discontinuation are detailed in the Supplementary material (Figure S1).
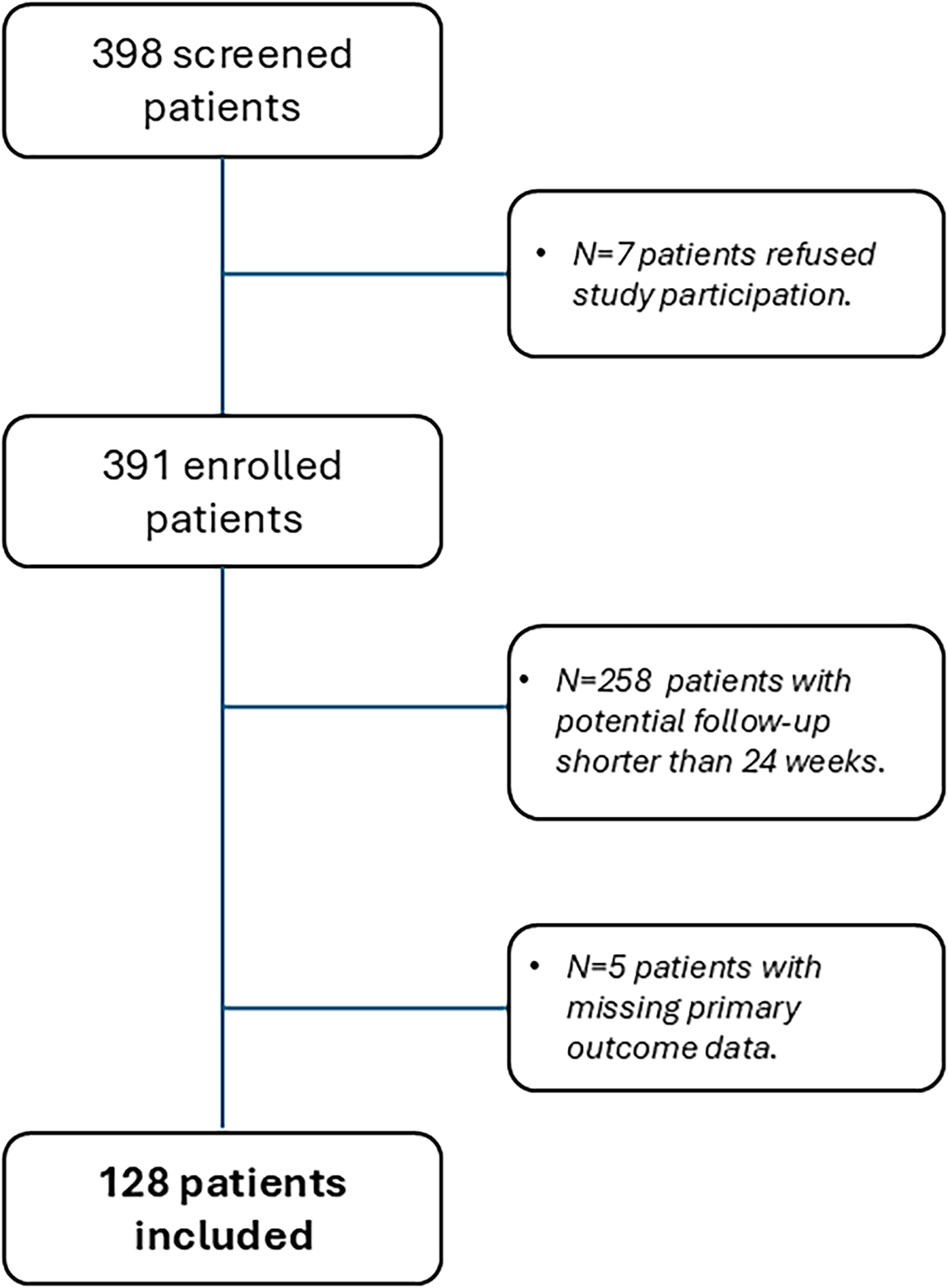
PRISMA diagram showing patients’ screening and enrollment
Baseline characteristics of the STAR-2 study cohort, comparing patients with episodic (EM) and chronic (CM) migraine
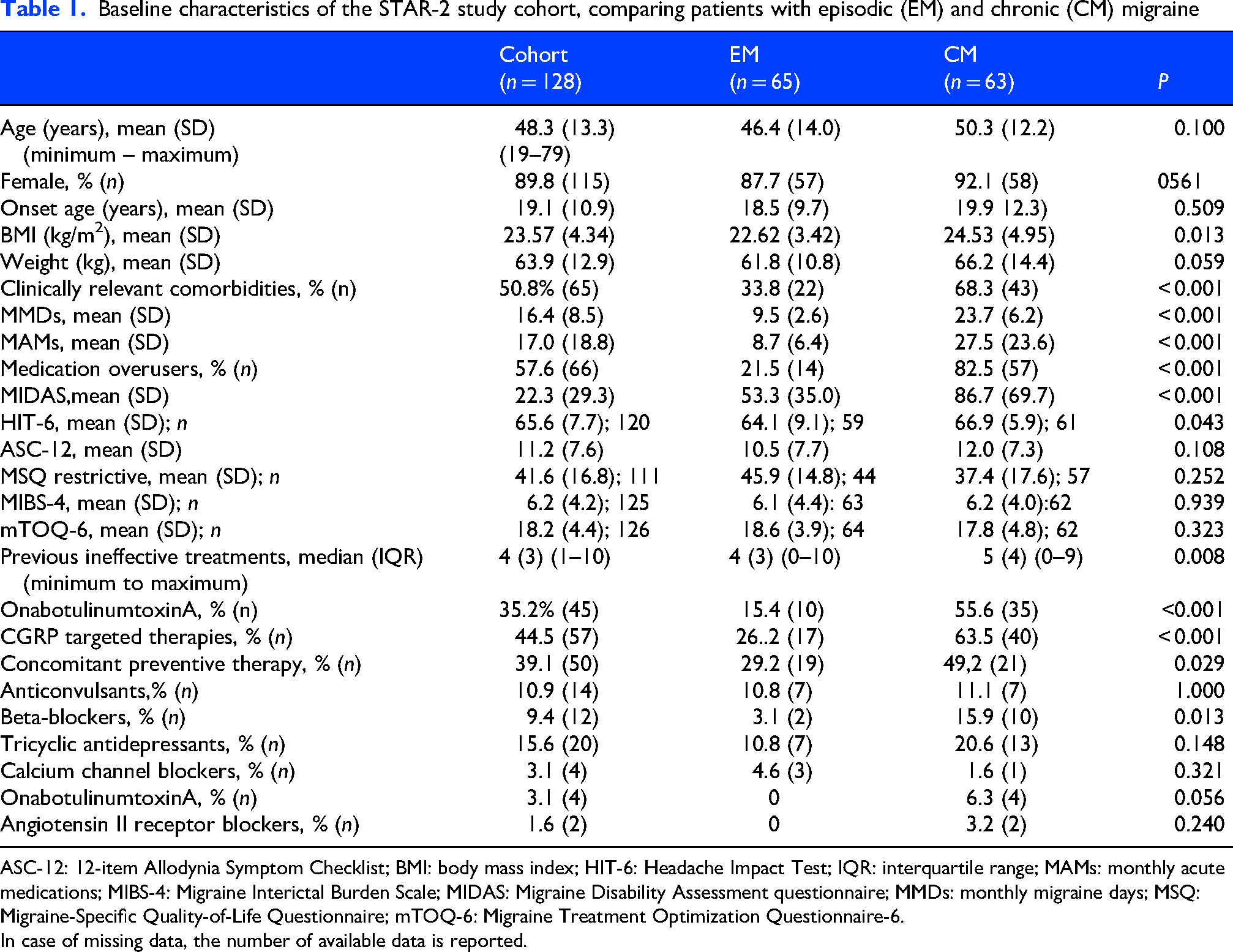
ASC-12: 12-item Allodynia Symptom Checklist; BMI: body mass index; HIT-6: Headache Impact Test; IQR: interquartile range; MAMs: monthly acute medications; MIBS-4: Migraine Interictal Burden Scale; MIDAS: Migraine Disability Assessment questionnaire; MMDs: monthly migraine days; MSQ: Migraine-Specific Quality-of-Life Questionnaire; mTOQ-6: Migraine Treatment Optimization Questionnaire-6.
In case of missing data, the number of available data is reported.
