
Abstract
Methylmercury (MeHg) is a potent hepatotoxin with a complex mechanism of inducing liver injury. Ferroptosis, an iron-dependent form of non-apoptotic cell death, is implicated in various toxicological responses, but its role in MeHg-induced liver damage remains under investigation. In this study, we established an acute liver injury (ALI) model in mice via gavage of MeHg (0, 40, 80, 160 μmol/kg). Histopathological analysis revealed dose-dependent liver damage, corroborated by elevated serum biochemical markers, confirming MeHg-induced hepatotoxicity. MeHg exposure raised MDA levels, inhibited SOD and GSH activity, and downregulated CAT expression. Increased iron accumulation and elevated transferrin receptor expression were observed, alongside decreased GPX4 and SLC7A11 levels, indicating ferroptosis involvement. Additionally, inflammation in MeHg-exposed livers was markedly intensified, as evidenced by increased MPO activity, upregulation of pro-inflammatory cytokines, and activation of the NF-κB/NLRP3 signaling pathway. The Keap1/NRF2/HO-1 oxidative stress response pathway was significantly activated, and p38/ERK1/2 MAPK signaling was notably increased. These findings suggested that MeHg induced acute liver injury through the interplay of ferroptosis, oxidative stress, inflammation, and MAPK signaling pathways, providing a scientific basis for future exploration of the mechanisms underlying MeHg-induced hepatotoxicity and potential therapeutic strategies.
Introduction
Methylmercury (MeHg) is a highly neurotoxic compound formed by the methylation of inorganic mercury by microorganisms in aquatic environments (Pan et al., 2022). Owing to its high lipophilicity, MeHg readily accumulates in the tissues of aquatic organisms, becoming biomagnified as it moves up the food chain. Humans primarily encounter MeHg through the consumption of contaminated fish and seafood, which poses significant health risks (Harada, 1995). It has been well-documented that MeHg severely impairs brain development and causes damage to other organs and systems, including the liver, kidneys, reproductive, and immune systems (Hong et al., 2012; Mela et al., 2007). Among these, the liver—an essential detoxification organ—acts as the primary site for MeHg metabolism and accumulation (López-Berenguer et al., 2020; Tian et al., 2023).
Cell death, whether in physiological or pathological conditions, is an inevitable aspect of life and marks the endpoint of cell lifespan. Ferroptosis, a recently characterized form of programmed cell death, is distinguished by iron-dependent lipid peroxidation (Liang et al., 2023) and plays a critical role in ischemic and hypoxic damage within the nervous system and liver (Dietrich and Bradley, 1988; Gai et al., 2019). The liver, the main iron storage organ, is highly susceptible to iron overload under the toxic effects of various drugs and chemicals, leading to oxidative damage mediated by ferroptosis (Xing et al., 2022; Yang et al., 2022). MeHg toxicity is primarily associated with glutathione depletion and reactive oxygen species (ROS) generation, causing oxidative stress damage in the body, potentially mediated by the Fe2+ ion (LeBel et al., 1992). Moreover, MeHg disrupts the balance of antioxidant defenses in the liver by interfering with key enzymes such as glutathione peroxidase (GPx) and superoxide dismutase (SOD) (Antunes dos Santos et al., 2018). The Keap1/NRF2/HO-1 signaling pathway is considered a crucial mechanism regulating oxidative stress (Hybertson et al., 2011; Jiang et al., 2024). Moreover, previous studies have indicated that the p38-Nrf2-HO-1 axis regulates ferroptosis (Li et al., 2023; Yang et al., 2021). Additionally, studies have found that ferroptosis occurs in various liver diseases, such as hepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma (Pan et al., 2021; Wu et al., 2021). However, it remains unexplored whether ferroptosis is involved in MeHg-induced liver dysfunction.
Inflammation is a protective mechanism triggered by tissue injury and exposure to toxic substances. MeHg exposure has been shown to induce inflammatory responses, and it activates pro-inflammatory signaling pathways, such as the NF-κB/NLRP3 inflammasome axis (Ye et al., 2023). NF-κB, a key regulator of inflammation (Yu et al., 2020), is closely linked to the development of ferroptosis (Chen et al., 2022, 2023). Additionally, activation of MAPK pathway-dependent inflammation has been associated with ferroptosis, with one study noting that iron overload suppressed ERK1/2 and p38 phosphorylation, increasing oxidative stress and exacerbating muscle damage (Chen et al., 2023).
Building on these findings, this study aimed to investigate the effects of MeHg exposure on the liver in mice and the associated molecular mechanisms, focusing on ferroptosis, oxidative stress, inflammatory responses, and the MAPK signaling pathway. By elucidating these mechanisms, this research seeked to enhance our understanding of MeHg toxicity and to identify potential therapeutic targets to mitigate its harmful effects on the liver.
