
Abstract
To move towards clinical applications, tissue engineering (TE) should be validated with human primary cells and offer easy connection to the native vascularisation. Based on a sheet-like bone substitute developed previously, we investigated a mesenchymal stem cells/endothelial cells (MSCs/ECs) coculture to enhance pre-vascularisation. Using MSCs from six independent donors whose differentiation potential was assessed towards two lineages, we focused on donor variability and cell crosstalk regarding bone differentiation. Coculture was performed on calcium phosphate granules in a specific chamber during 1 month. MSCs were seeded first then ECs were added after 2 weeks, with respective monocultures as control groups. Cell viability and organisation (fluorescence, electronic microscopy), differentiation (ALP staining/activity, RT-qPCR) and mechanical cohesion were analysed. Adaptation of the protocol to coculture was validated (high cell viability and proliferation). Activity and differentiation showed strong trends towards synergistic effects between cell types. MSCs reached early mineralisation stage of maturation. The delayed addition of ECs allowed for their attachment on developed MSCs’ matrix. The main impact of donor variability could be here the lack of cell proliferation potential with some donors, leading to low differentiation and mechanical cohesion and therefore absence of sheet-like shape successfully obtained with others. We suggest therefore adapting protocols to cell proliferation potentials from one batch of cells to the other in a patient-specific approach.
Introduction
Bone tissue engineering, among other regenerative medicine approaches, aimed at developing new solutions to treat injured bone tissues and fill critical size defects that cannot healed naturally.1,2 Cells of interests, namely osteoblasts and/or mesenchymal stem cells (MSCs), are cultured in vitro on a scaffold to form hybrid implantable substitutes with same organisation and functions as the native tissue. 3 Such substitutes could be an alternative to allografts (suffering from risks of rejects and transmission of infection), 3 autografts (limited stock, donor site morbidity),4,5 and biomaterials used alone (limited bioactivity).6,7 However, a current drawback in tissue-engineered substitutes preventing strong clinical outcomes is their poor connexion to the existing vascular network after implantation.8,9 Native bone is indeed a composite tissue containing a high density of blood vessels allowing for the diffusion of interstitial liquid, waste and nutrients from and to the cells.6,8 If there is no blood capillaries in the close environment of cells (150–200 μm), death of the implanted rebuilt tissue can occur at mid or short term.6,10 To achieve translational progresses, it is therefore mandatory to enhance the vascularisation potential of bone substitutes to allow for easy connection in vivo.
One of the main approaches to perform pre-vascularisation is the combination of endothelial cells (ECs) with the primary cells of interest during the development of the implantable substitutes in vitro through coculture protocols.11–15 Important cross-talks have been reported between ECs and MSCs including mutual beneficial effects and synergies on their respective growth and differentiation,16–19 in particular through a virtuous circle involving the vascular endothelial growth factor (VEGF, produced by MSCs and promoting angiogenesis) and the bone morphogenetic protein-2 (BMP-2, produced by ECs, promoting bone differentiation while stimulating in turn VEGF production by MSCs). 20 Although this method has been widely investigated, consensus are still lacking for many parameters of the coculture protocols, such as ECs:MSCs cell ratio, medium composition over time, seeding density or scaffold structure and composition.13,21–23 It is also still discussed if cells should be cultured together at day 1 or the addition of one cell lineage delayed, as suggested by some studies.13,24 Here, we hypothesised that delayed addition of ECs would allow first for proper osteogenesis from the MSCs before creating a pro-angiogenic environment, and we hypothesised that the added ECs could then benefit from the new matrix produced by the differentiated MSCs to better anchor to the scaffold (as protein coating such as collagen are often required to expand ECs in 2D culture). Optimising these aspects would be very promising to benefit from the mutual synergetic effects that could appear between ECs and MSCs, in particular promoting the MSC differentiation into osteoblasts, resulting in faster and more reliable processes. This would in turn decrease the overall costs of such cell-based therapies as less consumables and work force would be required.
In a previous study, we developed a novel biohybrid bone substitute specifically designed to meet clinicians’ expectations for the regeneration of the maxillo-facial area 25 : pre-osteoblastic cells (MC3T3-E1) were cultured on a monolayer of calcium phosphate granules with various sizes in a parallelepiped culture chamber for 4 weeks. On granules with optimal size (80–200 µm), cells were able to grow and differentiate to form a continuous tissue surrounding the granules, providing mechanical cohesion to the cell/material composite. We obtained therefore, after opening the chamber, a sheet-like substitutes formed by both the newly formed cell matrix and the scaffold with good mechanical cohesion. 25 This allowed the substitute to be folded or stretched with easy handling. This original shape would be beneficial for maxillo-facial surgeries as clinicians could adapt it manually to the specific geometry of each patient’s face but from a single standard fabrication protocol. 25 Both biological and mechanical properties of this substitute were characterised, optimised and validated with a pre-osteoblastic cell line.
To bring this innovative process a step further towards a clinically relevant solution, our objective here was therefore to perform a proof-of-concept study evaluating if the sheet-like substitute could be adapted to a ECs/MSCs coculture with similar performance. The co-culture approach would also allowed validating its in vitro pre-vascularisation potential and confirming assumptions on ECs and MSCs interactions. As a translational research approach, we used human primary mesenchymal stem cells obtained from bone marrow of six different donors as a first batch. After validating their potential to differentiate towards two lineages (osteogenic and adipogenic fates), we focused on the evaluation of bone differentiation within our system. The comparison of variations between donor sources in a tissue-engineering process previously validated was therefore also a main objective of this study. Both lineages were cultured inside the specific culture chamber on calcium phosphate granules over 1 month, following optimisation of the technical parameters in our previous study. 25 We used a delayed coculture approach, seeding first MSCs before addition of ECs. We focused on the evaluation of cell viability, morphology and organisation of the rebuilt tissue through microscopy as well as early (alkaline phosphatase staining and activity) and late (gene expression of specific markers) differentiation of MSCs to osteoblasts, with a special care to the comparison of the substitute’s cohesion between donors. Survival of the endothelial population was also evaluated through fluorescence microscopy observations and specific gene expression. Cross-analysis was then performed in regard of the donor variability, to highlight the most affected parameters and suggest ways of optimisation towards patient-specific protocols.
