
Abstract
Paraquat (PQ), a widely used herbicide, induces severe pulmonary fibrosis through complex mechanisms that are not fully understood. This study employed an integrated computational approach combining network toxicology, molecular docking, dynamics simulations, and AlphaFold2-based protein design to systematically investigate PQ-induced pulmonary fibrotic processes. We identified 111 common targets from pulmonary fibrosis and PQ toxicity databases, among which AKT1, IL6, TP53, and CASP3 were recognized as core targets. Functional enrichment analyses revealed significant involvement of oxidative stress, inflammatory response, and apoptotic pathways. Molecular docking demonstrated strong binding affinity between PQ and key targets (docking scores < −5 kcal/mol), while molecular dynamics simulations confirmed stable interactions with favorable binding energies (< −12 kcal/mol). Furthermore, AlphaFold2 predicted three novel proteins exhibiting even higher binding affinity to PQ than natural targets. These findings reveal that PQ might promote pulmonary fibrosis by stably binding to core proteins and activating critical pathological pathways, providing valuable insights for developing targeted biomarkers and therapeutic strategies against PQ-induced lung injury.
Introduction
Paraquat (PQ), a broad-spectrum herbicide valued for its multifunctionality, is extensively utilized in numerous countries (Tang et al., 2024). Its active ingredient, 1,1′-dimethyl-4,4′-bipyridinium, is highly effective yet poses substantial risks to both terrestrial and aquatic ecosystems (Huang et al., 2019). PQ contamination leads to various environmental issues, including pollution of water resources, soil residue accumulation and acidification, disruption of food chains, and bioaccumulation of toxins (Jain et al., 2025). Of particular concern is its severe impact on human health, where PQ exposure can cause lung injury and multi-organ damage, with a majority of fatalities resulting from progressive pulmonary fibrosis (Suntres, 2018). These serious health implications underscore the potential of PQ to contribute to public safety incidents (Shi et al., 2023).
The toxicological mechanisms of PQ have been partially characterized in several studies. For example, patients who died from PQ poisoning exhibited increased lymphocyte counts, elevated neutrophil counts, and a higher CD4+/CD8+ T cell ratio, along with a reduced natural killer cell percentage, suggesting that these immunological markers may be associated with mortality in acute PQ poisoning (Dong et al., 2025). Acute PQ exposure at a dose of 45 mg/kg induced pyknosis, edema, and apoptosis in rat substantia nigra neurons. Moreover, alterations in microglial phenotypic differentiation were found to be dependent on both the exposure dose and duration (Zhang et al., 2022). PQ induces pulmonary tissue damage by triggering multiple forms of cell death. It regulates epithelial-mesenchymal transition progression, which contributes to autophagy-mediated pulmonary fibrosis (Liu et al., 2022). Additionally, PQ decreases forkhead box F1 expression, induces oxidative stress and promotes apoptosis in lung injury through the mitochondrial-related pathway (Liu et al., 2022). Furthermore, PQ has also been implicated in ferroptosis. Key mechanisms involve disruption of mitochondrial homeostasis, induction of autophagy, activation of the NCOA4-FTH1 axis, and modulation of the Nrf2 antioxidant pathway, collectively leading to lipid peroxidation and subsequent ferroptosis (Du et al., 2024). However, the mechanisms underlying the entire poisoning process of PQ have not been fully elucidated.
In this study, we employed an integrated approach combining network toxicology, molecular docking, molecular dynamics simulations, and AlphaFold2 predictions to elucidate the molecular mechanisms of PQ-induced pulmonary fibrosis, with particular emphasis on its multi-target and multi-pathway biological effects. The findings provide a foundation for the development of biomarkers and anti-fibrotic therapies targeting PQ-induced pulmonary fibrosis.
Materials and methods
Disease targets identification
Using “pulmonary fibrosis” as the keyword, potential targets were retrieved from the GeneCards database (relevance score >30; https://www.genecards.org/), the Online Mendelian Inheritance in Man (OMIM) database (https://www.omim.org/), and the Comparative Toxicogenomics database (CTD; https://ctdbase.org/). The final set of disease-associated targets was obtained by integrating the results from these three databases (database access date: March 25, 2025).