In the whole group, MMDs decreased from 16.4 ± 8.5 at baseline to 8.0 ± 8.4 at T3 (p < 0.001) and 8.6 ± 9.2 at T6 (p < 0.001 vs. baseline; p = 0.265 vs. T3). Subjects with EM had a reduction in MMDs from 9.5 ± 2.6 at T0 to 4.5 ± 5.3 at T3 (p < 0.001) and to 4.1 ± 4.5 at T6 (p = 0.560 compared to T3). Subjects with CM had a reduction from 23.7 ± 6.2 at T0 to 11.6 ± 9.4 at T3 (p < 0.001) and to 12.3 ± 10.5 at T6 (p = 0.055 compared to T3) (Figure 2A).
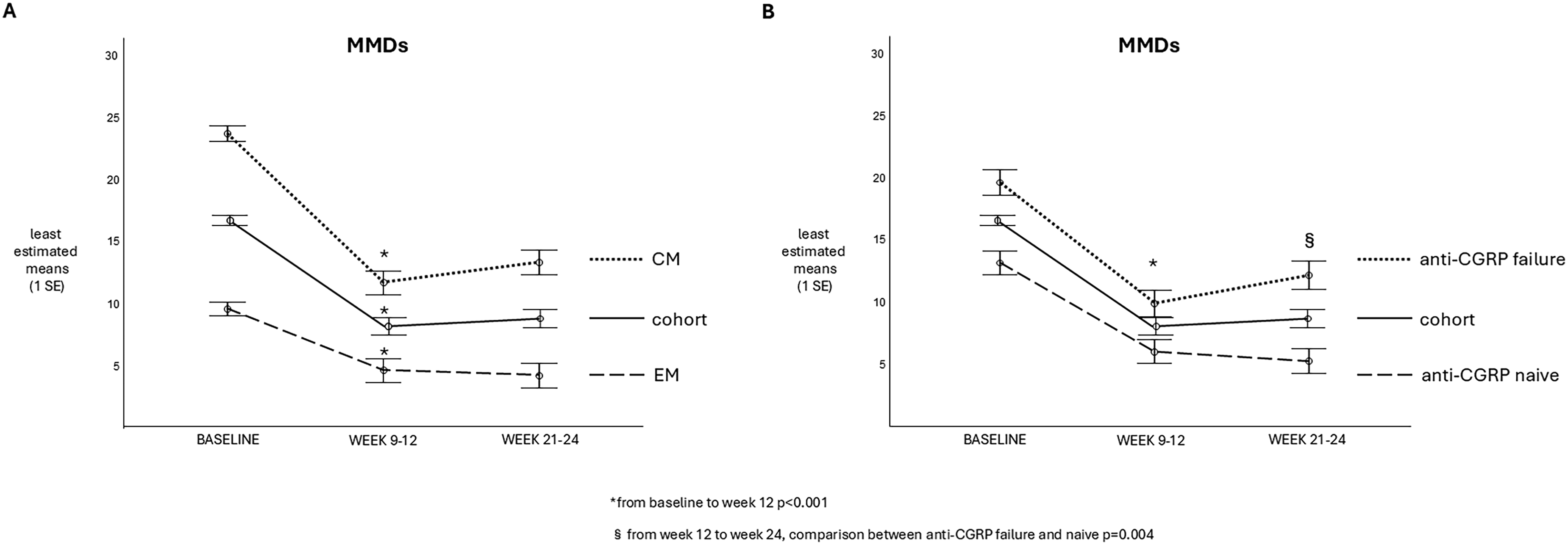
Monthly migraine day (MMD) variations from baseline to 9–12 and 21–24 weeks of atogepant therapy. (a, left) In the whole cohort (black line), episodic migraine (EM) patients with (dotted line) and chronic migraine (CM) (dashed line). (b, right) In the whole cohort (black line), in anti-calcitonin gene-related peptide (CGRP) naïve patients (dotted line) and in those with previous experience of ineffective anti-CGRP mAbs (dashed line).
