
Abstract
The present study was undertaken to investigate the protective effect of Pleurotus florida lectin (PFL) against arsenic-induced cytotoxicity and oxidative damages in freshly isolated splenocytes of rodents. Our finding indicated that arsenic caused reduction in cell adhesion, morphological alterations, cell proliferation, nitro blue tetrazolium (NBT) index, superoxide dismutase (SOD) activity, catalase (CAT) activity and relative mRNA expression of SOD2 in relation to housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and increased production of nitric oxide (NO), protein carbonyl (PC) and lipid peroxidation levels (LPO) assembled to play key factors for cytotoxicity and oxidative stress. PFL normalized cellular damages and enhanced SOD production pathway relating to gene expression. Further studies are needed to address effective phytochemicals of the edible mushroom and their mechanism.
Introduction
Inorganic arsenic, a common constituent of the earth’s crust, is widely distributed in the soil and water (Morton and Dunnette, 1994). Contamination of arsenic in drinking water is a major health problem throughout the world. Inorganic arsenic has a pronounced acute toxicity in human and experimental animals. Epidemiological studies provide clear evidence that it is also a human carcinogen (Morton and Dunnette, 1994). Human exposure to arsenic in drinking water has been associated with cancers (lung, bladder and skin) and other chronic disease such as dermal, cardiovascular, neurologic and diabetic effects (Abernathy et al., 1999). Recent studies have shown that arsenic-induced oxidative stress could be a reversible mechanism for some of the toxic effects of arsenic in cultured cells and in experimental animals (Shi et al., 2004).
Antioxidants have been found beneficial to mitigate chemical-induced oxidative damages. L-ascorbate, a water-soluble vitamin, is one of the biological antioxidants that protects tissues from oxidative damage (Ramanathan et al., 2002). It is previously reported that treatment with herbal products/extracts could be a better option in finding suitable treatment against acute or chronic arsenic poisoning (Gupta and Flora, 2006). Pleurotus florida is one of the important ingredients of Indian traditional folk medicines. It is believed to consist of important dietary and medicinal properties, which are used against a number of ailments and diseases (Wang et al., 1998). A variety of compounds with important pharmacological properties have been isolated from mushroom, which include polysaccharides, polysaccharopeptides, polysaccharide proteins with immunoenhancing and anticancer properties (Wang et al., 1996; 2002). Lectin produced from Pleurotus florida is able to interfere with tumor initiation through a variety of mechanism, such as enhancing the host’s antioxidant capacity in upregulating antioxidant enzymes involved in the metabolic transformation and detoxification of mutagenic compounds (Dalloul et al., 2006). In the present study, it was planned to determine the effect of Pleurotus florida lectin (PFL) on arsenic-induced alteration of splenocytes functions in rat.
Materials and methods
Reagents and chemicals
Diphelenetriamine penta acetic acid (DTPA) (99% pure), pyrogallol (>98%), sodium dodecyl sulfate (>99%), BSA (97%), triton X-100 and thiobarbuturic acid (99%) were purchased from Sigma chemicals, USA, sodium meta arsenite (98%), William’s medium E, RPMI 1640, penicillin, streptomycin, glutamine, collagenase and ascorbic acid (98%) were procured from HiMedia, India. Mushroom was purchased from local market. All other chemicals and solvents were of analytical grade.
Animals
Adult Wister male rats (70–80 g) were used as donor of splenocytes. Animals were housed 3 per cage, submitted to a 12-h light and 12-h dark cycle and had access to water and standard rat chow ad libitum. All the experimental protocols were performed according to Animal Ethics committee, Indian Veterinary Research Institute.
Preparation of mushroom lectin
Lectin was prepared as per the method described previously (Dalloul et al., 2006). Fifty (50) g of crashed mushroom (Pleurotus florida) were dissolved in 20 mM Tris HCl buffer (pH 8.0) and then kept for 18 h at 4ºC with frequent swirling. The resulting suspension was filtered and 156.50 g of ammonium sulfate were added to make 50% saturation of ammonium sulfate. After allowing it to stand overnight at 4ºC, the resulting precipitate was separated by centrifugation at 8000g at 4ºC for 20 min. In order to obtain 100% saturation, 313 g of ammonium sulfate was then added to the supernatant. After standing overnight at 4ºC, the resulting precipitate (from 100% ammonium sulfate precipitation) was separated by centrifugation. The precipitate was then dissolved in 50 mM NaCl, 50 mM Tris-HCl buffer (pH-8.0), dialyzed against distilled water for 5 to 7 d at 4ºC with frequent changing of distilled water. After dialysis, the dialysate was freeze-dried to yield crude lectin (0.64% w/w). The crude lectin was grayish brown in color, freely soluble in distilled water as well as saline and tissue culture media viz. William’s medium E but insoluble in organic solvent viz. ethanol, acetone, chloroform or diethyl ether.
Separation of splenic macrophages
Spleen was isolated from rat, macerated and the cells were repeatedly aspirated with a sterile Pastuer pipette until a single-cell suspension was obtained. Suspension was then transferred to sterile tubes and kept on ice. The supernatant was then transferred to sterile tubes and kept on ice to settle the cell debris. The supernatant was then layered over 3 ml of histopaque (density 1.077) (Sigma, USA) and then centrifuged at 1,500 rpm for 30 min (Sikorsi et al., 1991). After centrifugation, the cells were collected and washed with DPBS. Then the cell pellet was resuspended in RPMI-1640 containing 20 mM HEPES (pH-7.2), 1 mg/ml BSA and were allowed to adhere on plastic surface for 1 h in a 37°C incubator. The nonadherent cells were removed and collected by repeated aspiration with Pasteur pipette. Cells were washed and finally resuspended in culture media (RPMI + BSA) at a density of 2 × 105/ml. More than 95% cells were viable as determined by Trypan Blue dye exclusion technique (Sengupta and Bishayi, 2002). After the establishment of monolayers, the medium was removed and replaced with fresh medium. Isolated splenocytes were seeded into 24 wells tissue culture plate (Axygen, India) and incubated at 37ºC with 5% CO2 tension for 24 h. The following groups were considered for this experiment, cultured for 24 h and was maintained in triplicate.