PQ targets identification
The SMILES data of the PQ molecule was obtained from the PubChem database and subsequently submitted to the SEA database (https://sea.bkslab.org/) for target prediction. Targets with a p-value of less than 0.05 were selected. Additional target prediction was conducted using the SuperPred database (https://prediction.charite.de/subpages/target_prediction.php), retaining those targets with a probability score above 0.6. Further targets were identified via the CTD database (https://ctdbase.org/), applying an inference score threshold of greater than 10. Finally, the toxicant-related targets derived from these three databases were integrated to form a comprehensive target list.
Common targets analysis
The selected pulmonary fibrosis-related targets and PQ-related targets were input into the Venn diagram tool (Venny 2.1) to identify common targets. These overlapping targets represent key candidates for further investigation into the mechanisms of PQ-induced toxicity and disease, and warrant deeper analysis using network toxicology approaches to more accurately assess the compound’s toxicological risks.
Construction of the protein-protein interaction (PPI) network
The common targets of pulmonary fibrosis and PQ were imported into the STRING database (v12.0, https://string-db.org/cgi/input.pl) to construct a PPI network, with the organism specified as “Homo sapiens.” Based on the resulting PPI network, the cytoHubba plugin in Cytoscape software was used to filter and visualize the key nodes in the network using the Degree and Maximal Clique Centrality (MCC) algorithms, respectively.
Construction of the disease-toxicant-target network
To elucidate the complex interactions among pulmonary fibrosis, PQ, and their corresponding targets, a disease-toxicant-target network was constructed using the identified pulmonary fibrosis and PQ associated targets. This network was subsequently imported into Cytoscape 3.10.0 for visualization and topological analysis.
Gene ontology (GO) enrichment analysis
GO enrichment analysis was performed on the intersection of key targets for pulmonary fibrosis and PQ, including biological processes (BP), molecular functions (MF), and cellular components (CC). Enrichment items with an adjusted p-value <0.05 were filtered using the STRING database (v12.0). Bubble plots were subsequently generated in R software (version 4.4.0) with the clusterProfiler, enrichplot, and ggplot2 packages.
Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis
KEGG pathway enrichment analysis was conducted on the common targets associated with both pulmonary fibrosis and PQ using the STRING database (v12.0). Terms with an adjusted p-value <0.05 were considered statistically significant. Bar plots and bubble charts were generated in R software (version 4.4.0) with the clusterProfiler package.
Molecular docking analysis
The top ten key targets identified by two network toxicology algorithms were selected as key proteins for molecular docking with PQ. Crystal structures of these proteins and their corresponding PDB IDs were retrieved from the Protein Data Bank (PDB) database. The three-dimensional structure of PQ was downloaded from the PubChem database and energy-minimized using the MMFF94 force field. Molecular docking was performed using AutoDock Vina 1.2.3 software. Prior to docking, all receptor proteins were preprocessed with PyMOL 2.5.5 to remove water molecules, salt ions, and other hetero molecules. Subsequently, the docking box was defined using the center_of_mass.py plugin in PyMOL, which positioned the box center at the active site with an edge length of 22.5 Å. Additionally, ADFRsuite 1.0 software was used to convert both the small molecules and proteins into the PDBQT format required for docking. During docking, the global search detail was set to 32, while all other parameters remained at default values. The docking poses were visualized using PyMOL, and the conformation with the highest binding affinity was selected for subsequent molecular dynamics simulations.
Molecule dynamics simulation
Based on the docking results, the complex formed between PQ and the core target protein was used as the initial structure for all-atom molecular dynamics simulations. Simulations were performed using AMBER 22 software. Before the simulation operations, the charge of PQ was obtained through the antechamber module and Gaussian 09 software using the Hartree–Fock SCF/6-31G* calculation. The small molecule PQ and the core target protein were treated using the GAFF2 small molecule force field and the ff14SB protein force field, respectively. Each system was prepared with the LEaP module: hydrogen atoms were added, a truncated octahedral TIP3P water box was placed with a 10 Å buffer around the solute, and Na+/Cl- ions were added to neutralize the system charge. Finally, the topology and parameter files required for simulation were generated.