Methods
Isolation and expansion of cells
Proliferation medium consisted in αMEM (Sigma, USA) supplemented with 10% foetal bovine serum (FBS, Eurobio, France), 2 mM L-glutamine (Gibco, USA) and 1% penicillin/streptomycin (Gibco, USA). Differentiation medium consisted in DMEM high glucose (Sigma, USA) with 10% FBS (Eurobio, France), 2 mM L-glutamine (Gibco, USA), 1% penicillin/streptomycin (Gibco, USA), 10 mM β-glycerophosphate (Sigma, USA), 50 µg/mL ascorbic acid (Sigma, USA), 0.1 µM dexamethasone (Sigma, USA). The endothelial medium consisted in EGM-2 medium (Lonza, Switzerland).
MSCs: Bone marrow aspirates were obtained by standard puncture and aspiration from the iliac crest of healthy human donors after receiving informed consent. The protocol was approved by the Amiens University Hospital Ethics Committee. Bone marrow aspirates were obtained from six donors (three males and three females, aged 35–56 years). After centrifugation, the buffy coat layer (mononuclear cells) was isolated and plated in T175 culture flasks with proliferation medium supplemented with 2 ng/mL basic fibroblast growth factor (TebuBio, France). Cells were seeded at 1 × 105 mononuclear cells/cm2 and cultured in 5% CO2 atmosphere humidified at 37°C. Medium was refreshed every 4 days, three times and then every week. Then, adherent cells were enzymatically removed with 0.25% trypsin—EDTA (Sigma, USA) at 37°C and seeded at 1500 cells/cm2 in T175 culture flasks. Cells cultured in proliferation medium were used between passage 4 and 5. Differentiation capacity of MSCs was successfully tested prior to use (two-lineage assay, bone and adipose tissue after 21 days of culture).
ECs: Human umbilical cord bloods were collected from full-term births with informed consent. The protocol was approved by the Amiens University Hospital Ethics Committee. Umbilical cord bloods were diluted at a ratio of 1/1 (v/v) in PBS. The cells suspensions were added to Ficoll Plus solution (Sigma, USA) followed by centrifugation at 1800 rpm for 30 min. Peripheral blood mononuclear cells were washed twice with PBS, and then suspended in endothelial medium. Cell suspensions were seeded at a density of 1.25 × 106 cells/well into 24-well plates. Medium was refreshed every day during the first week and then every 2 days. Cells were used between passage 4 and 5.
Biomaterials
Sterile calcium phosphate granules (20% hydroxyapatite (HA) Ca10 (PO4)6 (OH)2, 80% b-tricalcium phosphate (TCP) Ca3 (PO4)2) were kindly provided by Biomatlante, France.
Culture protocols
We used the Clinicell25® culture cassette (Mabio-International, Figure 1(a)) as a specific flat cell culture chamber for the development of the tissue-engineered substitute. It is a gas-permeable, plasma-treated parallelepiped chamber with a 24-cm2 culture surface for a 10-mL volume filled with medium. After 1 month of culture, the upper side of the chamber was opened to remove the tissue samples and to perform analyses. Based on previous optimisation, 25 400 mg of calcium phosphate granules with a 80–200-mm diameter were inserted as scaffold material inside the culture chamber, to obtain a single monolayer covering the whole surface. To minimise ion release from the granules during cell culture, they were incubated in culture chamber 48 h in proliferation medium before cell seeding.
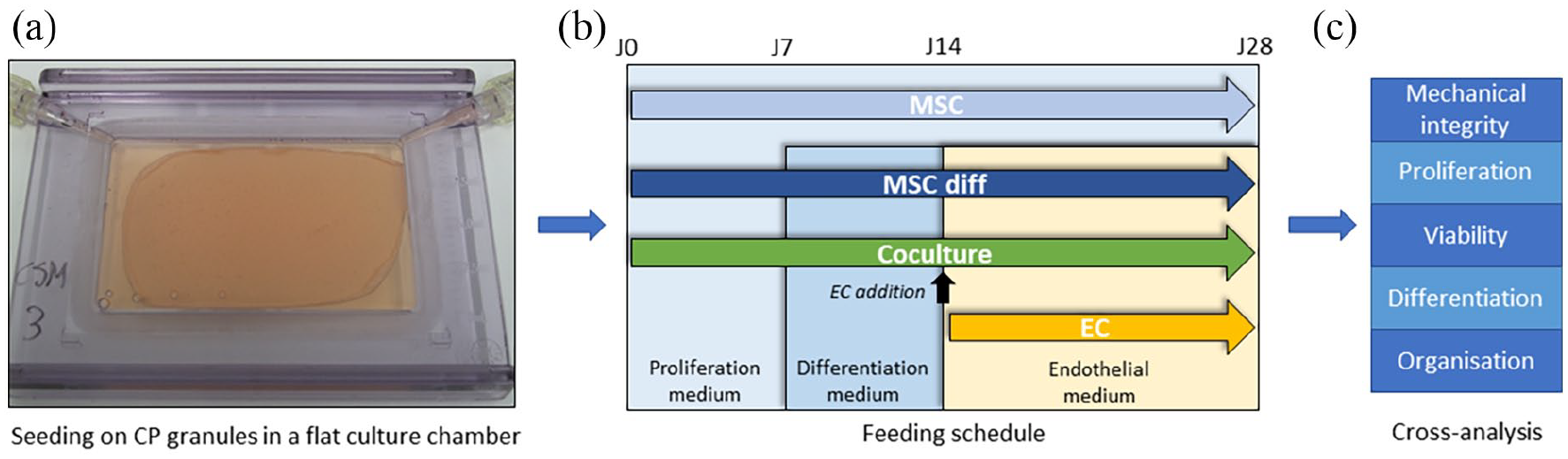
Study design. (a) MSCs are seeded in a specific culture chamber and attach on a calcium phosphate (CP) granules monolayer, forming a sheet-like substitute over time (here: sample from MSC group after 1 week), (b) several feeding schedules, mono- and co-culture protocols are compared and (c) the rebuilt tissue is removed from the chamber after 1 month to perform cross-analysis between groups and between donors regarding mechanical integrity of the substitutes, cell proliferation, cell viability, cell differentiation and tissue organisation.