Overall, 64.8% of patients achieved 50%RR at T3 and 61.7% at T6 (70.8% at T3 and 69.2% at T6 in the EM group, and 58.7% at T3 and 54.0% at T6 in the CM group). The differences in 50%RR between T3 and T6 were not significant (p > 0.400 for all comparisons) (Figure 3A). Notably, in the whole cohort, 81.9% of responders at T3 maintained the 50%RR also at T6, while 24.4% of patients who were non-responders at T3 became 50%RR responders at T6.
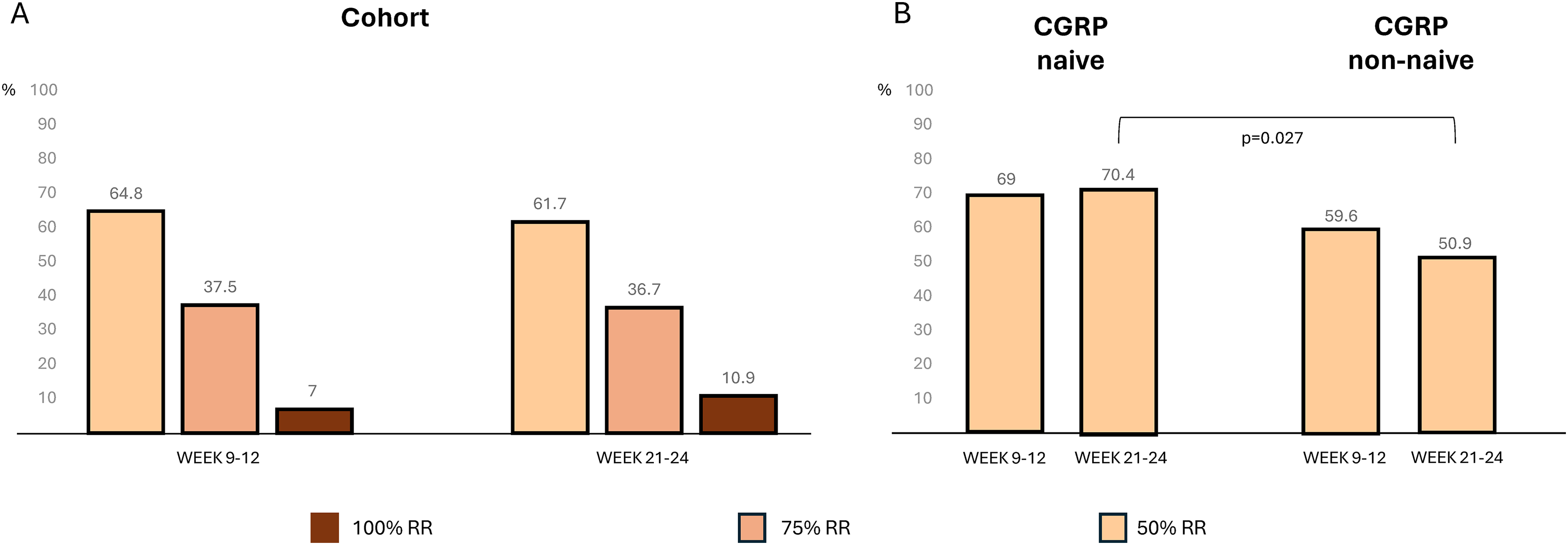
(a, left) Percentages of patients achieving monthly migraine day (MMD) response rates (50%, 75% and 100%) in the whole cohort, at 9–12 weeks and 21–24 weeks after atogepant treatment. (b, right) MMD 50% response rate (RR) in patients with (calcitonin gene-related peptide (CGRP) failure, right) and without (CGRP naïve, left) previous experience of ineffective anti-CGRP monoclonal antibodies.
Key secondary outcome
Figure 4 displays categories of migraine control at different time points. At T3, according to the IHS position statement categories, 39.1% of the entire cohort achieved at least optimal migraine control (20.6% in CM and 56.9% in EM; p < 0.001 from baseline for all groups). At T6, we observed at least optimal migraine control in 39.1% of the whole cohort, 20.6% in CM and 56.9% in EM. Notably, a higher proportion of patients achieved migraine freedom at T6 (10.9%) compared to T3 (7%). However, while the rate of migraine freedom in patients with EM more than tripled from T3 to T6 (rising from 4.6% to 15.4%), it decreased among those with CM, from 9.5% to 6.3%.

Percentages of patients whose migraine control is classified according to the International Headache Society proposed categories: at baseline of the 24-week STAR study, at 0–12 weeks and 13–24 weeks after atogepant treatment, in the whole cohort, chronic migraine (CM) and episodic migraine (EM) patients.
At least optimal control of migraine was inversely associated with the presence of a clinically relevant comorbid condition (OR = 0.371; 95% CI = 0.178–0.773; p = 0.011), previous failure of anti-CGRP mAbs (OR = 0.165; 95% CI = 0.072–0.377; p < 0.001), and of BoNTa (OR = 0.421; 95% CI = 0.196–0.816; p = 0.949). Likewise, the presence of concomitant preventive therapy at T3 (OR = 0.307; 95% CI = 0.115–0.817; p = 0.019) was inversely associated with optimal outcome.