Treatment group

Cytotoxicity study
In vitro cell adhesion assay
Cells were seeded separately for treated and control (HC) group in 96-well microtitre plate and allowed to adhere for different times after 24 h of incubation. With time, the wells were washed with HBSS, then 100 μl of 0.5% crystal violet, in 12% neutral formaldehyde and 10% ethanol was added to each well and incubated for 4 h to fix and stain the cells. Wells were then washed and air-dried for 30 min. Crystal violet was extracted from the adhered macrophage in the wells by lysing with 1% SDS in HBSS. Absorbance was measured spectrometrically (Beckman DU 640 B) at 570 nm. Cell adhesion was expressed as the increased absorbance at 570 nm (Sengupta and Bishayi, 2002).
Morphological alteration of macrophages
One hundred (100) μl of Hank’s balanced salt solution with bovine serum albumin (HBSS-BSA) were taken and fixed in an equal volume of 2.5% glutaraldehyde in HBSS. After 10 min, the cells were centrifuged at 2000g for 5 min. The supernatant was removed and the pellet was resuspended in HBSS. Smears of cells were drawn on glass slides, air-dried, fixed in methanol and stained with Giemsa. The cells were observed under oil immersion microscope (Olympus, Bx60, Japan, 100×). Any cell deviating from spherical outline was scored as polarized and this was expressed as a percentage of the total number of cells counted (Sengupta and Bishayi, 2002).
Cell proliferation index
Splenocytes were plated on flat-bottomed 48-well tissue culture plate (2 × 105) and allowed to adhere for 24 h at 37ºC. After incubation, the cells were washed twice with warm phosphate-buffered saline (pH 7.4) in order to remove the nonadherent dead cells. The cell proliferation was determined by MTT (3-(4,5-dimethythiazol-. 2-yl)-2,5-diphenyl tetrazolium bromide) assay (Ohta et al., 2004) at 570 nm (Beckman DU 640 B) and the cell proliferation index (CPI) was expressed as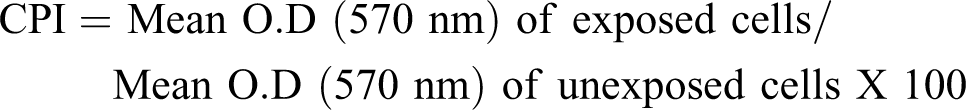
Nitric oxide release assay
Splenocytes were suspended in DPBS-BSA and centrifuged at 8000g for 20 min. Cell free supernatant was collected in a separate microcentrifuge tube and was used for NO release assay. Each of the cell-free supernatant (50 μl) was reacted with Griess reagent (100 μl) containing 1 part of 1% sulfanilamide in 5% phosphoric acid, and 1 part of 0.1% N-C-1 napthyl ethylene diamine dihydrochloride was added and incubated for 10 min at room temperature. Readings were taken in a spectrophotometer (Beckman DU 640 B) at 550 nm and compared to a NaNO2 standard curve and the amount of NO release in the supernatant was measured in each set (Czuprynski et al., 1984).
Nitro blue tetrazolium reduction assay
The activity of nitro blue tetrazolium (NBT) reduction was quantitatively measured according to the method of Imaizumi and Breitman (1986). A million of splenocytes in 0.5 ml of PBS were added to 0.5 ml of 0.1% NBT solution and incubated while being shaken at 37°C for 25 min, after halting the reaction by immersion in an ice cold bath. Formazan was recovered as a pellet by centrifugation at 1000g for 10 min. The formazan was washed twice with 0.1 N HCl. Three milliliters of N, N dimethylformamide and 3 μl of 1 N NaOH were used to extract the formazan from the pellet. The optical density (OD) of extract was immediately read at a wave length of 710 nm (Beckman DU 640 B). The formazan yield was calculated according to observation that 1 nmole/ml of formazan should correspond to an OD of 0.051 ± 0.003.
Oxidative stress
Superoxide dismutase activity
The activity of superoxide dismutase was measured according to the method of Marklund and Marklund (1974) Aliquots of cell lysates were used to measure superoxide dismutase (SOD) by quantifying the inhibition of pyrogallol autoxidation at 420 nm. The reaction mixture consisted of 2.0 ml 50 mM Tris-HCl (pH 8.2) and 20–30 μl of cell lysates. The reaction was initiated by adding 20 μl pyrogallol (10 mM, pH 7.4). Activity was expressed as the amount of enzyme capable of inhibiting pyrogallol oxidation rate by 50%, which is equal to 1 unit per mg protein.
Catalase activity
Catalase (CAT) was assayed by the method as described earlier (Cohen et al., 1970) as enzyme-catalyzed decomposition of H2O2. This activity was measured at wave length of 240 nm after appropriate dilution and the values were expressed in units/mg protein.
Protein carbonyl assay
Protein carbonyl (PC) levels were measured according to the standard method (Reznick and Packer, 1994) based on spectrophotometric (Beckman DU 640 B) detection of the reaction of 2,4-dinitrophenylhydrazine with PC to form protein hydrazones. Briefly, after the precipitation of protein with an equal volume of 1% trichloroacetic acid (TCA), the pellet was resuspended in 10 mM DNPH (2,4-Dinitrophenylhydrazine) in 2 N HCl or with 2 N HCl as control blank. After washing with 1:1 ethanol/ethylacetate, the final pallet was dissolved in 6 M guanidine hydrochloride. The carbonyl group was determined from the absorbance at 370 nm. The result was expressed as micromoles of carbonyl groups per milligram of protein, with a molar extinction coefficient of 21.5 nmol/l/cm.