Materials and methods
Reagents and antibodies
Methylmercury chloride (MeHgCl) was provided by Tansoole (CAS: 115–09-3, Shanghai, China), dissolved in dimethyl sulfoxide (DMSO) (CAS: 67–68-5, Macklin, China), and finally diluted in physiological saline. The concentration of DMSO in the experiments was approximately 0.08%. Antibodies against glutathione peroxidase 4 (GPX4) and 4-hydroxynonenal (4-HNE) were purchased from Abcam (Cambridge, UK). Antibodies from Cell Signaling Technology included HO-1, IκB, ERK1/2, p-ERK1/2, p38, p-p38, and p65(Boston, USA). Antibodies against Nrf2, p-IκB, p-p65, and transferrin receptor (TfR) were obtained from Wanlei Bio (Shenyang, China). Keap1 antibody was purchased from Bioss Biotechnology (Beijing, China). GAPDH antibody and NLRP3 antibody were acquired from Boster (Wuhan, China).
Animals and treatments
Male BALB/c mice (6-8 weeks old, 20-25 g), specific-pathogen-free, obtained from the Guangdong Medical Laboratory Animal Center, were acclimatized for 1 week. Twenty mice were randomly divided into four groups: control (0.08% DMSO) and MeHgCl (40, 80, 160 μmol/kg), administered via gavage once at the specified concentrations, followed by normal feeding for 7 days. The mice were euthanized and necropsied for blood and liver tissue collection after 1 week.
Histopathological analysis
Liver tissues were immediately fixed in 10% formalin solution upon collection. After dehydration through graded ethanol, tissues were embedded in paraffin, sectioned into 5 μm slices, and mounted on slides. Deparaffinization was performed using xylene and different concentrations of ethanol, followed by hematoxylin and eosin (H&E) staining. Tissue pathological changes were observed under an optical microscope (Eclipse e200, Nikon, Japan).
Serum biochemical index determination
Blood samples were kept overnight at 4°C, followed by centrifugation to separate serum. Using reagents from Mindray Bio-Medical Electronics (Shenzhen, China), serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) were determined using a Clinical Fully-Auto Biochemistry Analyzer BS240 according to the manufacturer’s instructions.
Detection of biochemical indicators
Liver tissues were homogenized in physiological saline to obtain tissue homogenates of appropriate concentrations. Following the instructions of the assay kit, all from Nanjing Jiancheng Bioengineering Institute (Nanjing, China), activity of superoxide dismutase (SOD, CAT: A001-1), glutathione (GSH, CAT: A006-2-1), myeloperoxidase (MPO, CAT: A044-1-1), and level of malondialdehyde (MDA, CAT: A003-1) and iron (A039-2-1) in liver tissues were measured.
Gene expression analysis
Total RNA from kidney tissues was extracted using TRIzol reagent, and its concentration and OD260nm/OD280nm ratio were measured. RNA was reverse transcribed into cDNA using a reverse kit following the manufacturer’s instructions (TransGen Biotech, Beijing, China). The mRNA expression levels of target genes were quantified using a fluorescence quantitative PCR instrument (CFX, BIO-RAD, USA) according to the instructions of the SYBR reagent kit from TransGen Biotech (Beijing, China). GAPDH was used as an internal reference gene, and relative expression of target genes was calculated using the 2−ΔΔCT method.
Western Blot
Liver tissue lysates were centrifuged at low temperature for 10 minutes at 10000 × g, and the supernatant was used to determine protein concentration with a BCA assay kit. Protein samples were separated by 10% SDS-PAGE, transferred to PVDF membranes, blocked with 5% non-fat milk for 4 hours, and then incubated overnight at 4°C with primary antibodies. After washing, membranes were incubated with HRP-conjugated goat anti-rabbit secondary antibody (CAT: SA00001-2, Proteintech Group, USA) or HRP-linked anti-mouse secondary antibody (CAT: 7076s, Cell Signaling Technology, USA) for 1 hour, washed with Tris-buffered saline three times, and developed using enhanced chemiluminescence (ECL Plus) for protein blot image analysis.
Statistical analysis
All data were analyzed by Graph Pad Prism 9. The Shapiro-Wilk test was used to determine the normality of the sample. Differences among multiple groups were assessed by one-way analysis of variance (ANOVA) followed by Dunnett or Turkey’s multiple comparison tests as applicable. The data were expressed as mean ± SEM. (*p < 0.05 was considered significant. **p < 0.01, ***p < 0.001, ****p < 0.0001, “ns” means not significant).