The coculture protocol and feeding schedule are reported in Figure 1(b). In first phase, after gently elimination of chamber medium, 2 × 106 MSC were injected in the culture chamber. MSCs were cultured in proliferation medium over the first week and in osteogenic differentiation medium over the second week. ECs were then added (2.5 × 106 cells) with endothelial medium and cells cocultured for two additional weeks. Medium was refreshed twice per week. Three control groups were used in the same culture chamber with scaffolds, (1) monoculture of MSCs in proliferation medium for 4 weeks (‘MSC’ group), (2) monoculture of MSCs in proliferation medium for 1 week, differentiation medium for 1 week and endothelial medium for 2 weeks, that is the same feeding schedule as the coculture samples (‘MSC diff’ group) and (3) monoculture of ECs in endothelial medium for 2 weeks (Figure 1(b)). For each independent donor, three chambers per group were seeded as technical replicates.
Cell viability and organisation
After 1 month in the chamber, cell viability was estimated by a Live/Dead® kit (Invitrogen, USA) according to the manufacturer’s protocol on sections of the tissue-engineered substitutes. Calcein AM (1 µM) and Ethidium homodimer-1 (EthD-1, 1 µM) fluorescent dyes were respectively employed to stain viable and dead cells. The samples were observed using fluorescence microscopy (Leica microsystems, Germany).
Samples were also analysed using immunofluorescence staining. Briefly, after washing with PBS, sections of the tissue-engineered substitutes were fixed with 4% paraformaldehyde for 10 min (Agar Scientific, UK) then rinsed and permeabilised with 0.5% Triton X-100 (VWR, UK) for 10 min. They were then immersed in the same Triton solution supplemented with bovine serum albumin (Sigma, USA) for 20 min. Murine anti-connexin 43 (Cx43) primary antibodies (Invitrogen, USA) and donkey anti-mouse 488 secondary antibodies (Invitrogen, USA) were used to highlight gap junctions between cells. A rhodamine/phalloidin solution (Invitrogen, USA) was used to visualise actin filaments.
Tissue morphology and density were observed using scanning electron microscopy (Philips XL30 ESEM-FEG, The Netherlands). The samples were immersed in Rembaum solution for 24 h, rinsed with demineralised water and then coated with gold before observations.
Cell differentiation
Section of the tissue-engineered substitutes were fixed with 2% formalin after 1 month in the culture chamber and stained for alkaline phosphatase (ALP) coloration using SIGMAFAST™ BCIP®/NBT (Sigma, USA). ALP activity was determined using the kit ‘Alkaline Phosphatase Activity Colorimetric Assay’ (Biovision, USA), according to the manufacturer’s recommendations. The level of ALP activity was normalised by the amounts of total protein in the cell lysates (nmol/min/mg protein). Details of the total protein mass measurements are available as Supplemental Information (Supplemental Figure S1).
Differentiation was also assessed through the expression of genes of interest quantified by reverse transcription-polymerase chain reaction (RT-qPCR). Total RNA was isolated using Tri Reagent (Sigma, USA). RNA concentrations were measured by NanoPhotometer® Pearl (Implen, Germany). The reverse-transcription PCR reactions were performed with 1 µg total RNA using High capacity cDNA reverse Transcription kit (Applied Biosystem, USA). The reaction was performed for 10 min at 25°C, followed by 2 h at 37°C, and 5 min at 85°C. Real time-PCR was performed using SYBR PrimeScript RT-PCR kit, according to the manufacturer’s recommendations. All amplifications were normalised by GAPDH expression. Results were analysed using the comparison Ct (2-DDCt) method, and expressed as fold-change compared with MSC control group. Primers sequences used in this study are reported in Supplemental Table S1.
Statistical analysis
All results were obtained on samples seeded with MSCs from six independent donors, as stated earlier. Cells obtained from one specific donor were used in at least three technical replicates for each analysis method. The focus was then given to the comparison between donors and variability analysis. Statistical significance between groups (MSCs, MSCs diff, ECs or coculture) was estimated after calculating the mean of all donors using two-way ANOVA with Tukey’s test, however no significance was found due to this variability (p > 0.05). Normal distribution of the data was successfully checked through a Kolmogorov-Smirnov normality test.
Results
Cell viability and organisation
The cocultured tissue-engineered substitutes were first evaluated with fluorescence microscopy and SEM at the end of the 1-month culture. Samples were also taken out of the culture chambers and handled to assess integrity and cohesion (i.e. handling with forceps was possible without disrupting the sample). Observations of Live/Dead staining (Figure 2(a)) showed a very good overall viability in all samples regardless of the donor source. Only a few dead cells were found (red dots) locally in the cell tissue covering the phosphate calcium granules. No difference of viability was observed between the six donors of MSCs. These observations revealed that cells were able to attach to the granules and to overlay them with a 3D organisation. The cells organised together and formed a tissue that encompassed the granules, although cell density could slightly differ from one donor to the other.