Lipid peroxidation assay
The role of lipid peroxidase was assayed by studying the level of formation of malondialdehyde (MDA), which is an indicator of lipid peroxidation (LPO). Quantitative measurement of LPO was performed following the thiobarbituric acid (TBA) test (Wills, 1987). The amount of MDA formed was quantitative with TBA and used as an index of LPO. The results were expressed as nmol MDA/mg protein using molar extinction coefficient (1.56 × 105) cm2/nmol.
DNA fragmentation assay
DNA fragmentation was assessed as per standard methodology (Maroufi et al., 2005). In brief, the cells (2 × 105/ml) were resuspended in hypotonic lysis buffer (0.2% Triton X 100, 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, pH 8.0) and centrifuged for 10 min at 13,000g at 4°C (preparation B). The supernatant containing small DNA fragments was collected carefully and transferred to a tube (preparation A). Lysing buffer, 0.5 ml was added to the pellet containing preparation B; 0.5 ml of 25% trichloroacetic acid (TCA) was added to the A and B preparations and vortexed vigorously. The tubes were placed at 4°C and left the precipitate overnight. The precipitates were centrifuged for 10 min at 13,000g. The supernatants were aspirated and discarded. Eighty (80) μl of 5% TCA was added to each pellet. The DNA was hydrolysed by heating for 20 min at 83°C in a water bath. Diphenylamine solution, 160 μl, was added to the test tubes and to a blank containing 80 μl 5% TCA. All tubes were vortexed and then left overnight at room temperature. In order to read the OD, the collected supernatants were transferred to a 96-well plate and the optical densities were read at 620 nm by enzyme-linked immunosorbent assay (ELISA) reader (Multiskan EX, Thermosystems, USA). The percentage of fragmented DNA was calculated according to the following formula: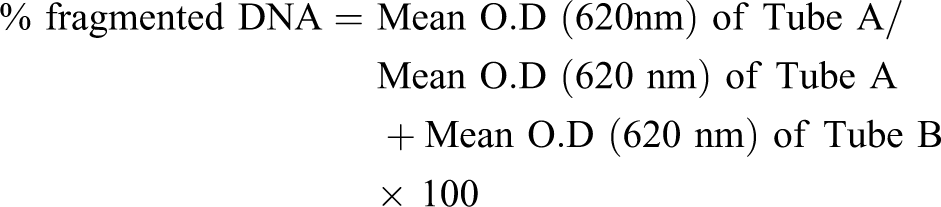
Primers
For partial expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and SOD2 gene, the following primers were designed. The sequence of primers, source, and size of the fragments are given below.

RNA isolation from splenocytes
RNA was isolated from splenocytes using TRI Reagent (Sigma). In brief, the cultured splenocytes (2 × 105) were homogenized with TRI Reagent in a sterilized glass-Teflon tissue homogenizer. Phase separation was performed using chloroform. Following centrifugation aqueous phase was collected for RNA precipitation. Precipitated RNA was washed with 70% ethanol. RNA pellet was air-dried and dissolved in nuclease water and DNAse (Promega) was added and kept for 30 min at room temperature. Excess DNAse was inactivated using DNAse stop solution (Promega) and simultaneous heat treatment.
Reverse transcription polymerase chain reaction (RT-PCR) of GAPDH and SOD2 gene expression and quantitation
RT-PCR was performed using RT-complete (Bio enzyme) as per manufacturer’s protocol. Briefly, in 45 µl of RT complete reaction mixture both the primers (0.5 μM each) were added, RNA template 500 ng and nuclease-free water to make a total volume of 50 μl. The mixture was placed at 55°C for 30 min. PCR was performed using a Gene Amp PCR system 9700 (Applied Biosystem) with the following program of denaturation at 95°C for 5 min, 35 cycles of denaturation at 95°C for 45 s, annealing at 61°C for 45 s for GAPDH and SOD2 and elongation at 72°C for 45 s followed by final elongation of 10 min at 72°C. The amplified products were visualized in ethidium bromide stained 1.5% agarose gel. Banding intensity with respect to each gene was quantitated using GelQuant Software (DNR Bio-imaging System, Israel).
Statistical analysis
The results are expressed as mean ± SD. The statistical significance was analyzed using one-way ANOVA followed by Dunnett’s post hoc test for comparison between HC group and the others (GraphPad Prism 4.03 for Windows, GraphPad Software, USA) and further p < 0.05 was considered to be statistically significant.
Results
Effect of PFL on cell adhesion of splenocytes
The development of an effective inflammatory response involves the adherence of immunocompetent cells near the site of inflammation. In vitro cell adherence assay may reflect the in vivo capacity for cellular adherence upon exposure to arsenic. After 24 h of incubation, the cell adhesion of arsenic-treated group (AS) significantly (p < 0.001) decreased gradually with respect to control. And AS + AA- and AS + ML-treated cells showed significantly (p < 0.001) increased level of cell adhesion at 30 min and 60 min, respectively, of additional incubation (Figure 1).
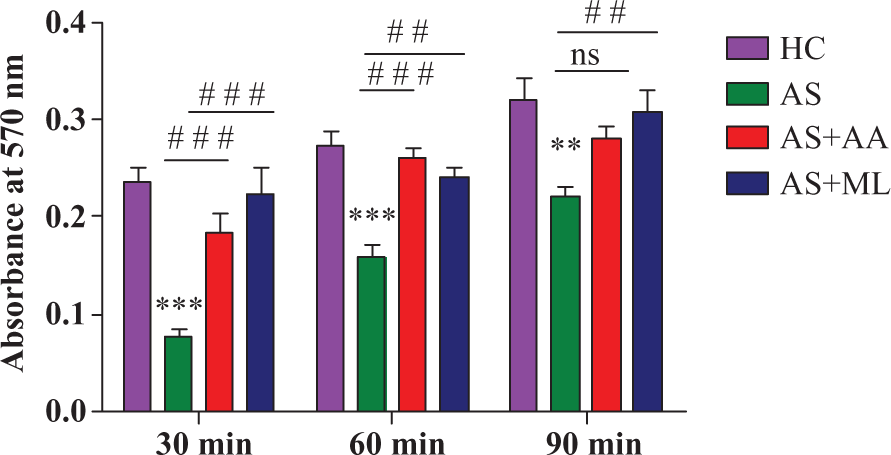
Cell adhesion in different groups of splenocytes at 30 min, 60 min and 90 min of additional incubation after 24 h of exposure to different treatments. Values are expressed as mean± SEM for n = 3. ** and*** indicate significant difference at p < 0.01 and p < 0.001, respectively, when compared to control. # # and # # # indicate p < 0.01 and p < 0.001 when compared to 5 µM arsenate.