Results
MeHg-induced acute liver injury in mice
We investigated the effect of MeHg exposure on the liver in mice. Histopathological examination of liver tissue revealed that the liver structure and morphology appeared normal in the control group, while in the MeHg-exposed groups, liver tissues exhibited significant swelling and degeneration, disordered hepatic cord arrangement, and partial cell atrophy and necrosis, with severity increasing in a dose-related manner (Figure 1(a)). MeHg exposure significantly elevated the levels of serum ALT, AST, and ALP, which also increased in a dose-dependent manner (Figure 1(b)). In summary, MeHg exposure induced acute liver injury in mice in a dose-dependent manner. The effect of MeHg treatment on acute liver injury in mice. (A) The liver tissue sections (5 μm) were stained with hematoxylin-eosin (H&E) and photographed by an optical microscope (100× and 400×). (B) ALT, AST, and ALP kits were used to compare the severity of liver damage in mice treated with different treatments. Data were shown as mean ± SEM (*p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001, “ns” means not significant). Abbreviations: MeHg, methylmercury; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase.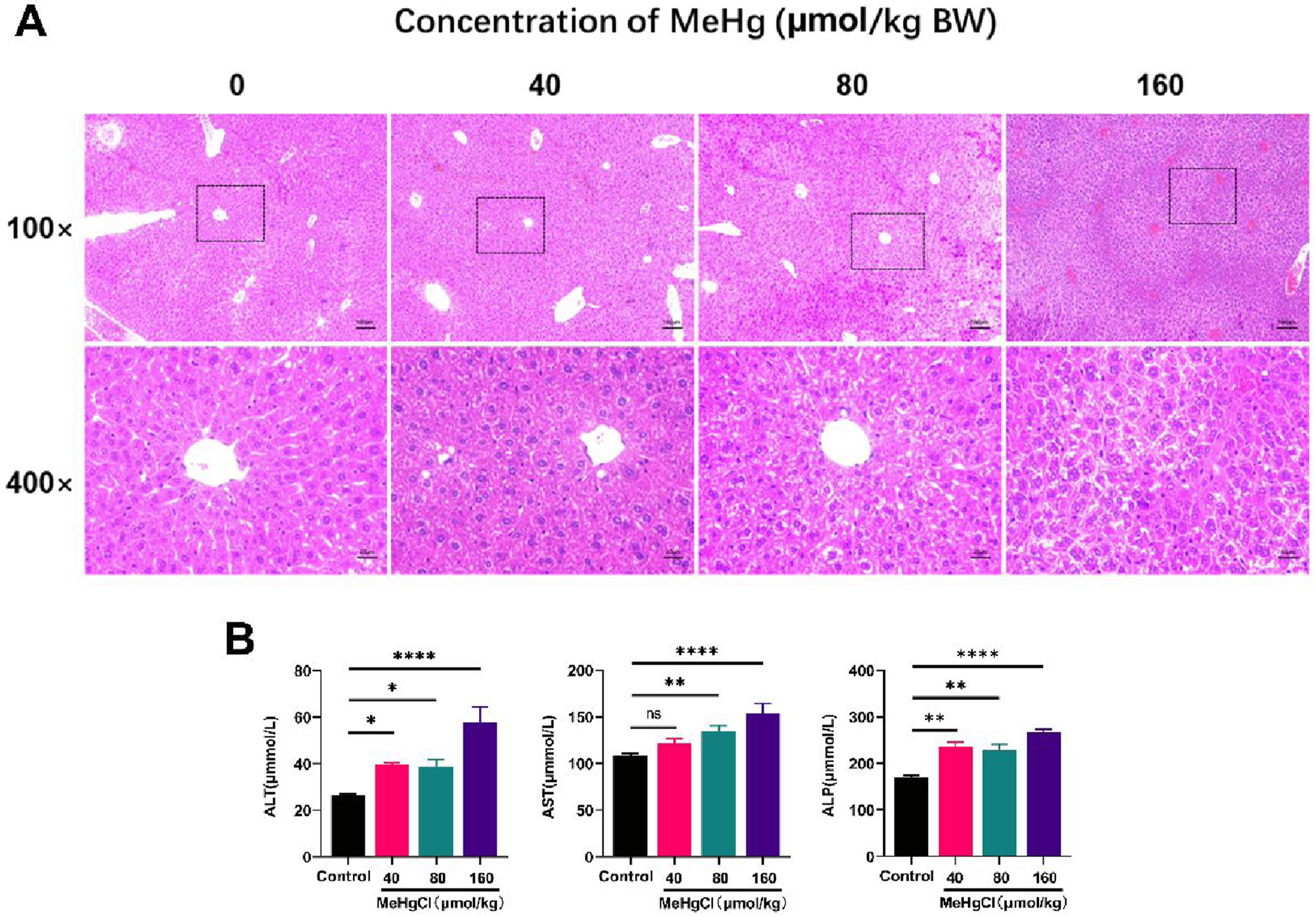
Involvement of ferroptosis in MeHg-induced acute liver injury