When including these variables into the binary regression model, baseline CM (p = 0.020) and prior failure with anti-CGRP mAbs (p = 0.001) were the only factors that remained inversely associated with optimal control at T6 (see Supplementary material, Table S2).
Other secondary outcomes
Baseline MAMs (available for 105 subjects) decreased from 17.0 ± 18.9 to 8.7 ± 11.8 drugs taken at T3 (p < 0.001) and to 8.10 ± 15.0 at T6 (T3–T6 comparison p = 0.157). Consistently, the number of patients with medication overuse declined from 67 patients (52.3%) at T0 to 25 (19.5%; p < 0.001) at T3 and to 26 (20.3%) at T6 (T3–T6 comparison: p = 0.827).
As detailed in Table 2, from T0 to T6, PROM scores from the HIT-6, MIDAS, ASC-12 and MIBS-4 questionnaires significantly decreased (p < 0.005) while the MSQ role-function restriction score increased (p < 0.001). Notably, the most pronounced changes in these measures between T0 and T3, except for the MIBS-4 score, which continued to decline from T3 to T6 (p < 0.001). Interestingly, the mTOQ-6 score decreased from baseline to T3 (p = 0.002) and then increased from T3 to T6 (p < 0.001).
Patient-reported outcomes at baseline and at 12 and 24 weeks of observation
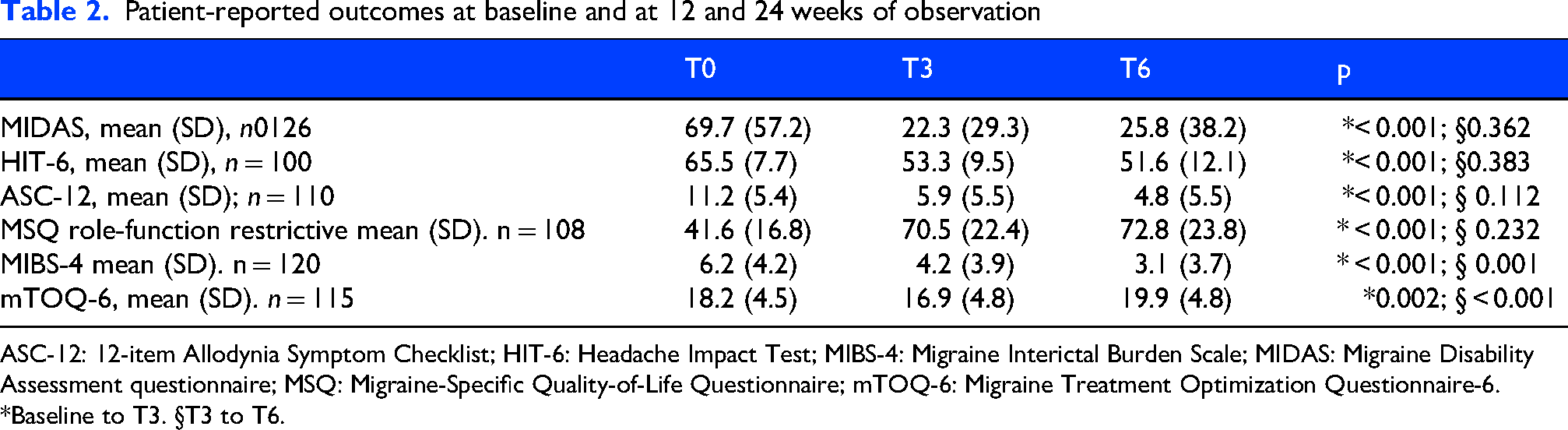
ASC-12: 12-item Allodynia Symptom Checklist; HIT-6: Headache Impact Test; MIBS-4: Migraine Interictal Burden Scale; MIDAS: Migraine Disability Assessment questionnaire; MSQ: Migraine-Specific Quality-of-Life Questionnaire; mTOQ-6: Migraine Treatment Optimization Questionnaire-6.
*Baseline to T3. §T3 to T6.
At T6, the MIDAS, HIT-6, MSQ, MIBS-4, MSQ (p < 0.001) and ASC-12 (p = 0.008) scores significantly differed across IHS position statement categories of migraine control, with better scores observed in the migraine freedom and optimal control categories. Table 3 details the paired comparisons across the IHS position statement categories of migraine control at T6.
Patient-reported outcomes at 24 weeks compared by categories of migraine control outlined by the IHS position statement
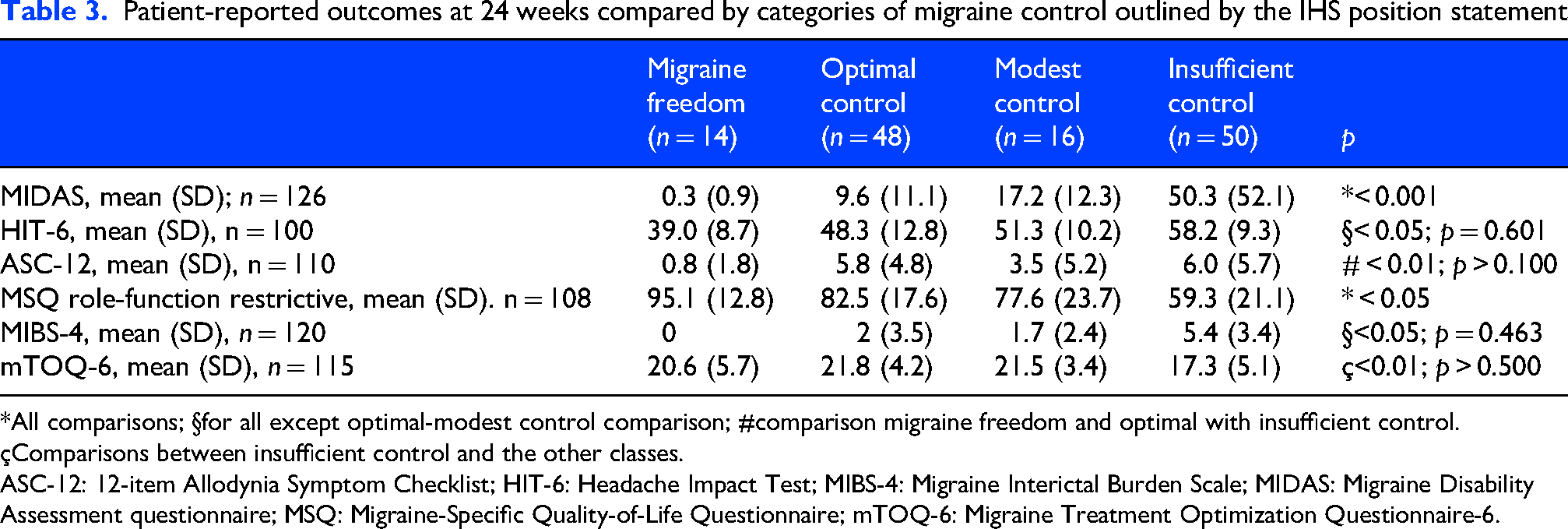
*All comparisons; §for all except optimal-modest control comparison; #comparison migraine freedom and optimal with insufficient control.