Effect of mushroom lectin on morphological alteration of splenocytes
Enhanced functional step up in the activated macrophages is accompanied by a morphological alteration of the target cells. In order to address whether there was any changes in the number of deformed cells that is inactivated cells following arsenic exposure, shape change assay was performed. As shown in Figure 2, it was observed that exposure to arsenic led to morphological changes in splenocytes since more deformed cells were obtained in the AS group (p < 0.05) with respect to the HC group. Ascorbic acid and PFL significantly (p < 0.05) decreased the deformed cell as compared to AS group.
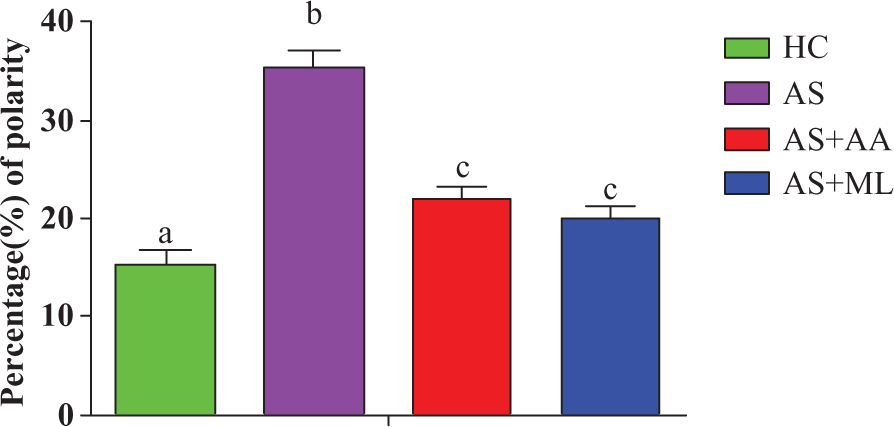
Morphological alteration in different groups of splenocytes. Values are expressed as mean ± SEM for n = 3. Data are expressed in percentage.
Effect of PFL on CPI of splenocytes
The present study indicated that AS group showed significantly (p < 0.05) higher levels of CPI when compared to HC group. The statistical results revealed that both ascorbic acid and PFL significantly (p < 0.05) restored CPI (p < 0.05) reduced by arsenic exposure in rat splenoctyes after 24 h of incubation (Figure 3).
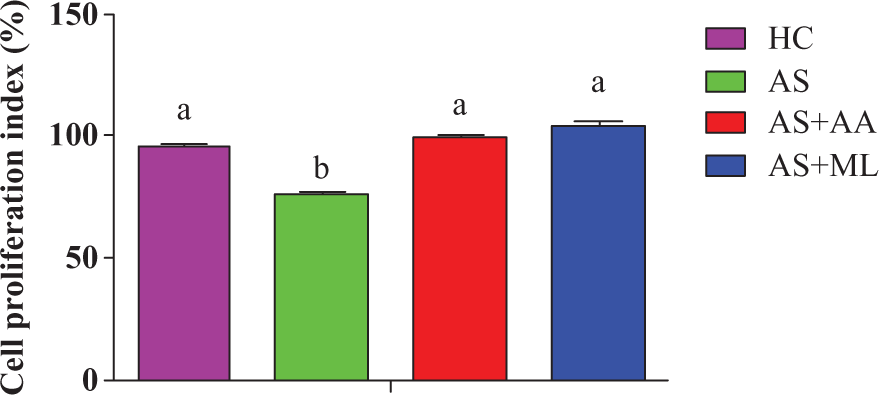
Cell proliferation index (CPI) in different groups of splenocytes. Values are expressed as mean ± SEM for n = 3. Different letters indicate significant difference between groups (p < 0.05).
Effect of PFL on NO production of splenocytes
Results in Figure 4 showed that nitric oxide (NO) production in AS splenocytes was significantly (p < .05) elevated rather than the HC group. The results also revealed that ascorbic acid- and PFL-treated group exhibited lowered level (p < 0.05) of NO production when compared to AS splenocytes.
Nitric oxide (NO) production in different groups of splenocytes. Values are expressed as mean ± SEM for n = 3. Different letters indicate significance difference between groups (p < 0.05).
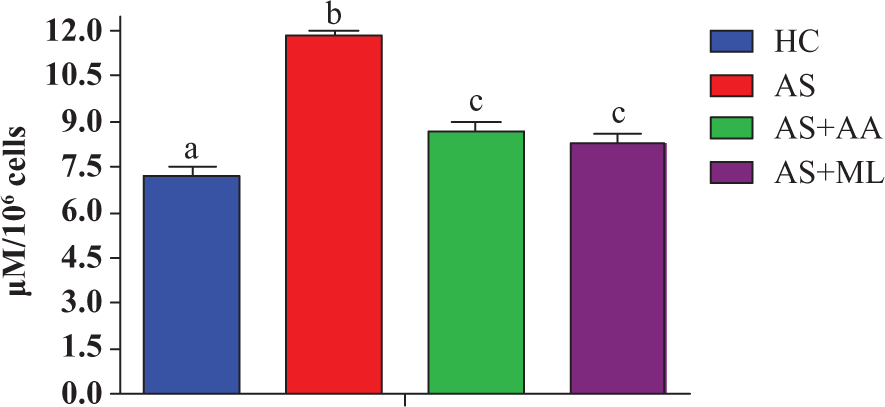
Effect of PFL on phagocytic activity of splenocytes
The formazan yielded from splenocytes treated with arsenic was significantly (p < 0.05) decreased than HC group. Both ascorbic acid and PFL significantly (p < 0.05) augmented phagocytic activity during the period of exposure (Figure 5).