We investigated the potential role of ferroptosis in methylmercury-induced acute liver injury. Results indicated that MeHg treatment significantly increased MDA content (Figure 2(a)), suppressed SOD (Figure 2(b)) and GSH (Figure 2(c)) activities, and reduced CAT mRNA expression levels. Following MeHg exposure, results demonstrated a significant elevation in total iron content (Figure 2(e)) and TfR1 protein expression levels (Figure 2(f)), both showing a dose-dependent relationship compared with the control group. These findings strongly suggest the involvement of ferroptosis in MeHg-induced acute liver injury. MeHg exposure induces hepatic ferroptosis in mice. (A) The concentration of MDA in mouse liver induced by MeHg. (B) Impact on GSH activity, an antioxidant enzyme. (C) Assessment of SOD activity. (D) Relative expression levels of catalase (CAT) gene analyzed in the liver. (E) Measurement of relative total iron content in the liver. (F) Western Blot and quantitative analysis results of TfR1 protein expression in the liver. Data were shown as mean ± SEM (*p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001, “ns” means not significant). Abbreviations: MeHg, methylmercury; MDA, malondialdehyde; GSH, glutathione; SOD, superoxide dismutase; Tf receptor, transferrin receptor.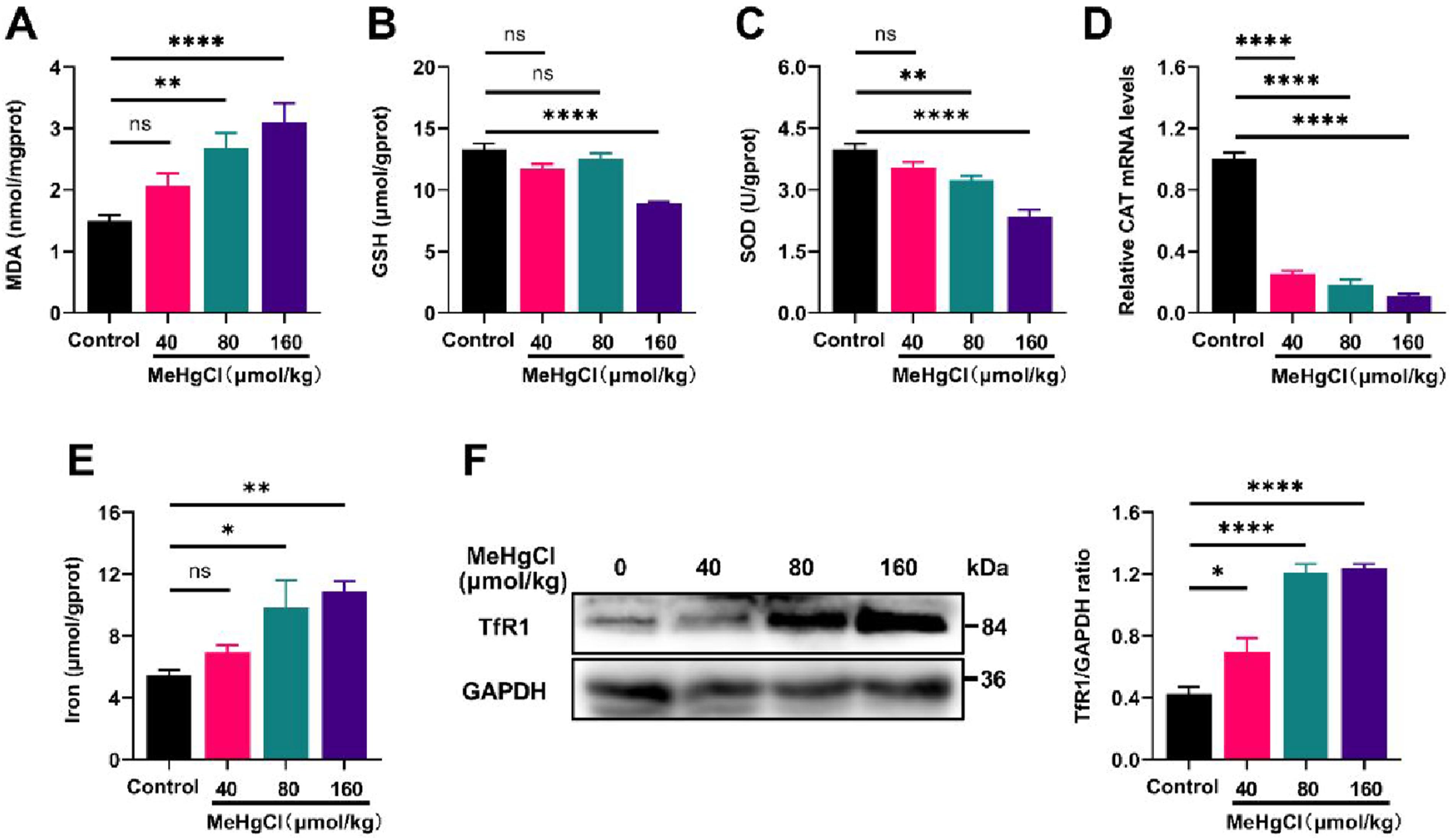