The molecular dynamics simulation was carried out according to the following procedure. Before running, the system undergoes energy optimization using 2,500 steps of the steepest descent method and 2,500 steps of the conjugate gradient method. Subsequently, the system was gradually heated from 0 K to 298.15 K over 200 ps at constant volume and constant heating rate. After reaching the target temperature, a 500 ps NVT (canonical ensemble) simulation was conducted to equilibrate the solvent distribution. This was followed by NPT (isothermal-isobaric) conditions, a 500 ps equilibrium simulation of the entire system. Finally, a 100 ns production simulation was performed in the NPT ensemble with periodic boundary conditions.
The following parameters were applied throughout the simulations: a cutoff distance of 10 Å was used for non-bonded interactions; long-range electrostatic interactions were treated with the Particle Mesh Ewald method; hydrogen bonds were constrained using the SHAKE algorithm; and temperature was regulated with the Langevin algorithm (collision frequency γ = 2 ps-1). The pressure was maintained at 1 atm, the integration time step was set to 2 fs, and trajectories were saved every 10 ps for subsequent analysis.
MM/GBSA binding free energy calculation
The binding free energy between the core targets and PQ was calculated using the MM/GBSA method. Long molecular dynamics simulations might adversely affect the accuracy of MM/GBSA calculations. Therefore, 90–100 ns MD trajectories were used for the calculations in this study, with the specific formula as follows:
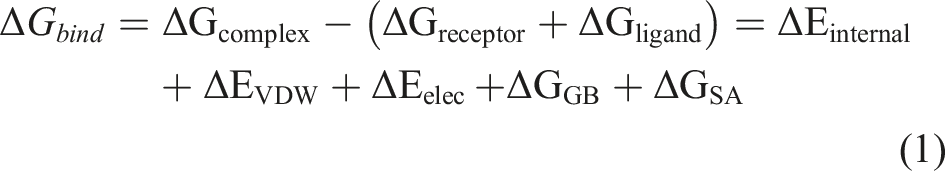
Drug design based on AlphaFold2
The computational experiments were performed on a hardware configuration equipped with 2 × NVIDIA A100 GPUs and 1 × 32-core AMD EPYC 7543 CPU. The workflow consisted of two steps: (1) generating the main chain under the condition of PQ; and (2) conducting sequence design followed by computational validation.
Ligand preparation
PQ dichloride (N,N′-dimethyl-4,4′-bipyridinium, PQ2+) was drawn in Maestro 13.7 (Schrödinger, LLC) and geometry-optimized at the B3LYP/6-31G level using Gaussian 16 Rev. C.01. Fifty low-energy conformers were enumerated with OMEGA 4.0.0.2 and the conformer with the lowest MMFF94S energy was retained. AM1-BCC charges were assigned via QUACPAC 2.6. The resulting PQ.mol2 file defined the ligand throughout the pipeline.
Pocket specification
From a structural survey of PQ-binding proteins (PDB IDs 4RUM, 6J9K), we extracted the minimal pharmacophore: two π-rich residues (Trp/Tyr/Phe) and one anionic residue (Asp/Glu) within 6 Å and 3.5 Å, respectively, of the ligand. A target pocket volume of 240 ± 30 Å3 was set based on POVME 3.0 calculations on these templates.