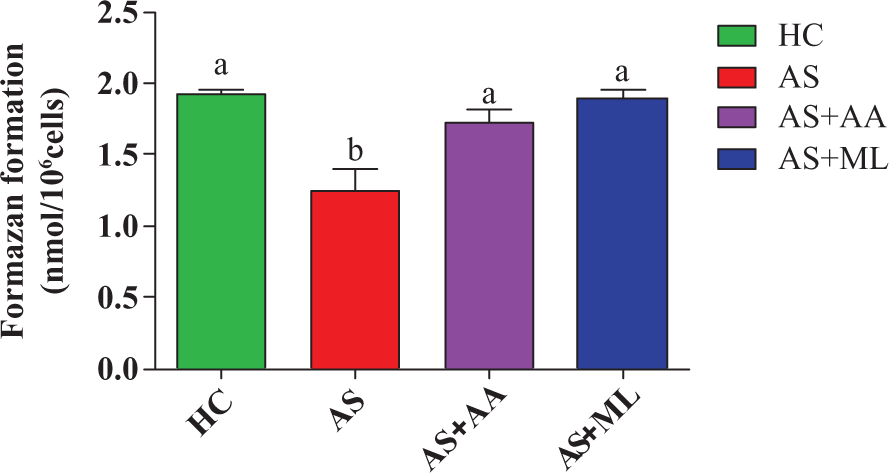
Nitro blue tetrazolium (NBT) reduction in different groups of splenocytes. Values are expressed as mean ± SEM for n = 3. Different letters indicate significant difference between groups (p < 0.05).
Effect of PFL on SOD activity of splenocytes
Results of our present work indicated that arsenic significantly (p < 0.05) decreased the SOD activity in splenocytes after 24 hr of exposure. Both ascorbic acid and PFL significantly (p < 0.05) restored the inhibition of SOD activity reduced by arsenic exposure in splenoctyes of rat (Figure 6).
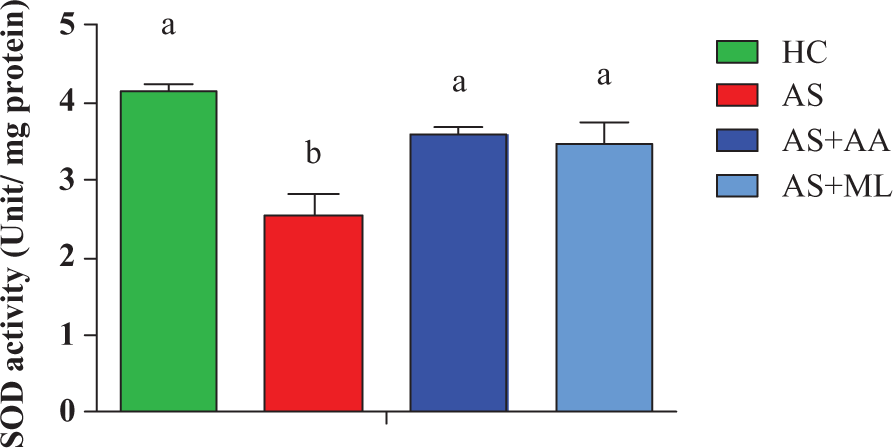
Superoxide dismutase (SOD) activity in different groups of splenocytes. Values are expressed as mean ± SEM for n = 3. Different letters indicate significant difference between groups (p < 0.05).
Effect of PFL on CAT activity of splenocytes
Values of CAT activity of splenocytes of both control and treated animals are given in Figure 7. Exposure of arsenic for 24 h caused significant (p < 0.05) reduction in CAT activity in AS group. However, further feeding of AS caused significant reduction in the level of the enzyme. A significant (p < 0.05) protection was observed when ascorbic acid and PFL were coadministered with arsenic, respectively (Figure 7).
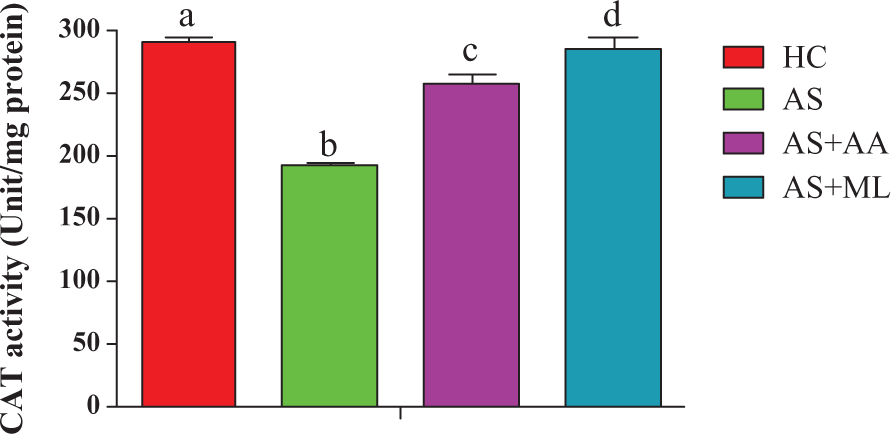
Catalase (CAT) activity in different groups of splenocytes. Values are expressed as mean ± SEM for n = 3. Different letters indicate significant difference between groups (p < 0.05).