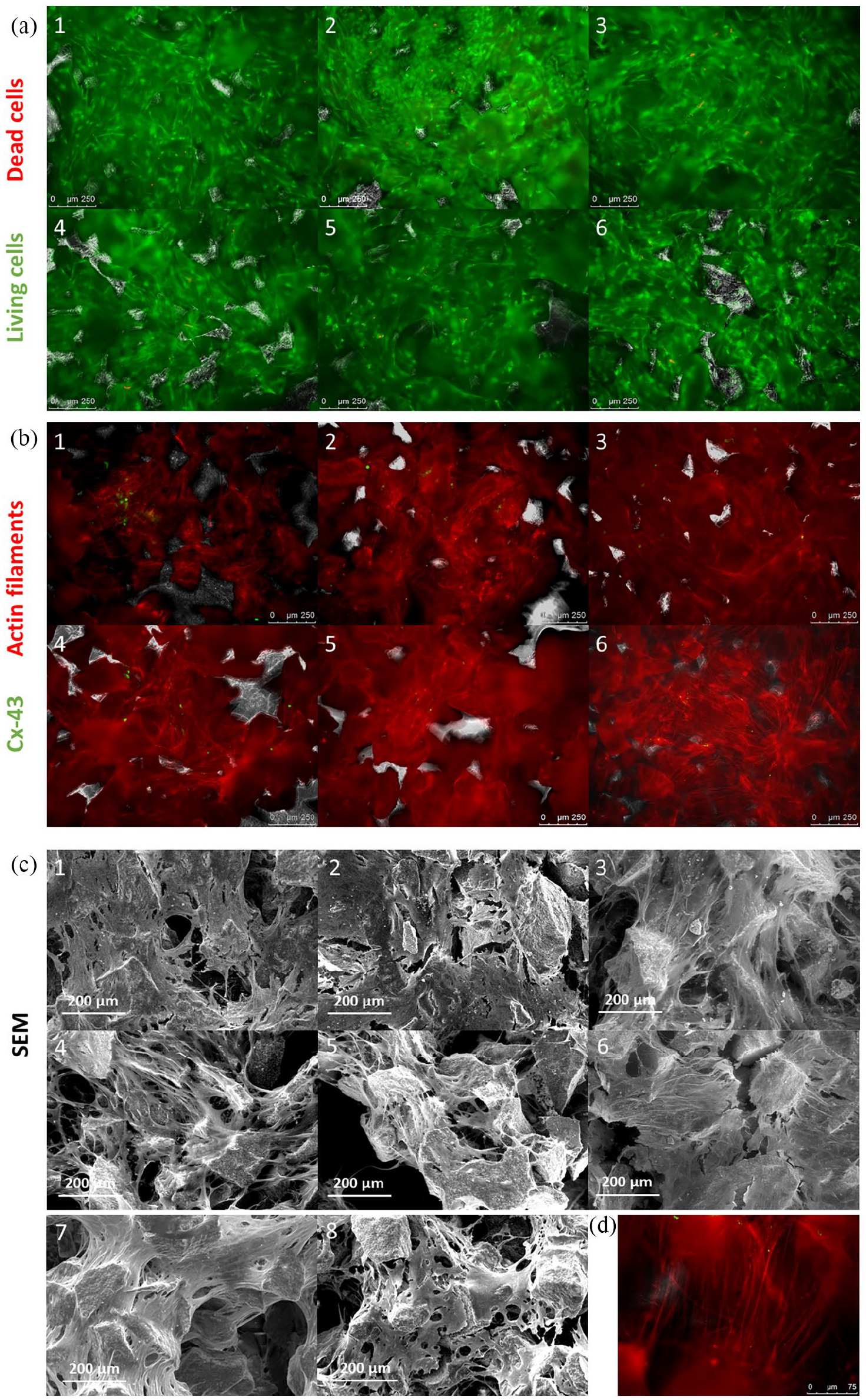
(a) Cell viability in sheet-like cocultured substitutes after 1 month. Live/Dead fluorescence staining of MSCs and ECs cocultured on BCP granules. Calcein AM in green (living cells) and EthD-1 in red (dead nuclei). (b) Cell organisation in sheet-like substitutes after 1-month culture. Actin filament and Cx-43 staining of MSCs and ECs cocultured on BCP granules. Rhodamine-phalloidin in red (actin filaments) and Cx-43 immunofluorescence in green (gap junctions). (c) Cell morphology in sheet-like substitutes after 1-month culture observed with Scanning electron microscopy. Donors 1–6; 7: MSC; 8: MSC diff. (d) Illustration of the overall morphology of actin filaments in the samples at higher magnification, here for donor 2. Same staining conditions as panel B.
To confirm this, we investigated further the developed cell tissue with other microscopy modalities. Indeed, slight differences were noticed regarding cell density after analysis of the actin filament staining and SEM observations (Figure 2(b) and (c)). On the one hand, the samples obtained with cells from donors 1 to 4 showed a continuous cell tissue penetrating the porous structure of the granules and filling the gaps between them with many bridges formed by the actin filaments (Figure 2(d)). Cells were spread with standard morphology, similar to MSCs monocultures on the same biomaterial although cell density could be slightly lower (Figure 2(c)), an expected result as the MSC group was maintained in proliferation culture medium. Cx43 expression (presence of gap junctions) was frequently noticed, highlighting early cell organisation, although no pattern or specific location could be noticed. This resulted in intermediate (donors 1 and 2) to good (donors 3 and 4) mechanical cohesion when substitutes were handled. It has to be mentioned that donor 4 showed higher heterogeneity of the cell tissue, and granules in some areas were not covered by cells. The mechanical integrity was therefore very strong at some locations while granules were still moving freely at others. Cx43 was however clearly present where the tissue successfully developed. On the other hand, substitutes obtained with cells from donors 5 and 6 showed mitigated results regarding density and homogeneity of the cell tissue. Although viability was good as mentioned earlier (Figure 2(a)), cells poorly proliferated in some areas, resulting in holes between the granules, as shown in particular for donor 5 (Figure 2(b)), some granules are not covered by cells as showed by the dark smooth red autofluorescence instead of bright actin filaments) which led to low mechanical cohesion (immediate disruption upon handling). This sample also showed very low expression of Cx43, barely noticeable (Figure 2(b)).