Backbone generation with RFdiffusion-All-Atom
Backbones were generated using the RFdiffusion-All-Atom checkpoint rfdiff_allatom_beta.pth (commit 1d3f87f). A contig string of (“10-30/A,” “PQ,” “60-90/A”) instructed the model to generate an N-terminal segment (10–30 residues), the ligand, and a C-terminal segment (60–90 residues). Hot-spot residues A27, A34 and A38 were specified to bias the network toward forming a binding pocket. They occupy critical positions at the protein-ligand interaction interface. These three residues are all located within the core region of the substrate-binding pocket, with their side-chain atoms positioned less than 4 Å from the ligand. They are characterized by pronounced steric hindrance and strong charge complementarity. One hundred independent trajectories were sampled deterministically (temperature 0.0). The resulting PDB files were filtered for pocket volume (200–280 Å3) and aromatic residue count (≥3 within 8 Å of PQ2+), yielding 42 candidates.
Sequence design with LigandMPNN
Sequences were designed with LigandMPNN checkpoint ligandmpnn_v1. Each backbone was processed with the command:
python design.py --pdb backbone.pdb --ligand PQ.mol2\
--out_fasta seq.fasta --temperature 0.1\
--pack_side_chains true
Five sequences with the highest log-likelihood scores were retained for each backbone.
In-silico validation
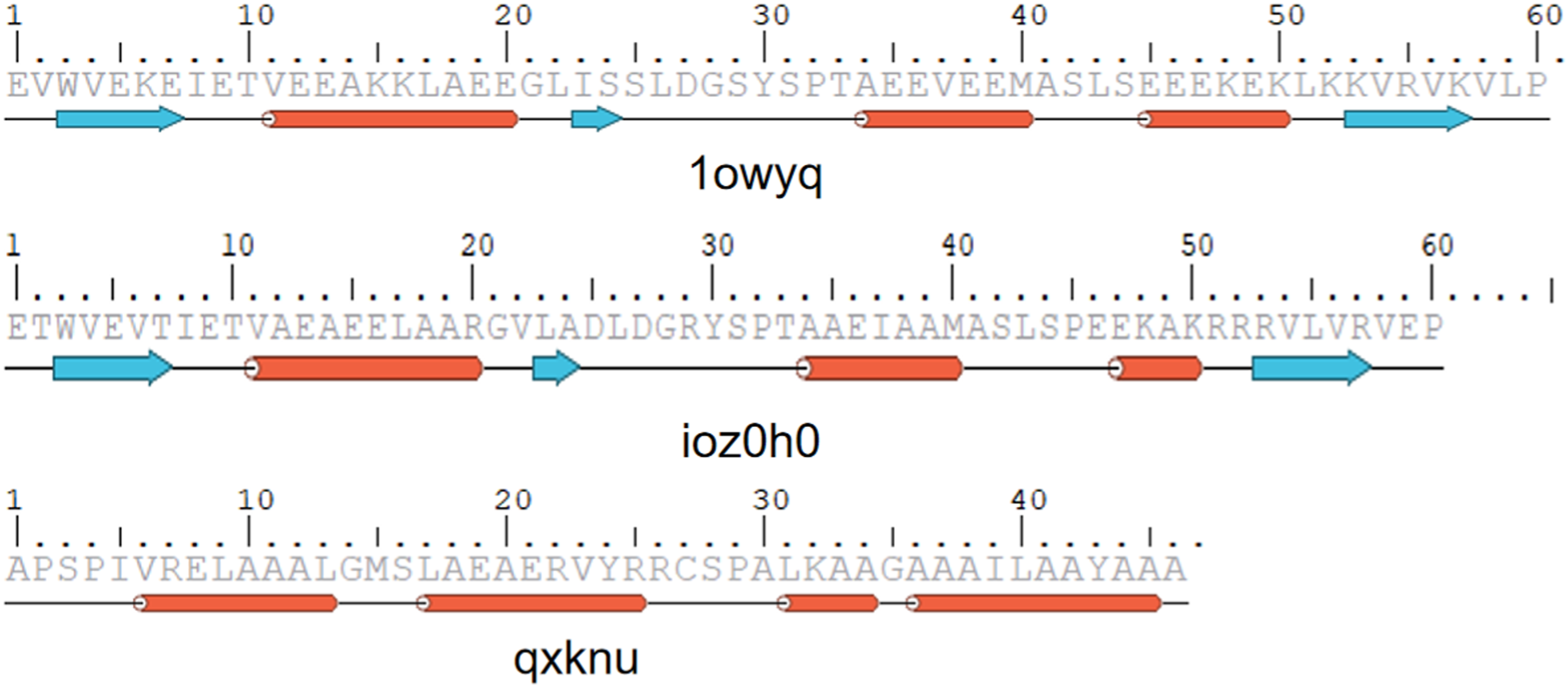
Complex structures were predicted with AlphaFold2-Multimer (ColabFold 1.5.5) using amber_relax=True. Designs exhibiting interface predicted aligned error (iPAE) > 10 Å or pLDDT <70 were discarded. The remaining models were relaxed in Rosetta 2024.27 with coordinate constraints (FastRelax, ref2015 score function). Binding energy (ΔGbind) was calculated with InterfaceAnalyzer; only designs with ΔGbind ≤ −6.0 kcal/mol were kept. Finally, pocket backbone RMSD between the RFdiffusion model and the AlphaFold2 prediction had to be ≤1.0 Å. These filters left three designs (1owyq, ioz0h0, qxknu).