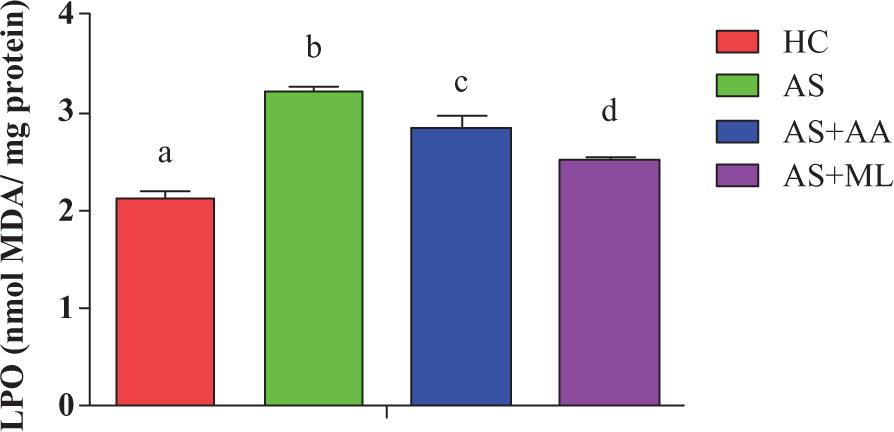
Effect of PFL on PC level in splenocytes
Figure 8 shows protein oxidation in terms of PC in all control and experimental groups exposed to arsenic and cotreatment of ascorbic acid and PFL with arsenic, respectively. Arsenic exposure for 24 h caused a significant (p < 0.05) level of PC when compared to HC group. Both ascorbic acid and PFL significantly (p < 0.05) reduced the level of PC when compared to AS group.
Protein carbonyl (PC) content in different groups of splenocytes. Values are expressed as mean ± SEM for n = 3. Different letters indicate significance difference between groups (p < 0.05).
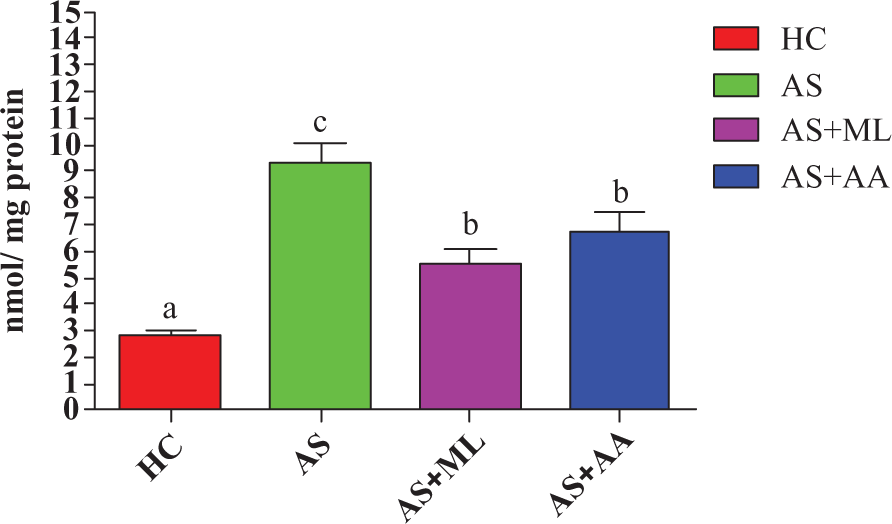
Effect of PFL on lipid peroxide level in splenocytes
Data on LPO in terms of TBA formation of splenocytes in control and experimental groups are presented in Figure 9. A significant increase (p < 0.05) in TBA level was noted in AS groups in comparison to HC group. But significant (p < 0.05) ameliorative potential of both ascorbic acid and PFL relating to the level of LPO was observed after 24 h of incubation than AS group.
Malondialdehyde (MDA) level in different groups of splenocytes in vitro. Values are expressed as mean ± SEM for n = 3. Different letters indicate significant difference between groups (p < 0.05).
Effect of PFL on DNA fragmentation in splenocytes
Arsenic exposure produced significant (p < 0.05) increase in the percentage of DNA fragmentation in splenocytes of rat after 24 h of incubation. But both ascorbic acid and PFL significantly (p < 0.10) prevented arsenic-induced DNA damage during the treatment period (Figure 10).
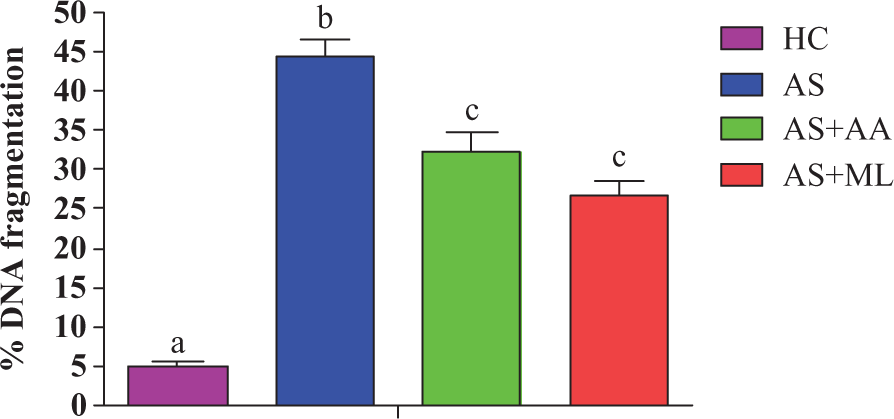
Percentage of DNA fragmentation in different groups of splenocytes. Values are expressed as mean ± SEM for n = 3. Different letters indicate significant difference between groups (p < 0.05).
Effect of PFL on SOD2 gene expression of splenocytes
Our result depicted that treatment with arsenic caused significant (p < 0.05) decrease in mRNA expression of SOD2 gene in splenocytes when compared with HC group. Both ascorbic acid and PFL significantly (p < 0.05) protected arsenic-induced mRNA expression of SOD2 gene during the time of exposure (Figure 11(a) to (c)).