Cell differentiation
ALP is an early marker of osteoblast differentiation. 26 ALP staining and activity measurement on samples removed from the culture chamber after 1 month can be seen in Figure 3. First, this confirmed the differences of integrity depending on the culture group (Figure 3(a)). As it could be expected, ECs didn’t produce substantial extracellular matrix, preventing the calcium phosphate granules to be bound together. Mechanical cohesion was similar between differentiated and non-differentiated MSCs, but the combination of both lineages in coculture led to improved integrity for some donors, in particular donor 3. We can also notice on these results the differences reported earlier with microscopy analysis depending on the donor source, with donor 5 showing poor mechanical cohesion and donor 4 high heterogeneity.
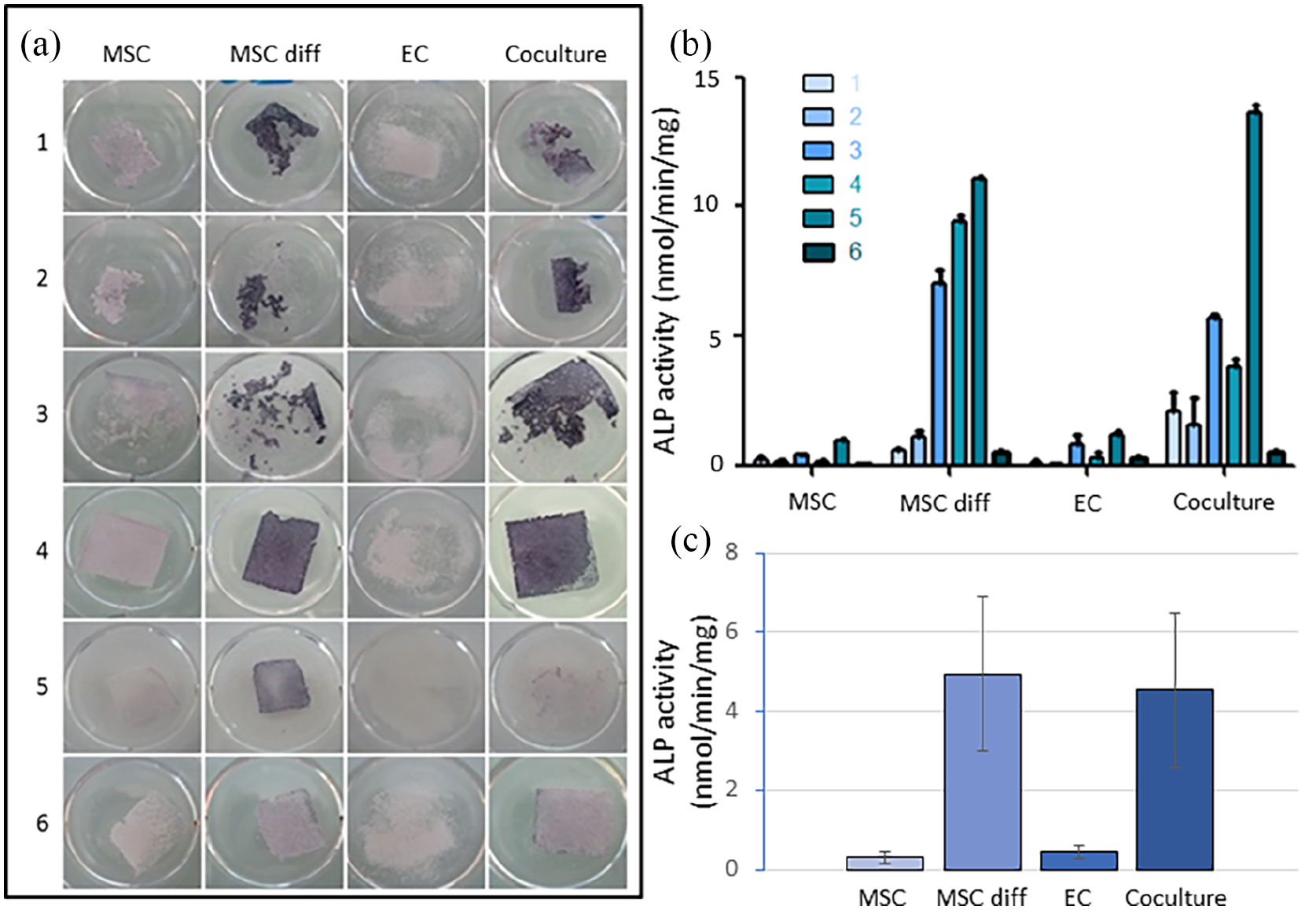
(a) Alkaline phosphatase staining and (b) alkaline phosphatase activity normalised by the amounts of total protein in the cell lysates (nmol/min/mg protein) of sheet-like substitutes (monocultures and cocultures) after 1-month culture for donors 1–6 (mean and standard deviation of technical replicates). (c) Mean of the results from panel B for each study group (mean and standard error of mean, no statistical significance (p > 0.05)).
Purple colour on tissue samples highlighted the presence of ALP (Figure 3(a)) and the activity of the produced enzyme was evaluated quantitatively (Figure 3(b)). ALP was differently expressed depending on donor although overall very high levels could be achieved in both MSC diff and coculture groups for most of them. These results were shown by both methods with good consistency, ALP activity highlighting quantitatively the important increase in these two groups (up to 15–20 fold, including when considering the mean result per group as seen in Figure 3(c)). As expected, non-differentiated MSCs and ECs didn’t produce this bone marker. Donor 6 showed the lowest expression in all groups.
The expression of genes of interest was then quantified with RT-qPCR to assess late bone differentiation (Figure 4(a), Runx2, ALP, Col1a2, OCN, OPN and BSP) as well as presence and activity of endothelial cells at the end of the coculture (Figure 4(b), Vegfr2, Von Willebrand factor). Runx2, a transcription factor, is considered as the first and most specific marker of early osteoblast differentiation, 26 followed by increase in the expression of ALP, OPN and the non-specific Col1a2. All of these three markers reach a peak of production as osteoblast differentiation occurs, followed by as slight decrease as cells start to mineralise.26,27 BSP and OCN are then the latest markers to be expressed, highlighting the osteoblast maturation.28,29 OPN would be the only marker to be still expressed by osteocytes. 30 Results were normalised by the expression obtained with non-differentiated MSCs cultured alone to highlight the variations through coculture. Vegfr2- and Von Willebrand factor-related genes were selected as markers of the presence of endothelial cells.31,32 Results of bone-related markers in monoculture of ECs are not reported in the figure as they led to null expression or background noise only, as expected.