Results
Network toxicology identifies the core targets
A total of 1,500 pulmonary fibrosis-related targets were retrieved from the GeneCards, OMIM, and CTD databases after removing duplicates, while 212 PQ-related targets were collected from the SEA and CTD databases. Venn analysis identified 111 common targets (Figure 1(a)). The PPI network obtained from the STRING database consisted of 108 nodes and 2,567 edges (Figure 1(b)). This network was visualized using Cytoscape, with node color and size scaled according to their degree values: larger and darker nodes indicate higher degree (Figure 1(c)). Additionally, a disease-toxicant-target network was constructed using Cytoscape 3.10.0 (Figure 1(d)). The top 10 hub targets identified by the Degree algorithm were AKT1, TP53, CASP3, IL6, TNF, BCL2, IL1B, JUN, HIF1A, and HMOX1 (Figure 1(e)). The top 10 targets obtained using the MCC algorithm were AKT1, IL6, TNF, JUN, CASP3, IL1B, TP53, NF-κB1, HIF1A, and TGFB1 (Figure 1(f)). Based on the combined results from both algorithms, AKT1, TP53, IL6, and CASP3 were selected as the four core target genes with the highest potential regulatory significance in PQ-induced pulmonary fibrosis. Network toxicology analysis. (a) Venn diagram of pulmonary fibrosis- and PQ-related targets. (b) The PPI network obtained from the STRING database. (c) The visualization of PPI network by Cytoscape software. (d) A component-disease-target network diagram was constructed using Cytoscape 3.10.0 software. (e) The top 10 targets obtained using the degree method with the cytoHubba plugin. (f) The top 10 targets obtained using the MCC method.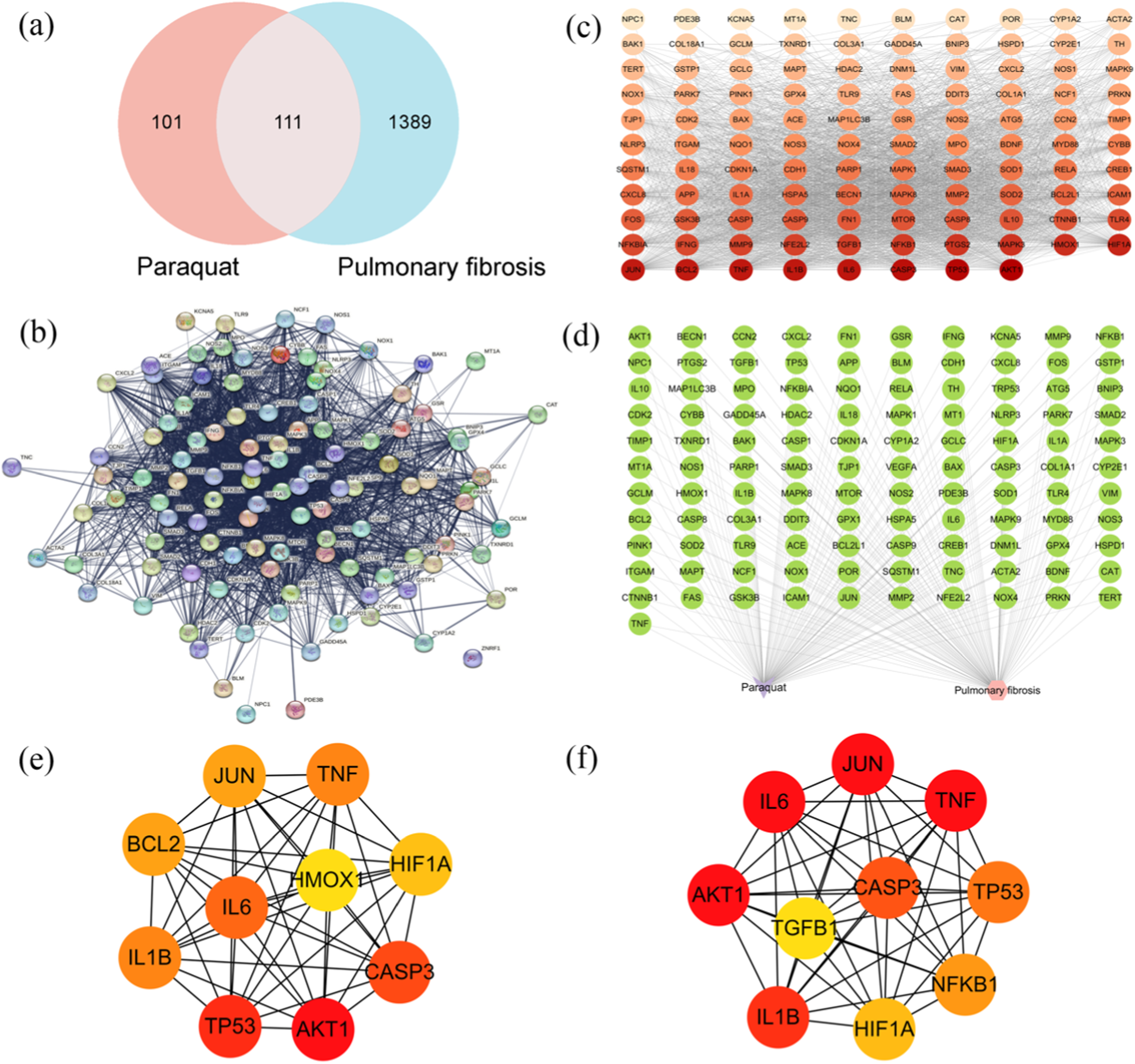
GO and KEGG pathway enrichment analysis
GO analysis enriched a total of 1410 biological processes related to 78 molecular functions and 21 cellular components. Several terms including regulation of apoptotic signaling pathway, response to lipopolysaccharide, response to oxidative stress, and reactive oxygen species (ROS) metabolic process were enriched. Furthermore, cellular component terms highlighted membrane raft, membrane microdomain, and organelle outer membrane. Molecular functions