To further elucidate this mechanism, we examined the expression of ferroptosis regulatory factors and biomarkers. The results revealed that MeHg exposure inhibited GPX4 protein expression, a critical regulator of ferroptosis (Liu et al., 2023b), while promoting the protein expression of 4-hydroxynonenal (4HNE), a significant lipid peroxidation marker associated with ferroptosis (Liu et al., 2023a) (Figure 3(a)). Additionally, MeHg treatment reduced mRNA expression levels of GPX4 (Figure 3(b)) and SLC7A11 (Figure 3(c)). These results indicated the substantial role of ferroptosis in MeHg-induced acute liver injury. Effects of Methylmercury on Ferroptosis Markers in Mouse Liver Tissue. (A) Western Blot and quantitative analysis results of GPX4 and 4-HNE protein expression. (B) Analysis of relative expression levels of the GPX4 gene. (C) Analysis of relative expression levels of the SLC7A1 gene. Data were shown as mean ± SEM (*p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001, “ns” means not significant). Abbreviations: MeHg, methylmercury; GPX4, glutathione peroxidase 4; 4-HNE, 4-hydroxynonenal; SLC7A11, solute carrier family 7 member 11.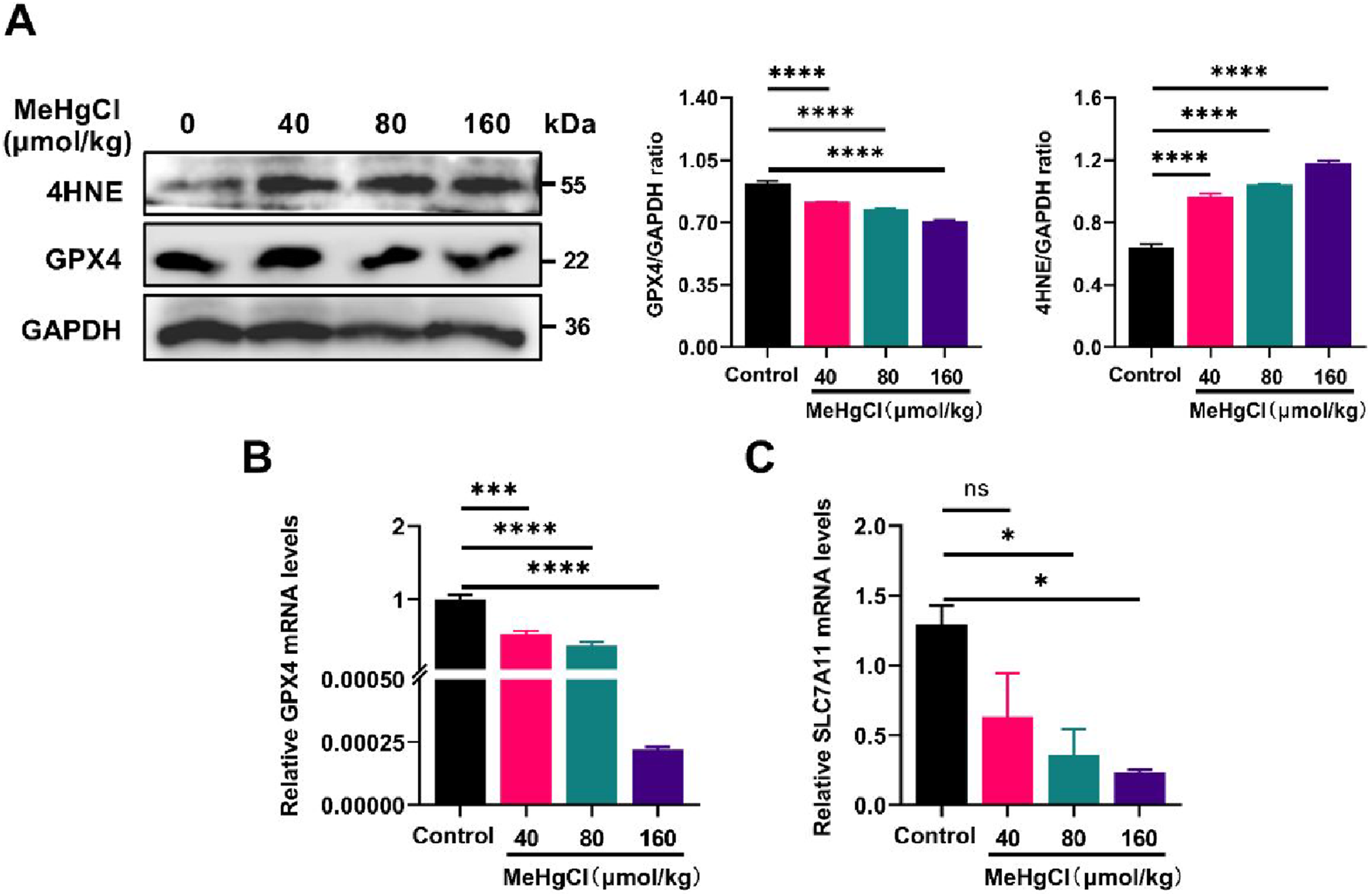
MeHg regulated the Keap1/Nrf2/HO-1 axis and activated p38/ ERK phosphorylation