çComparisons between insufficient control and the other classes.
ASC-12: 12-item Allodynia Symptom Checklist; HIT-6: Headache Impact Test; MIBS-4: Migraine Interictal Burden Scale; MIDAS: Migraine Disability Assessment questionnaire; MSQ: Migraine-Specific Quality-of-Life Questionnaire; mTOQ-6: Migraine Treatment Optimization Questionnaire-6.
Response to atogepant in anti-CGRP mAbs naïve patients and patients with previous failure of anti-CGRP mAbs
Fifty-seven (44.5%) reported the previous use of at least one anti-CGRP mAb,
During the first 3 months of atogepant treatment, MMD reduction in anti-CGRP naïve patients (−7.3 ± 0.9) was comparable to participants who previously did not benefit from anti-CGRP mAbs (−9.9 ± 1.1; p = 0.071). From the third to the sixth month of treatment, the change in MMDs was influenced by previous failure of anti-CGRP treatment (p = 0.004): naïve patients presented a further decrease in MMDs (−0.7 ± 0.7), while non-naïve patients showed a slight increase (+2.3 ± 0.8) (Figure 2B).
While at T3, the 50%RR was not significantly different between the anti-CGRP naïve (69.0%) and the non-naïve group (59.6%; p = 0.352), at T6, the 50%RR was higher in patients who had not previously experienced ineffective treatment with anti-CGRP mAbs (70.4% vs. 50.9%, p = 0.027) (Figure 3B).
Adverse events and tolerability
Forty-eight (n = 48) patients (37.5%) reported AEs related to atogepant at T3, while 27 (21.1%) reported these at T6 (p = 0.012) (see Supplementary material, Figure S2). At T3, the most common adverse events were constipation (18.0%), nausea (9.4%), decreased appetite (4.7%) and fatigue (3.1%). None of these AE differed by body weight (body mass index (BMI) dichotomized as above or below 25 kg/m2). At T6, constipation was reported by 9.4% of subjects, nausea by 4.7%, decreased appetite by 6.3% and fatigue by 3.1%. Notably, one patient at T6 reported telogen effluvium (0.8%) (see Supplementary material, Figure S2).
From baseline to T6, AEs induced treatment discontinuation in four (3.1%) patients (two for nausea and two for somnolence/weakness)
In the overall cohort, we observed a mean weight reduction of 0.8 ± 3.1 kg from T0 to T3 (mean percentual weight loss 1.3%) (p < 0.001), while the weight remained stable from T3 to T6. At T6, 31 patients (24.2%) had gained weight, while 58 (45.3%) presented weight loss; in particular, 15 participants (11.7%), presented clinically meaningful (> 7%) weight reduction. Interestingly, in the whole group, the extent of weight loss did not significantly differ (p = 0.560) in patients who were overweight (1.6%) at baseline (i.e. BMI ≥ 25 kg/m2) compared to those with normal weight (1.2%) (Figure 5A). In those who lost weight, the mean percent variation was 5.7% (minimum 0.9 – maximum 18.4) in the overweight group and 5.3% (minimum 1.19 – maximum 16.7) in the normal-weight subjects (p = 0.760). In patients experiencing significant weight loss, weight decreased progressively during each time point (p < 0.001) (Figure 5B). Notably, there was no association between the occurrence of clinically meaningful weight loss and 50%RR (p = 0.330), nausea (p = 0.256), perceived loss of appetite (p = 0.532) or different classes of concomitant therapies (p > 0.100, consistently)

Weight (kg) variations from baseline to 12 and 24 weeks of atogepant therapy. (a, left) In the whole cohort (black line), in patients with (dashed line) and without (dotted line) baseline over-weight. (b, right) In patients with (dashed line) and without (dotted line) clinically meaningful weight loss.
Discussion
The STAR study, for the first time in a real-life setting, showed that atogepant is effective, safe, and well-tolerated in preventing migraine in EM and CM patients during 12 weeks of therapy. 15 Here, we showed the persistence of clinical benefits up to 24 weeks, as assessed by reductions in MMDs and PROMs. More specifically, the main clinical effect achieved in the first 12 weeks of therapy persisted through the 24-week observation period.
The rapidity of the effect of blocking the CGRP pathway on clinical outcomes has been previously observed with the use of anti-CGRP mAbs, which were reported to achieve the main effect in the first trimester of treatment, with only a small percentage of patients (13.1%) obtaining a significant benefit later. 33 The rapidity of action of atogepant is even more expected, as the time to peak plasma concentration is approximately 2 h, and the steady-state concentrations are typically reached as early as day 3 of daily dosing (6). Indeed, in the ADVANCE trial, the benefits of atogepant were evident as early as the first full day after treatment administration. 34
While the pharmacokinetic properties of atogepant largely support its rapid onset of action, sustained response may depend on factors such as migraine fluctuations and secondary central desensitization. In our cohort, 81.9% of subjects who were responders at T3 maintained a 50%RR also at T6. Moreover, approximately a quarter of subjects who did not respond at T3 had a 50%RR at T6.