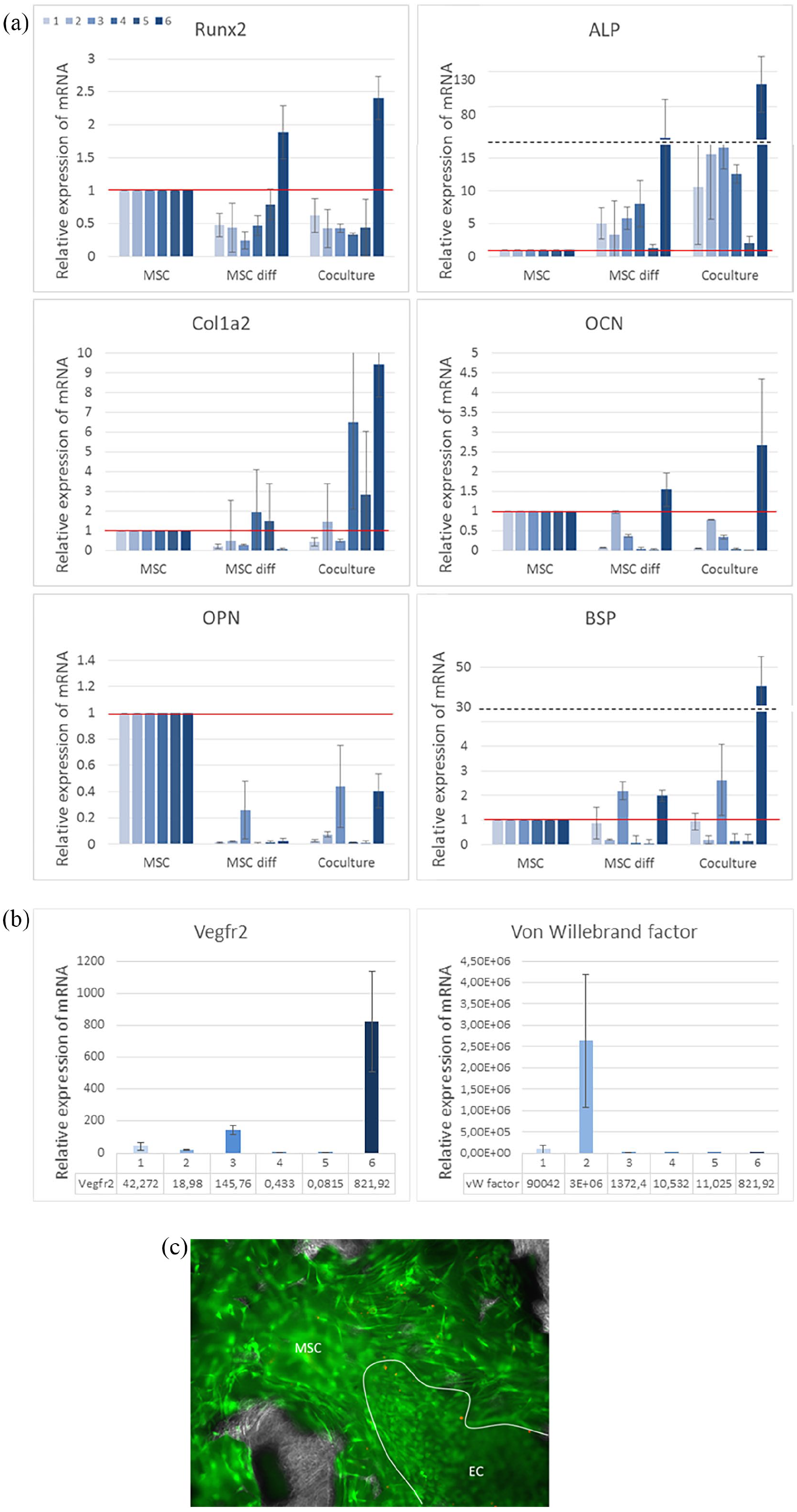
(a) Bone-related gene expression in sheet-like substitutes (control monocultures and cocultures) after 1 month. Detailed results of six MSC donors, no statistical significance when comparing the mean of each study group (p > 0.05). Relative expression detected by qRT-PCR with GAPDH as housekeeping gene normalised by results of non-differentiated MSCs (red line = 1). Dashed line: scale break. (b) Confirmation of endothelial cells presence at the end of the 1-month co-culture. Relative expression mRNA of Vegfr2- and von Willebrand factor-related genes detected by qRT-PCR in coculture samples, detailed results of six MSCs donors normalised by results of non-differentiated MSCs (=1), no statistical significance when comparing the mean of each study group (p > 0.05). For readability reasons (very high values), results of monocultures of ECs are not plotted. (c) Live/Dead fluorescence staining of MSCs and ECs cultured on BCP granules. Calcein AM in green (living cells) and EthD-1 in red (dead cells). The white line highlights a ECs colony (small round living cells on larger spread MSCs).
Expression of Runx2 and ALP-related genes, both early markers, showed different behaviours. Overall, a decrease in Runx2 expression was noticed in MSC diff and coculture groups compared to MSC (for all donors but donor 6) while ALP expression drastically increased. The expression of Col1a2, non-specific matrix protein, showed a clear trend for increase in coculture but with high variability. The same behaviour was noticed with the late differentiation markers OCN and BSP, showing high over-expression for some donors only (donor 6 in particular). The expression of OPN gene was never increased compared to MSC control. However, overall results showed a strong example of synergistic effects that can happen thanks to cell types crosstalk, as gene expressions in most cocultures samples were far higher than the sum of respective monocultures.