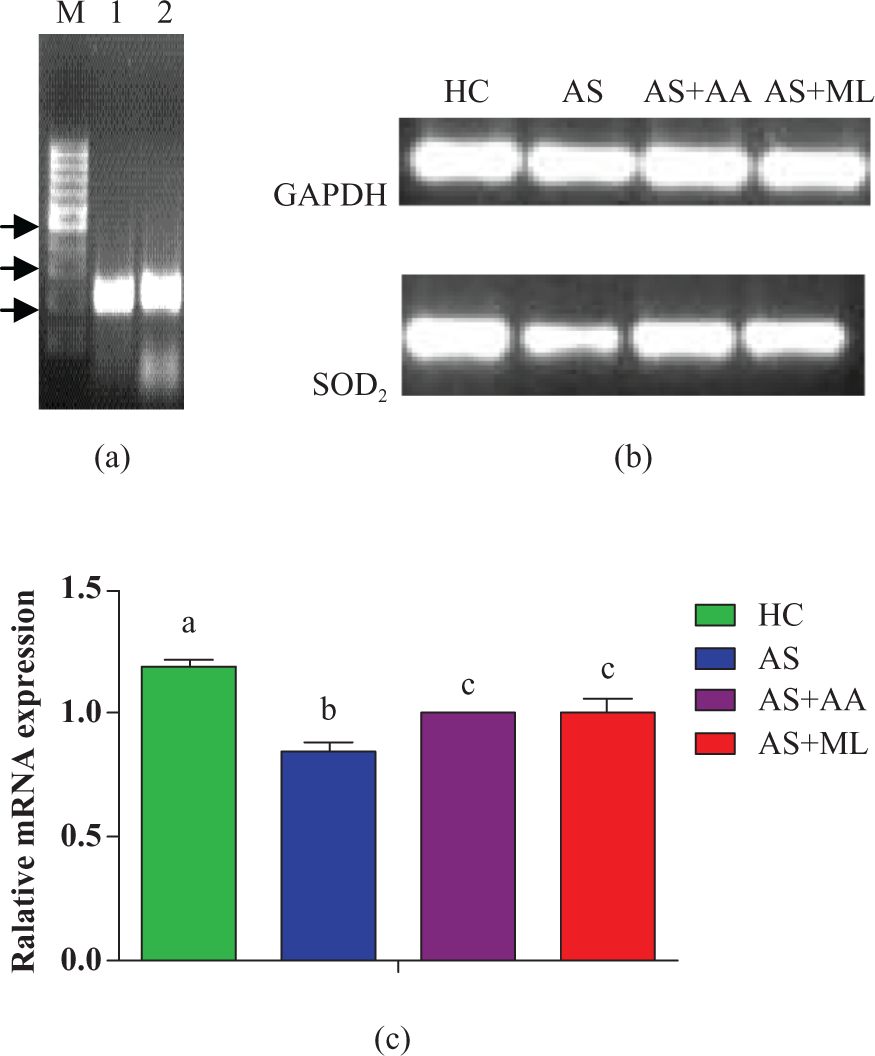
(a) mRNA expression of oxidative stress related and housekeeping genes. M-100 bp DNA ladder Lane 1 glyceraldehyde 3-phosphate dehydrogenase (GAPDH); Lane 2 superoxide dismutase 2 (SOD2) (arrows indicate from the bottom 200, 300, 500 bp fragment sizes). (b) The mRNA expression level of GAPDH and SOD2 genes in rat splenoctyes of different treatments after 24 h of incubation. mRNA level of GAPDH and SOD2 were detected by RT-polymerase chain reaction (PCR) using specific primers. (c) Relative mRNA expression of SOD2 quantified in relation to GAPDH mRNA (as endogenous control) in different treated groups. The data were expressed as mean ± standard errors (SE) n = 3. Significant differences with control were designated as a p < 0.05.
Discussion
The antioxidant and free radical scavenging activities of many substances have been assessed, and many substances that possessed immunomodulatory activities show antioxidant activity (Borchers et al., 2004). The cellular properties of splenocytes were becoming nonadhesive due to triggered effect of arsenic (Cheung and Cheung, 2005). Ascorbic acid- and PFL-treated splenocytes showed increased adhesion than AS group. Ascorbic acid plays an important role in the formation of the extracellular matrix, a substance necessary for cell adhesion (Han et al., 2004). PFL also has a great role for growth regulation, cell adhesion, cell migration, cell apoptosis and immune responses (Zheng et al., 2007).
The CPI (%) of arsenic-induced splenocytes was decreased, which corroborated with the earlier report (Rossi et al., 2001). Ascorbic acid and mushroom lectin were effective in preventing arsenic-induced cell cytotoxicity and both the agents were able to normalize the nitrite synthesis in splenoctyes. In addition, it is also reported that some of the mushroom lectins are capable of activating macrophages and lymphocytes (Zheng et al., 2007), whereas some of them possess potent mitogenic activities toward human lymphocytes and thus receive special attention. They are collectively designated as fungal immunomodulatory proteins (FIPs) (Ho et al., 2004).
Liu et al. (2001) were of the view that primary target in arsenic induced genotoxic response in mitochondria damage which in turn caused the release of superoxide anions that can react with NO to produce highly reactive peroxynitrites. NO is a highly reactive free radical that modulates tumorigenesis through its activity to regulate cell proliferation, cell death, migration and angiogenesis (Mukherjee et al., 2003). Arsenic generated nitrite oxide and reactive oxygen intermediate toxicity was demonstrated by Mukherjee et al. (2003) who recorded increased level of NO in islet cells of rat. Ascorbic acid reduced the NO production accelerated by arsenic. This reduction using ascorbic acid may be due to direct oxygen radical scavenging property or by decreasing the constituent levels of activated NF-β (nuclear factor) that respond to arsenic generated oxidative stress (Barchowsky et al., 1996). PFL normalize the NO production and it plays a great role by scavenging NO and peroxynitrite and also inhibits excessive production of NO by inducible form of NO synthase (iNOS) (Wang et al., 1996).