The impact of MeHg treatment on the Keap1/Nrf2/HO-1 signaling pathway, revealed that MeHg treatment led to an elevation in Nrf2 protein expression levels, a significant increase in KEAP1 protein levels, and a notable upregulation of HO-1 protein (Figure 4(a)). These findings indicated that MeHg activates the Keap1/NRF2/HO-1 signaling pathway, promoting oxidative stress in the liver. Effects of MeHg treatment on Nrf2 and p38/ERK1/2 MAPK Signaling Pathways. (A) Protein expression levels of Keap1, Nrf2, and HO-1, and their relative quantification analysis results. (B) Protein expression levels of ERK1/2, p-ERK1/2, p38, and p-p38, and their relative quantification analysis results. Data were shown as mean ± SEM (*p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001, “ns” means not significant). Abbreviations: MeHg, methylmercury; Nrf2, nuclear factor erythroid 2-related factor 2; Keap1, kelch-like ECH-associated protein l; HO-1, heme oxygenase 1; p-ERK, phosphorylation of p-ERK; p-p38, phosphorylation of p38.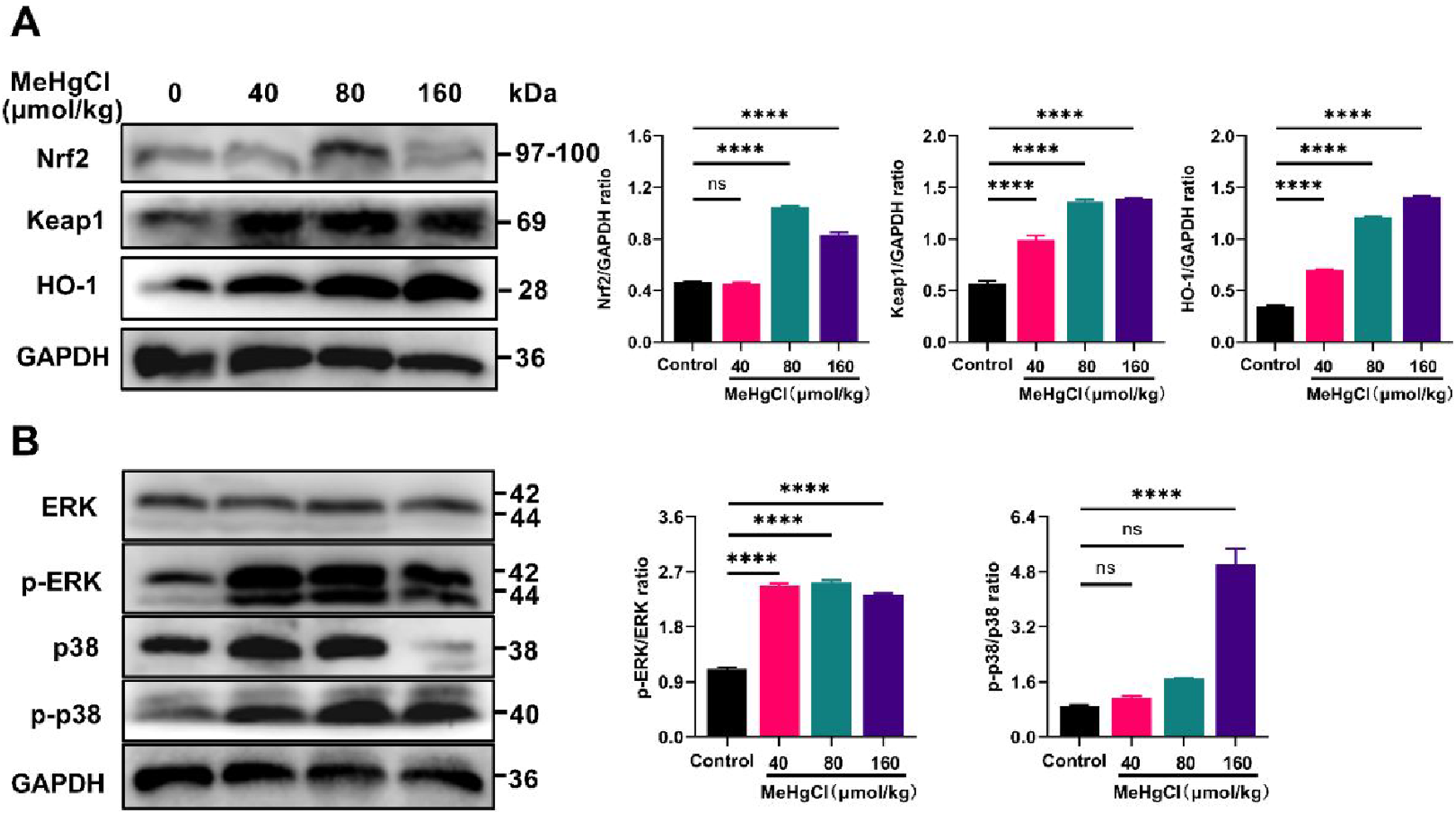
Moreover, the effect of MeHg exposure on the p38/ERK1/2 MAPK signaling pathway revealed that, compared with the control group, mice treated with MeHg exhibited significantly increased levels of p-ERK1/2 protein expression. Additionally, the expression of p-p38 was notably elevated in the 160 μmol/kg group, whereas no significant differences were observed in the 40 and 80 μmol/kg groups compared with the control group (Figure 4(b)).