Regarding endothelial-related genes (Figure 4(b)), over-expression of Vegfr2 and Von Willebrand factor genes was obtained in the cocultured chambers after 1 month for all donors, except 4 and 5 regarding Vegfr2. The other samples led to high or very high expression of this markers compared to non-differentiated MSCs, validating the presence of active ECs in coculture at the end of the culture process but also confirming a high variability in adhesion and MSCs/ECs crosstalk. As expected, monocultures of ECs led to extremely high expression for these two genes (not plotted in the figure for readability reasons), respectively 103–104 and 104–106 fold increases for Vegfr2 and Von Willebrand factor. Populations of ECs were observed attached to the tissue developed by the MSCs in some areas in substitutes from several donors as seen on Live/Dead images (Figure 4(c)), highlighting that cell-cell contact and mixing between both cell types on the scaffold was feasible with our seeding and feeding schedules.
Discussion
We aimed here at investigating the pre-vascularisation potential of a sheet-like bone substitute previously developed. 25 Thanks to a new step of coculture between human primary MSCs and ECs, we also used this culture system to assess the variability between donors. The coculture approach was expected to lead to tissue-engineered substitutes mimicking better the native structure of tissues towards easier connexions to the existing vascular network after implantation.11,33 However, cocultures could also be beneficial directly during the in vitro culture steps to improve and speed up cell growth and differentiation. Indeed, growth factors produced by MSCs after differentiation towards bone lineage can promote angiogenesis and ECs proliferation, such as VEGF secreted by osteoblasts in the presence of BMP-2. 20 Moreover, mutual beneficial effects exist as the ECs population can significantly increase proliferation and differentiation of MSCs.16,17 Cell-cell direct contact was here prioritised over paracrine signalling as MSCs and ECs can both form gap junctions.11,34,35 The ECs/MSCs coculture could therefore allow for the development of substitutes with better vascularisation potential than the respective monoculture, even with synergetic effects,8,16,20,36 as we noticed here for the expression of some genes of interest, especially Col1a2. However, the successful development of both cell types requires specific culture conditions, still discussed nowadays (cell ratio, simultaneous or delayed co-culture, medium composition, etc.16,20–22,24,37), and in particular 3D growth structures appear to be mandatory.20,24,38 We were thus expecting the calcium phosphate scaffolds used in our system to promote coculture growth and activity despite the absence of specific coating such as collagen, commonly used for primary EC monoculture in vitro.
Moving from cell line to human primary cells is not a straightforward step but requires adjustments and more complex protocols, in particular for initial seeding density and culture medium schedules,13,17,36 but the fundamental analysis of the coculture parameters was not the initial purpose of this study. We wanted to focus instead on the results variability with cells obtained from six donors treated and cultured independently. Selection of the optimal parameters for the tissue-engineering protocol presented here was therefore based first on a previous study using murine pre-osteoblastic cells, but also thanks to preliminary results and literature background.13,25 We decided then to work with a delayed addition of ECs to allow for a first phase where MSCs would be able to proliferate and generate extra-cellular matrix prior to ECs anchorage. We used then endothelial medium to supply the coculture with nutrients ensuring ECs survival, although osteogenic medium could also promote vascularisation. 39 Coculture samples were compared to the respective monocultures to highlight the effects of the crosstalk.
Microscopy observations showed that a continuous cell tissue was successfully developed in coculture samples, with good viability and early signs of cell organisation (gap junctions, endothelial cell islets attached on the MSCs matrix, Table 1). Cells spread and bonded the calcium phosphate granules together, in a similar way as it was achieved previously with a cell line monoculture. 25 This absence of cytotoxicity induced by the calcium phosphate-based scaffold and the coculture conditions was expected but this confirmation was a first crucial result towards the use of human primary cells. Ultimately, the goal of such hybrid tissue-engineered substitutes is the implantation to strengthen injured or lacking tissues, and optimal cell viability in contact with the biomaterial, as validated here, is therefore mandatory. The newly formed cell tissue however showed sometimes more cracks and non-covered areas in case of lower proliferation of MSCs (donor 5), leading to limited mechanical integrity, but coculture didn’t induce drastic decrease in proliferation potential of either cell types, in contrast with some literature results.40,41 As stated previously, one of the main advantages of the tissue-engineered substitute reported here is its sheet-like shape. 25 The mechanical cohesion of the samples showed here a certain variability. Some coculture samples offered very easy handling with forceps after being removed from the culture chamber while other were immediately completely disrupted. The mechanical cohesion level could be linked to a certain extent to the degree of over-expression of late markers (Col1a2, BSP, Table 1), but it could depend more likely on the proliferation potential of the MSCs source. High cell density with good organisation seemed directly correlated to high mechanical properties (Table 1). The levels of cell density and proliferation showed no clear correlation with the donor’s age as both the youngest (35) and the oldest (56) donors led to high cellular density. As female and male donor groups had the same average age (46 years), we can mention that the cells from all female donors showed high proliferation potential in contrast with male donor’s (one over three), but a larger study focusing on gender would be needed to highlight a clear trend. It has been previously shown that cells harvested even on elderly, osteoporotic female donors were able to proliferate and promote osteogenesis and angiogenesis in vitro. 42
Summary and comparison of the results obtained with six independent MSCs donors.

Bold and green: relevant results to consider further in vivo applications; italic and red: limited results; F: female; M: male; ++: results similar to or higher than the previous study using cell lines 25 ; +: results slightly lower than previous study; −: results much lower than previous study; —: absence of the studied marker.