A recent real-world study 16 reported the effectiveness and tolerability of atogepant, focusing on 100 Norwegian CM patients who completed 24 weeks of therapy. The results showed improvements in MMDs and monthly headache days at weeks 12 and 24 compared with baseline, with 50%RR of 45% and 53% at the two time points. In agreement with the above and another short-term real-world study, we reported the effectiveness of atogepant also in a percentage of patients who were non-responders to anti-CGRP mAbs.17,18 However, when assessing the MMDs from 12 to 24 weeks of treatment in patients naïve to anti-CGRP therapy compared to those with a previous unsatisfactory experience, we found that the former inclined to a further decrease in MMDs (−0.7 ± 0.7), while the latter showed a slight increase (2.3 ± 0.8) (Figure 2B). In the Norwegian study, the two groups, anti-CGRP naïve and non-naïve, presented a similar trend. 16 To clarify this issue, a longer follow-up is warranted, as long-term extensions of clinical trials did not enroll large cohorts of patients with prior failure of anti-CGRP mAbs. 35 One possible explanation is that, in a subset of patients, migraine attacks could become resistant to different proportions of the CGRP pathway inhibition.
In this view, a 50%RR assessment, although consistent with current IHS guideline standards for evaluating treatment efficacy versus placebo, cannot, by itself, comprehensively capture the overall effectiveness in alleviating the migraine burden in patients in real life. As an example, patients with CM and very frequent attacks before starting a treatment can present a very high disability migraine burden even when achieving a 50% reduction in MMDs. Although it provides a measurable and significant response to a treatment, the 50%RR is far from capturing the well-being of a person with migraine. Therefore, it should not be used as the sole instrument for evaluating treatment goals in clinical practice and real-world research.
In this context, the IHS proposed a new standard for therapy goals, aiming to picture the actual benefit that patients receive from the treatment. We applied them for the first time in a population treated with gepants. Compared to the study baseline (i.e. after previous therapies including oral preventives, BoNTA anti-CGRP mAbs), when no patients had an optimal control, we found that after 12 weeks as well as 24 weeks of atogepant, 39% of the entire cohort achieved at least optimal control of migraine; however, there was a large gap between EM (56.9%) and CM (20.6%). Along the same lines (Figure 4), the percentage of EM patients achieving migraine freedom tripled from T3 to T6 (up to 15.4%), whereas, in CM, it decreased. The different outcomes in EM compared to CM population support the idea that treating migraine earlier, before chronic transformation takes place and disability is established, is actually a game-changer. 36
On the other hand, while reaching at least an optimal migraine control in more than one-third of patients can be considered a relevant achievement in patients who have experienced multiple previous treatment failures, it also captures the residual burden of migraine despite the most innovative pharmaceutical management. 8 The presence of comorbid conditions and prior anti-CGRP mAb or BoNTa failure was also inversely associated with an optimal outcome, highlighting a group of patients whose management poses a real challenge. Those who do not benefit from all available classes of preventive treatment, defined as refractory migraine, have a poor prognosis and high disability, even when they receive care in tertiary headache centers. 37 A possible suggestion for clinical practice is to add preventive therapy to the treatment regimen, aiming to combine different mechanisms of action. 38 However, in the absence of ad hoc designed randomized trials, real-world observational studies assessing outcomes in patients on anti-CGRP mAbs combined with standard therapy suggest that concomitant therapies do not increase the magnitude of response.33,39,40 Thus, the residual burden of these patients is one of the most difficult migraine unmet needs to address. 41 While, in a more modern approach, treating migraine and comorbid conditions early will probably hinder the disease progression toward disability and chronification, more strategies should be implemented in resistant and refractory patients.36,42
In the present study, atogepant improvement of migraine burden was confirmed by several migraine-specific questionnaires. Allodynia severity and disability questionnaire scores decreased, with some fluctuations from baseline to T6, with the main variation occurring from baseline to T3 (Table 2), as observed for MMD reduction.
Several scales are used in clinical trials. real-world studies and clinical practice. Although none of these tools fully capture the entire experience of headache disability, and each score has its disadvantages, they are the only available instruments to depict the many aspects of the disease burden. 43 To better understand the impact of migraine on PROMs, we compared scores across the categories outlined in the IHS position statement at 24 weeks of observation. Allodynia and PROMs scores differed across categories (Table 3), with a significantly greater impact in the optimal than in the insufficient control group, confirming that adequate control of migraine days is indispensable for improving disability scores. Indeed, quality of life is particularly sensitive to improvements in MMD. The role function restrictive domain of the MSQ scale measures how migraine limits a patient's ability to engage in daily social, work, and personal activities, capturing the functional impact of migraine beyond pain, including on relationships, leisure and productivity (i.e. higher scores indicate better outcomes). The scores increased from baseline to T3 but were significantly different across the different categories, even between “migraine freedom” and “optimal control.” This aspect has significant social and economic implications, as an active and healthy individual uses fewer healthcare resources and is more productive. 44
In the STAR study, atogepant was well tolerated, with no serious AEs, and the rate of patients reporting AEs related to atogepant decreased from T3 to T6, suggesting that tolerance could occur over time. The low discontinuation rate (13.2%) was principally due to the lack of effectiveness (see Supplementary material, Figure S1). In our cohort, AEs were not influenced by BMI, suggesting that they are not related to the drug body distribution. 29
The magnitude of weight loss (1.3%) was slightly lower than that observed in RCTs, where the percentage change from baseline was around 2% at 24 weeks. Conversely, in RCTs, only 4.9–5.8% of patients had a reduction of 7% or higher, while, in our cohort, it was observed in 11.7% of participants. 45 This discrepancy is surprising, especially considering that RCTs included patients with a mean BMI higher than 25 kg/m2 (overweight) or 30 kg/m2 (obesity) 45 and one could expect that the weight loss is proportional to the baseline body weight.