MeHg enhanced hepatic inflammation via NF-κB/NLRP3 signaling pathway activation
It was found that MeHg treatment significantly increased MPO activity (Figure 5(a)), which is associated with inflammation and oxidative stress (Aratani, 2018). Additionally, the mRNA expression levels of pro-inflammatory cytokines TNF-α, IL-6, and IL-1β were significantly elevated, compared with the control group (Figure 5(b) to (d)). Western blot analysis revealed that MeHg exposure increased NLRP3 inflammasome expression and the phosphorylation levels of p65 and IκB (Figure 5(e)), indicating increased activity of the NF-κB/NLRP3 pathway in MeHg-treated mice. In summary, MeHg exacerbated liver inflammation and enhanced the production of pro-inflammatory mediators TNF-α, IL-6, and IL-1β, as well as MPO activity, through the activation of the NF-κB/NLRP3 inflammatory signaling pathway (Table 1). MeHg exposure exacerbates inflammation response in liver. (A) Measurement of myeloperoxidase (MPO) activity in the liver. (B-D) Gene expression analysis of the inflammatory cytokines IL-6, IL-1β, and TNF-α in the liver. (E) Western blot results and relative expression analysis of NLRP3 inflammasome, p65, pp65, IκB, and p-IκB in the liver. Data were shown as mean ± SEM (*p < 0.05; **p < 0.01, ***p < 0.001, ****p < 0.0001, “ns” means not significant). Abbreviations: MeHg, methylmercury; MPO, myeloperoxidase; TNF-α, tumor necrosis factor α; IL-1β, interleukin-1β; IL-6, interleukin-6; pp65, phosphorylation of p65; p-IκB phosphorylation of IκB. The primer sequences used in the study.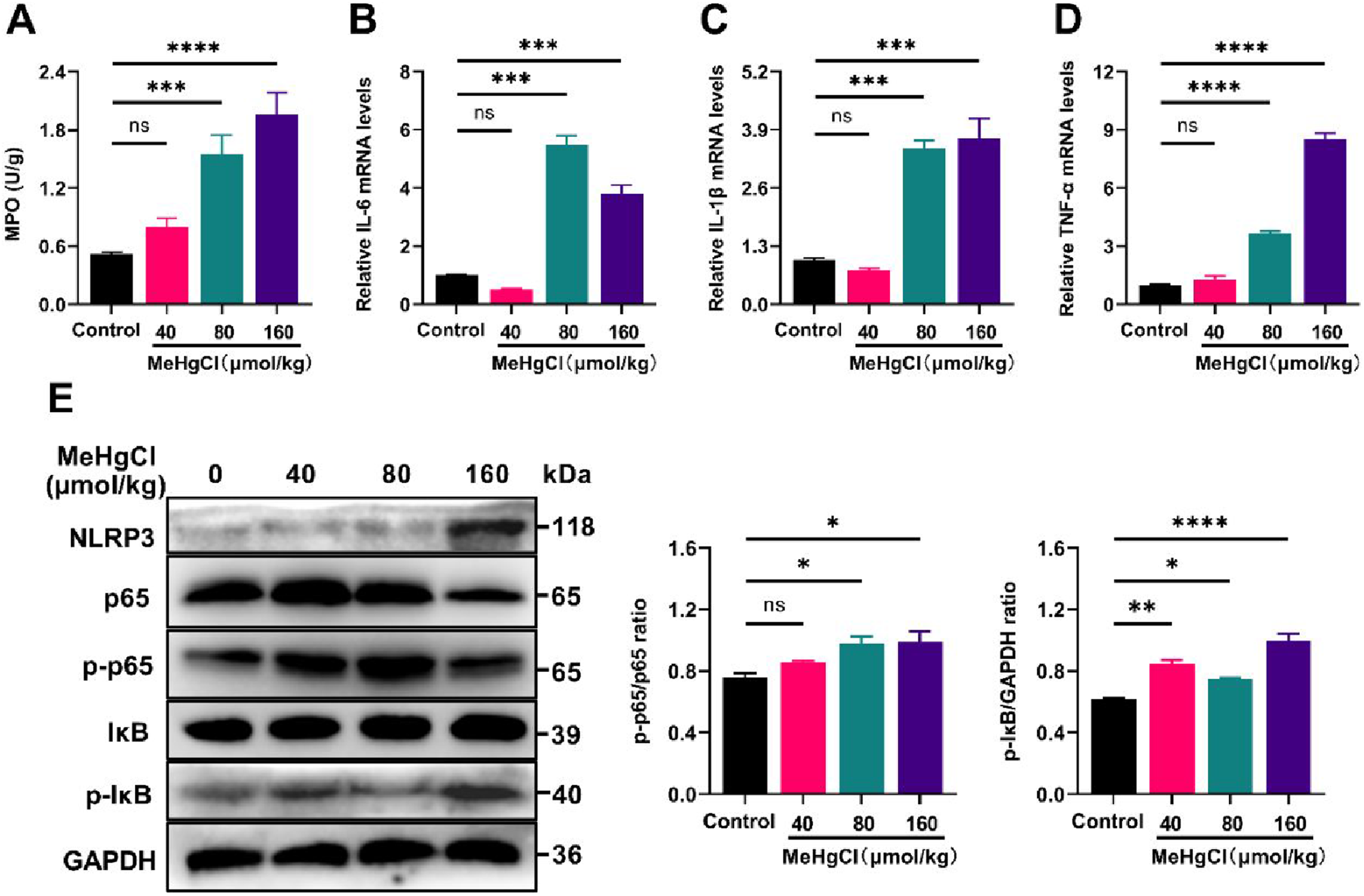

Discussion
This study provided preliminary evidence that MeHg exposure induces acute liver injury in mice, with the severity of damage increasing with higher doses. These findings align with previous research that identified MeHg as a potent hepatotoxin capable of causing liver dysfunction through oxidative stress, inflammation, and cell death (Mela et al., 2007). The data presented in this study further elucidates the molecular mechanisms behind this toxicity, focusing on ferroptosis, inflammation, oxidative stress, and the MAPK signaling pathway.