More importantly, overall, bone differentiation results in coculture showed systematically similar or higher trends than the monocultured groups of MSCs, with hints for strong synergistic effects (gene over-expression levels). The presence of ECs and the specific coculture feeding schedule didn’t jeopardise MSCs differentiation towards the bone lineage and even increased it for some donors, as it was expected thanks to the crosstalk between both cell types. Due to low Runx2 expression along with high ALP, moderate Col1a2 and occasional BSP/OCN over-expressions compared to the proliferating MSCs, we could assume that cocultured MSCs were starting to maturate and produce mineralised matrix.26–29 This was also consistent with the total absence of OPN. Moreover, even if the order of magnitude was drastically different from the EC monoculture, Vegfr2 and Von Willebrand factor expressions confirmed that a population of active ECs was still present in the coculture samples after 1 month of tissue development. Samples 4 and 5, which showed no over-expression of VEGFR2, also had the lowest or most heterogeneous cell densities with no expression of late bone-specific genes. Therefore, we hypothesised that important MSCs growth and matrix production was mandatory to allow the ECs to attach on the newly formed tissue and to survive in coculture without the need for a specific coating. This was confirmed by the absence of cohesion in the monoculture of ECs, with a very low number of cells after 1 month. This behaviour strengthened the rationale behind using a delayed coculture protocol and a specific feeding schedule, with a first step of MSCs development on the scaffold before addition of ECs. These results are however too limited to conclude on an actual pre-vascularisation of the more consistent substitutes, and further studies should focus on advanced angiogenesis markers (tube formation in particular through fluorescent angiogenesis assays). Using endothelial medium in cocultures for 2 weeks maintained the development of both lineages without stopping MSCs differentiation. Overall, our system allowed therefore for cell proliferation, viability and activity of MSCs and ECs in coculture over the 1-month culture period required for the development of the sheet-like substitute, paving the way to sheet-like bone substitute with enhanced vascularisation potential, which should be validated through in vivo assays in further studies.
Although our process was therefore successfully adapted from cell line monoculture to primary cell coculture, the important variability between donor sources prevented to guarantee a relevant mechanical cohesion for all samples. Power analysis was not performed directly as the expected effect size was completely unknown a priori, but we can assume that the sample size (number of independent donors) might have been too limited. Still, this proof-of-concept study highlighted in particular the potential role of a the proliferation level during the first expansion phase of the cells in vitro in contact with the scaffold. As a solution, the culture time in the chamber could be adjusted for further experiments in order to allow cells from each source to reach confluence state on the calcium phosphate granules. Indeed, variability in proliferation potential between donor sources was noticed during the tissue-engineering process, but also during the standard pre-amplification steps (expansion in T175 flasks) needed to obtain the initial number of MSCs to be seeded in the specific culture chamber. To allow for the development of a consistent cell tissue in the chamber, the culture time could vary from batch to batch, in a patient-specific approach, to be consistent with the pre-culture proliferation rate. This could lead to better homogeneity in the gene expression levels of late differentiation markers, and therefore consistently high mechanical cohesion. It has to be noticed that this can be however difficult to achieve due to more complex protocols and regulations. In particular, according to reports such as the Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue-Based Products: Minimal Manipulation and Homologous Use provided by the FDA (as consulted in June 2021), such processes might not be considered as minimally manipulated procedures anyway and would require investigations and approval, which is also a duty for researchers to ensure non-harmful new products. 43 Another solution would therefore be to use higher initial cell densities. Even for cell batches with high differentiation and proliferation potentials, 1 million cells per culture system could be considered as low density. 44 This would imply additional pre-culture expansion steps to reach the expected number of cells. Finally, although no clear trend appeared with our results, the donor age remains a parameter to consider. 45
In conclusion, we investigated in this paper how the tissue-engineering protocol leading to the development of a bone sheet-like substitute with easy handling for maxillo-facial regeneration could be adapted to the coculture of human primary MSCs and ECs, in a translational and proof-of-concept approach. From optimised technical parameters, both cell types were cultured together in a specific chamber on a monolayer of calcium phosphate granules to enhance pre-vascularisation potential with samples obtained from six independent donors. Activity and proliferation of both cell types were confirmed after 1 month, without detrimental effect from one on the other or from the feeding schedule, but rather synergistic effects compared to the respective monocultures. Cell viability was always very high, validating the absence of cytotoxicity from the scaffold and the tissue-engineering protocol for the use of human primary cells. Production of cell matrix by MSCs enhanced EC survival which, in turn, supported bone differentiation. MSCs were found to be in early mineralisation stage at the end of the protocol. More importantly, we highlighted that the important variability in proliferation potential and, in turn, gene expression, could have prevented the development of the sheet-like shape with enough mechanical integrity. A patient-specific approach would be to adapt culture time based on the cell proliferation rate. Further studies should therefore investigate this approach with more independent donors to increase the study’s power following this proof of concept, before first in vivo validation in a small animal model. Additional assays evaluating further cell functionality and phenotypic expression of the differentiated MSCs (such as actual production of osteocalcin or levels of VEGF and BMP-2 thanks to specific immunostaining in particular) could then be included to strengthen our understanding of the exact state of maturation of both cell types. The same goes for investigations of blood vessel-like structures thanks to the endothelial population. In addition, besides vascularisation, the interface between bone and other surrounding tissues (cartilage, tendon) could be recreated in such substitutes to enhance biointegration, in particular by the use of multilineage cultures with stem cell differentiation initiated by the biomaterial itself.
Supplemental Material
sj-pdf-1-jao-10.1177_03913988211051758 – Supplemental material for Donor variability alters differentiation and mechanical cohesion of tissue-engineered constructs with human endothelial/MSC co-culture
Supplemental material, sj-pdf-1-jao-10.1177_03913988211051758 for Donor variability alters differentiation and mechanical cohesion of tissue-engineered constructs with human endothelial/MSC co-culture by Timothée Baudequin, Marie Naudot, Sébastien Dupont, Sylvie Testelin, Bernard Devauchelle, Fahmi Bedoui, Jean-Pierre Marolleau and Cécile Legallais in The International Journal of Artificial Organs
Footnotes
Acknowledgements
The authors would like to thank Dr. Pierre Layrolle for providing the biomaterials (Biomatlante).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Picardie region and the Equipex FIGURES. This project was cofinanced by the European Union and the European Regional Development Fund. Timothée Baudequin acknowledges the financial support of the CNRS and the Collegium INSIS-UTC.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.