Similarly, in our cohort, weight reduction was not influenced by baseline BMI, suggesting that other factors (e.g. individual predisposition, metabolism, diet and maybe a return to an active lifestyle) also contribute to weight loss in treated patients. In RCTs, weight loss increased progressively from baseline to 24 weeks, then stabilized from 40–52 weeks. In the 24-week STAR population, the weight change occurred in the first 12 weeks and remained stable until 24 weeks (Figure 5A). However, those with clinically meaningful weight loss (≥ 7%) exhibited a progressive weight reduction (Figure 5B), which was neither related to nausea and perceived loss of appetite, nor with 50%RR, suggesting that a subset of patients is particularly prone to atogepant-related weight loss; however, the mechanisms are unclear.
To the best of our knowledge, STAR is the first study to evaluate the effectiveness of atogepant using the new outcomes outlined by the IHS position statement, 19 providing the highest level of data quality. Its strengths include a prospective, multicenter design that reflects routine clinical practice across several Italian headache centers and its integration with a nationwide registry of patients with headache disorders. Another strength is the prospective data collection over 24 weeks, which enabled assessment of both AE tolerance over time and response consistency. The study also followed guidelines for real-world preventive treatment studies 24 and was preregistered.
The main limitation of our study is its real-world observational design, which lacks a placebo control group and more detailed information, such as tablet counts, to assess treatment adherence. Moreover, we included only patients enrolled in selected Italian centers. These aspects may limit interpretation and make the results susceptible to uncontrolled confounding factors, and are prone to selection bias, potentially including patients with more severe or treatment-resistant migraine, not reflecting the general population of migraine patients.
Conclusions
The STAR study demonstrates that atogepant effectiveness is maintained over 24 weeks of treatment, with approximately one-quarter of patients becoming responders after 12 weeks. Besides, no serious safety concerns emerged in this real-world population. Additionally, the STAR study proves that the ambitious goals set by the IHS position statement on higher standards for migraine treatment can be achieved with atogepant in many patients, suggesting that anti-CGRP drugs should be offered earlier in the disease course. 46 Nonetheless, we highlight that, especially for patients with CM, a substantial unmet need may be present even with atogepant treatment, and that future studies should aim to define how we can achieve better outcomes.
Clinical implications
Atogepant effectiveness in real-life, assessed by reductions in MMDs and PROMs, is observed within the first 12 weeks and persists across 24 weeks of treatment.
Optimal control of migraine was achieved in 56.6% of EM but in 20.6% of CM patients
No signals of emergent adverse events are evident with 24 weeks of atogepant treatment.
Supplemental Material
sj-docx-1-cep-10.1177_03331024261435922 - Supplemental material for SafeTy and effectiveness of Atogepant accoRding to the IHS outcome categories: A multicentric, prospective observational study in real life (the 24-week STAR study)
Supplemental material, sj-docx-1-cep-10.1177_03331024261435922 for SafeTy and effectiveness of Atogepant accoRding to the IHS outcome categories: A multicentric, prospective observational study in real life (the 24-week STAR study) by Claudia Altamura, Luigi Francesco Iannone, Flavia Lo Castro, Gabriele Sebastianelli, Marilena Marcosano, Federico De Santis, Michele Corrado, Raffaele Ornello, Licia Grazzi, Danilo Antonio Montisano, Francesco De Cesaris, Antonio Munafò, Gianluca Avino, Michele Trimboli, Maria Albanese, Antonio Russo, Marcello Silvestro, Giorgio Dalla Volta, Marina Romozzi, Paolo Calabresi, Alberto Boccalini, Paola Merlo, Maria Pia Prudenzano, Maria Rosaria Valente, Innocenzo Rainero, Giada Giuliani, Marta Altieri, Luisa Fofi, Alberto Doretti, Gloria Vaghi, Francesca Pistoia, Delfina Ferrandi, Stefania Battistini, Gianluca Coppola, Simona Guerzoni, Simona Sacco, Cristina Tassorelli, Fabrizio Vernieri and in Cephalalgia
Supplemental Material
sj-docx-2-cep-10.1177_03331024261435922 - Supplemental material for SafeTy and effectiveness of Atogepant accoRding to the IHS outcome categories: A multicentric, prospective observational study in real life (the 24-week STAR study)
Supplemental material, sj-docx-2-cep-10.1177_03331024261435922 for SafeTy and effectiveness of Atogepant accoRding to the IHS outcome categories: A multicentric, prospective observational study in real life (the 24-week STAR study) by Claudia Altamura, Luigi Francesco Iannone, Flavia Lo Castro, Gabriele Sebastianelli, Marilena Marcosano, Federico De Santis, Michele Corrado, Raffaele Ornello, Licia Grazzi, Danilo Antonio Montisano, Francesco De Cesaris, Antonio Munafò, Gianluca Avino, Michele Trimboli, Maria Albanese, Antonio Russo, Marcello Silvestro, Giorgio Dalla Volta, Marina Romozzi, Paolo Calabresi, Alberto Boccalini, Paola Merlo, Maria Pia Prudenzano, Maria Rosaria Valente, Innocenzo Rainero, Giada Giuliani, Marta Altieri, Luisa Fofi, Alberto Doretti, Gloria Vaghi, Francesca Pistoia, Delfina Ferrandi, Stefania Battistini, Gianluca Coppola, Simona Guerzoni, Simona Sacco, Cristina Tassorelli, Fabrizio Vernieri and in Cephalalgia
Supplemental Material
sj-docx-3-cep-10.1177_03331024261435922 - Supplemental material for SafeTy and effectiveness of Atogepant accoRding to the IHS outcome categories: A multicentric, prospective observational study in real life (the 24-week STAR study)
Supplemental material, sj-docx-3-cep-10.1177_03331024261435922 for SafeTy and effectiveness of Atogepant accoRding to the IHS outcome categories: A multicentric, prospective observational study in real life (the 24-week STAR study) by Claudia Altamura, Luigi Francesco Iannone, Flavia Lo Castro, Gabriele Sebastianelli, Marilena Marcosano, Federico De Santis, Michele Corrado, Raffaele Ornello, Licia Grazzi, Danilo Antonio Montisano, Francesco De Cesaris, Antonio Munafò, Gianluca Avino, Michele Trimboli, Maria Albanese, Antonio Russo, Marcello Silvestro, Giorgio Dalla Volta, Marina Romozzi, Paolo Calabresi, Alberto Boccalini, Paola Merlo, Maria Pia Prudenzano, Maria Rosaria Valente, Innocenzo Rainero, Giada Giuliani, Marta Altieri, Luisa Fofi, Alberto Doretti, Gloria Vaghi, Francesca Pistoia, Delfina Ferrandi, Stefania Battistini, Gianluca Coppola, Simona Guerzoni, Simona Sacco, Cristina Tassorelli, Fabrizio Vernieri and in Cephalalgia
Footnotes
Acknowledgments
The “Società Italiana per lo Studio delle Cefalee” (SISC) is acknowledged for the “Registro Italiano delle Cefalee (RICe)”.