Ferroptosis, previously noted for its crucial role in cadmium-induced liver damage, involves oxidative stress and excessive inflammation (He et al., 2022). Similarly, this study confirmed that ferroptosis plays a prominent role in MeHg-induced liver damage in mice. MeHg exposure led to the inhibition of antioxidant enzymes such as GSH and SOD, as well as increased MDA production, a marker of lipid peroxidation. These changes, coupled with elevated iron levels and transferrin receptor expression, suggest iron overload and lipid peroxidation in the liver, key characteristics of ferroptosis (Liang et al., 2023). Based on these observations, it is hypothesized that ferroptosis contributes to the MeHg-induced liver dysfunction, mirroring earlier studies on hepatocytes, underscoring the critical role of ferroptosis in MeHg toxicity (Dong et al., 2022). Additionally, the study noted decreased expression of GPX4, a crucial enzyme in ferroptosis inhibition, along with reduced SLC7A11 levels and GSH activity, indicating that the System Xc-GSH-GPX4 pathway may play a role in promoting ferroptosis and lipid peroxidation during MeHg-induced acute liver injury.
Besides ferroptosis, inflammation was also significantly activated upon MeHg exposure. Similar to other heavy metals such as cadmium and nickel, MeHg exposure triggered the activation of inflammatory pathways, including the NF-κB/NLRP3 inflammasome signaling axis, which releases pro-inflammatory cytokines, exacerbating liver damage (Cai et al., 2021; He et al., 2022; Pan et al., 2024). The present study also emphasized the involvement of oxidative stress in MeHg-induced liver damage. The observed decrease in antioxidant enzymes like SOD and the accumulation of lipid peroxides such as MDA and 4-HNE suggest an imbalance between oxidation and antioxidation systems. The Keap1/Nrf2/HO-1 pathway’s active response indicates its role in regulating this balance. Nrf2 not only modulates the expression of key antioxidant enzymes to counteract oxidative stress (Ma, 2013), but also regulates SLC7A11 and GPX4, key targets in ferroptosis (Liu et al., 2023b), highlighting its role in the ferroptosis process (Dodson et al., 2019). Furthermore, the p38/ERK1/2 MAPK signaling axis is strongly associated with various toxic responses (Cheng et al., 2022a, 2022b), was significantly activated in our study following MeHg exposure. This finding aligns with previous research, indicating its involvement in the hepatotoxicity process. MAPK pathways, particularly p38 and ERK1/2, are known to regulate cellular responses to stress, including inflammation, oxidative stress, and ferroptosis (Cargnello and Roux, 2011; Liu et al., 2022). The strong correlation between the activation of these pathways and the severity of liver injury suggests their critical role in mediating MeHg’s toxic effects.
It is important to note that the absence of a ferroptosis inhibitor group leaves uncertainty regarding the interactions between ferroptosis and other signaling pathways. Nonetheless, the concurrent activation of ferroptosis, inflammation, oxidative stress, and MAPK signaling pathways strongly indicates that these mechanisms are interlinked and collectively contribute to MeHg’s hepatotoxic effects. In conclusion, this study showed that MeHg exposure led to acute liver injury via a combination of ferroptosis, oxidative stress, inflammation, and MAPK pathway activation. These findings deepen the understanding of the complex molecular mechanisms behind MeHg toxicity and highlight potential therapeutic targets for mitigating its harmful effects. Future studies should focus on elucidating the specific interactions among these pathways to develop strategies for reducing MeHg-induced hepatotoxicity.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (No. 31772721).