NBT level or phagocytic index is the factor in the activation of a wide variety of host defense mechanism, which includes phagocytosis, defective enzymes release and respiratory burst. Arsenic is a cytotoxic agent that causes decrease in phagocytic activity (Barchowsky et al., 1996). Ascorbic acid and PFL restored phagocytic activity inhibited by arsenic. Some of the mushroom lectins are capable of activating macrophages and lymphocytes and acts as antioxidants toward human lymphocytes (Wang et al., 1996; Zheng et al., 2007).
Free radical–induced oxidative stress has been implicated in the pathogenesis of a variety of clinical disorders, resulting usually from deficiency of natural antioxidant defense (Ramanathan et al., 2002). Diverse doses of arsenic have been used to induce arsenic toxicity in rats and to study therapeutic efficacy of antioxidant administration aftermath of arsenic exposure (Ramanathan et al., 2002). Enhanced production of free radicals and inhibition of antioxidant enzymes have been suggested as possible mechanism to explain arsenic-induced oxidative stress (Liu et al., 2001). SOD and CAT are the two most important radical scavenging enzymes and body’s secondary defense against oxygen enzyme metabolites produced due to transitional heavy metals (Nandi et al., 2005; Ramanathan et al., 2002). Both are two basic subcellular defenses of antioxidant enzymes that counteract free radicals produced during xenobiotic exposure.
The present study revealed that enzymes regulating oxidative stress were lower after arsenic exposure but were not lower in the arsenic plus ascorbic acid and AS- plus PFL-treated groups. The present study indicated that arsenic inhibited the activities of SOD and CAT leading to the accumulation of H2O2. Ascorbic acid is a standard antioxidant exhibiting a number of beneficial effects against various types of degenerative diseases (Chattopadhyay et al., 2001). PFL has a potential role to prevent oxidative stress and thereby ameliorates the toxicity of arsenic. Jose and Janardhanan (2000) also reported that edible mushroom Pleurotus florida has therapeutic use for prevention and control of cancer and cardiovascular diseases through its antioxidative role.
PC serves as a validated marker for protein oxidation. Reaction of free radicals with side chains of lysine, arginine, proline, theonine, and glutamic acid residues of protein leads to the formation of carbonyl derivatives (Stadtman and Berlett, 1997). Arsenic increased the level of the PC by inducing free radical generation or inhibition of antioxidant enzymes (Kadirvel et al., 2007). The observed increased level of PC formation in arsenic-exposed rats might be a result of either overproduction of ROS resulting from accumulation of antioxidants during chronic arsenic exposure. LPO is a basic cellular deteriorating process induced by oxidative stress and occurs rapidly in the tissue rich in highly oxidizable polyunsaturated fatty acids (Wang et al., 1996). Sodium arsenite treatment in the experiment resulted in a significant increase in the level of TBA in splenocytes. The elevation might be due to lower level of SH groups and antioxidant enzymes also observed in this study.
Mushroom could efficiently counteract arsenic-induced damage by quenching the generation of reactive oxygen species (ROS). Due to functional group (ricin B-like lectins), mushroom lectin is able to scavenge or neutralize free radicals by interacting with oxidative cascade, quenches oxygen and by chelating some metal ions inhibits peroxidation of membrane lipids, thereby maintaining membrane integrity and their functions (Josh and Janardhanan, 2000; Zheng et al. 2007). PC and LPO, which are produced due to arsenic exposure, are the biomarkers of oxidative stress restored by both AA and PFL. Lectin are very reactive compound present in mushroom reverse changes of ROS and oxidative stress (Josh and Janardhanan, 2000; Zheng et al. 2007). In addition, the present study was based on the fact that mushroom lectin acted as an antioxidant, thereby preventing the generation of ROS and oxidative stress.
From the present result, it is evident that arsenic-induced oxidative DNA damage in splenocytes of rat and ability to cause DNA damage without inducing direct mutation led to the concept that arsenic promotes DNA damage by inhibiting DNA repair (Zhao et al., 2003). Greater DNA percentage is susceptible to fragmentation on exposure to arsenic, and this is only one of the indications that might perhaps cause cell death in due course. But the present result revealed that ascorbic acid and PFL prevented DNA fragmentation. Zhao et al. (2003) also established that the decreased level of DNA fragmentation is due to accelerated production of DNA rep air efficiency in the damaged cells.
Semiquantitative gene expression showed significant downregulation of SOD2 gene expression following arsenic treatment. Increasing evidence from mammalian studies demonstrates that ROS are generated in response to exposure to inorganic forms of arsenic (Qian et al., 2003). Antioxidant-related genes (i.e. coding for SODs) play prominent roles in response to arsenate (Abercrombie et al., 2008; Kibriya et al., 2007). Ascorbic acid administration decreased the oxidative stress and might underlie the effect of ascorbic acid to increase the expression of SOD2 mRNA. It was further demonstrated that antioxidants have a capability to increase the expression of SOD2 gene in AS cell. In our present finding mushroom lectins prevent oxidative stress by restoring mRNA SOD2 expression and further demonstrated that PFL has a great role in elevating the expression of mRNA SOD2 gene related to oxidative stress.
In conclusion, it can be summarized that PFL like standard antioxidant viz. ascorbic acid could be a potential antioxidant in accelerating the functions of splenocytes and decreasing cytotoxicity and oxidative stress generated by arsenic. It is also suggested that ascorbic acid and mushroom lectin would be the benefactors to ameliorate arsenic-induced alterations of splenocyte functions in rat.
Footnotes
Funding
The financial assistance was provided by Indian Veterinary Research Institute and National Agricultural Innovation Project, ICAR, India.