ORCID iDs
Ethical considerations
The local Ethics committee approved the study as part of the Registro Italiano Cefalee (RICe) study (Studio RICe, 14591_oss CEAVC Studio RICe, 14591_oss and subsequent amendments).
Consent to participate
All patients provided their written informed consent before starting treatment with atogepant.
Author contributions
FV, CA, GS and LFI had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis; designed the study; and drafted the manuscript. CA performed statistical analysis. LFI and FV performed administrative and technical support. All authors recruited patients, and critically reviewed the manuscript, agreed to be fully accountable for ensuring the integrity and accuracy of the work, and read and approved the final manuscript submitted for publication.
Funding
This research did not receive any specific grants from funding agencies in the public, commercial or not-for-profit sectors.
Declaration of conflicting interests
CA is Associate Editor for Frontiers of Human Neuroscience and Frontiers in Neurology Headache and Neurogenic Pain section; she received travel grants and/or personal fees for advisory boards and speaker panels, from Novartis, Eli-Lilly,Lundbeck, Teva, Lusofarmaco, Laborest, Abbvie/Allergan, Almirall and Pfizer.
SG received fees and honoraria for advisory boards, speaker panels or clinical investigation studies from Teva, Eli Lilly/Organon, Pfizer, Lundbeck, Angelini and AbbVie.
LFI received financial support, consulting fees for the participation in advisory boards and support for attending meetings from: Teva, Eli Lilly, Lundbeck, Pfizer and AbbVie; he is Associate Editor for Frontiers in Neurology Headache and Neurogenic Pain section. GS received honoraria from AbbVie. RO reports personal fees from AbbVie, Bayer, Eli Lilly, Lundbeck, Novartis, Organon, Pfizer and Teva; he is Associate Editor for Arquivod de Neuropsiquiatria and Editorial Board Member for The Journal of Headache and Pain and Confinia Cephalalgica.
FP reports personal fees from Lundbeck and Organon.
GS received personal fees from AbbVie.
GV reports personal fees and non-financial support from TEVA.
SS reports compensation from Novartis for other services; compensation from Novo Nordisk for consultant services; compensation from Boehringer Ingelheim for consultant services; compensation from Teva Pharmaceutical Industries for consultant services; compensation from Allergan for consultant services; employment by Università degli Studi dell’Aquila; compensation from Novartis for consultant services; compensation from Allergan for consultant services; compensation from Pfizer Canada Inc for consultant services; compensation from Abbott Canada for consultant services; compensation from H. Lundbeck AS for consultant services; compensation from AstraZeneca for consultant services; and compensation from Eli Lilly and Company for consultant services.
SG has received fees and honoraria for advisory boards, speaker panels or clinical investigation studies from Teva, Eli Lilly/Organon, Pfizer, Lundbeck, Angelini and AbbVie.
CT in the past 3 years has received support (financial or drugs) from AbbVie and Novartis for an investigator-initiated trial; consulting fees for the participation in advisory boards for AbbVie, Dompé, Eli Lilly, Ipsen, Lundbeck, Medscape, Pfizer and Teva; honoraria for scientific lectures and presentations from AbbVie, Eli Lilly, Lundbeck, Pfizer and Teva; support for attending meetings from AbbVie, Dompé, Eli Lilly, Ipsen, Lundbeck, Pfizer and Teva; has been Principal Investigator in clinical trials sponsored by AbbVie, Biohaven, Eli Lilly, Ipsen, Lundbeck, Pfizer and Teva; and received grants from the European Commission, the Italian Ministry of Health and the Italian Ministry of University.
FV has received financial support from AbbVie, Angelini and Lundbeck for investigator-initiated trials; consulting fees for the participation in advisory boards from AbbVie, Angelini, Eli Lilly, Lundbeck, Organon, Novartis, Pfizer and Teva; honoraria for scientific lectures and presentations from AbbVie, Eli Lilly, Lundbeck, Novartis, Pfizer and Teva; support for attending meetings from Abbvie, Eli Lilly, Lundbeck, Pfizer and Teva; has been Principal Investigator in clinical trials sponsored by AbbVie, Eli Lilly, Lundbeck, Pfizer and Teva.
The other authors have no relevant financial or non-financial interests to disclose.
Data availability statement
Data supporting the findings in the present study are reported in the article and in the supplementary materials. The data collected and analyzed for the current study are available from the corresponding author on reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.