such as ubiquitin protein ligase binding, ubiquitin-like protein ligase binding, and cytokine receptor binding were also identified (Figure 2(a)). The KEGG pathway enrichment analysis identified a total of 163 signaling pathways, among which lipid and atherosclerosis and the AGE-RAGE signaling pathway in diabetic complications had the highest enrichment scores and number of targets. The classic inflammation and immune-related pathways, such as the TNF signaling pathway, apoptosis, the IL-17 signaling pathway, and the NOD-like receptor signaling pathway, were also significantly enriched (Figure 2(b)). GO and KEGG pathway enrichment analysis. Bubble size corresponds to the number of target genes; color represents the level of statistical significance. Enrichment analysis was performed using clusterProfiler with the human genome as background. (a) GO enrichment analysis. (b) KEGG pathway enrichment analysis.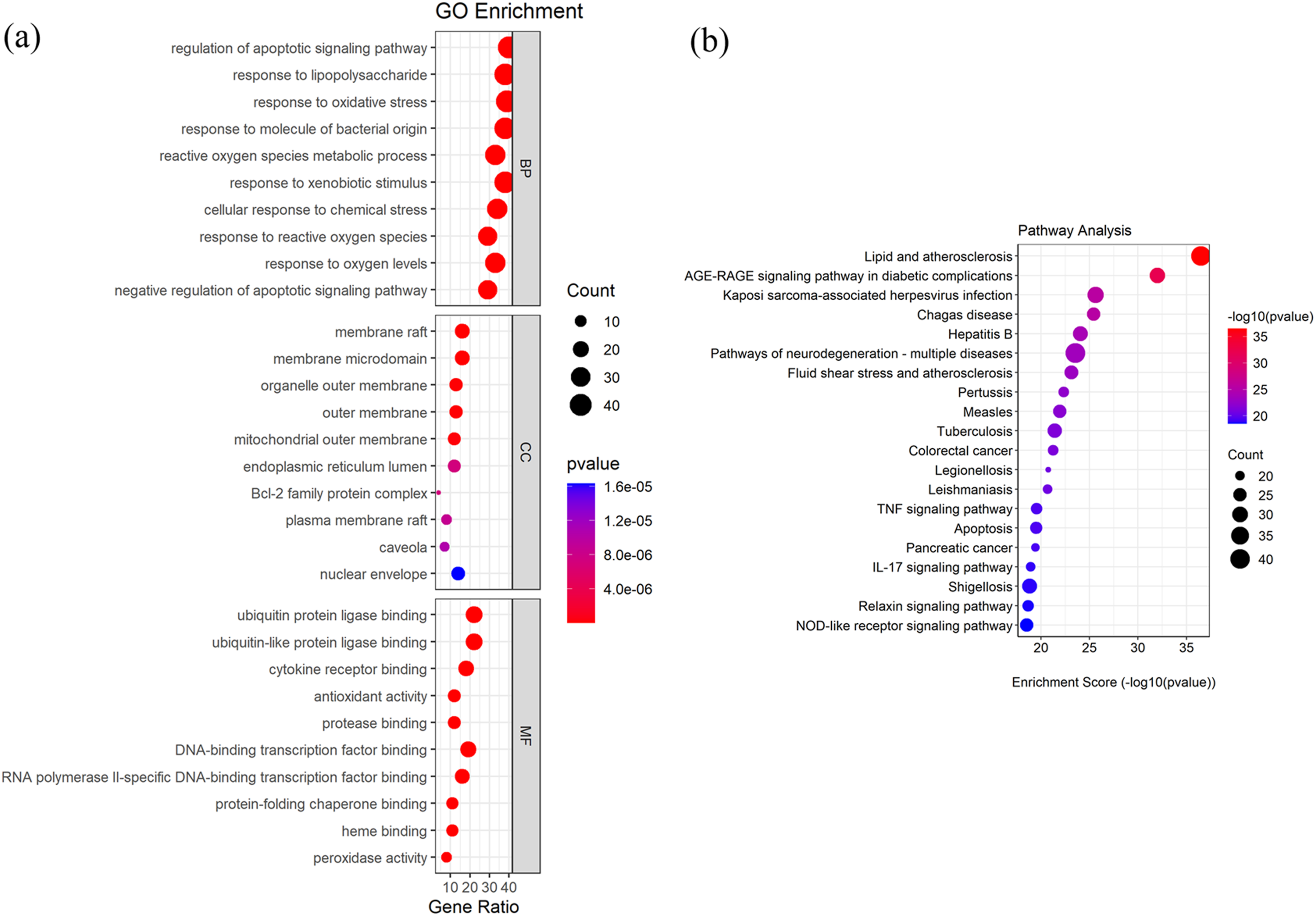
PQ strongly binds with AKT1, IL6, TP53, CASP3, TNF, and BCL2
Molecular docking information of PQ with target proteins. a
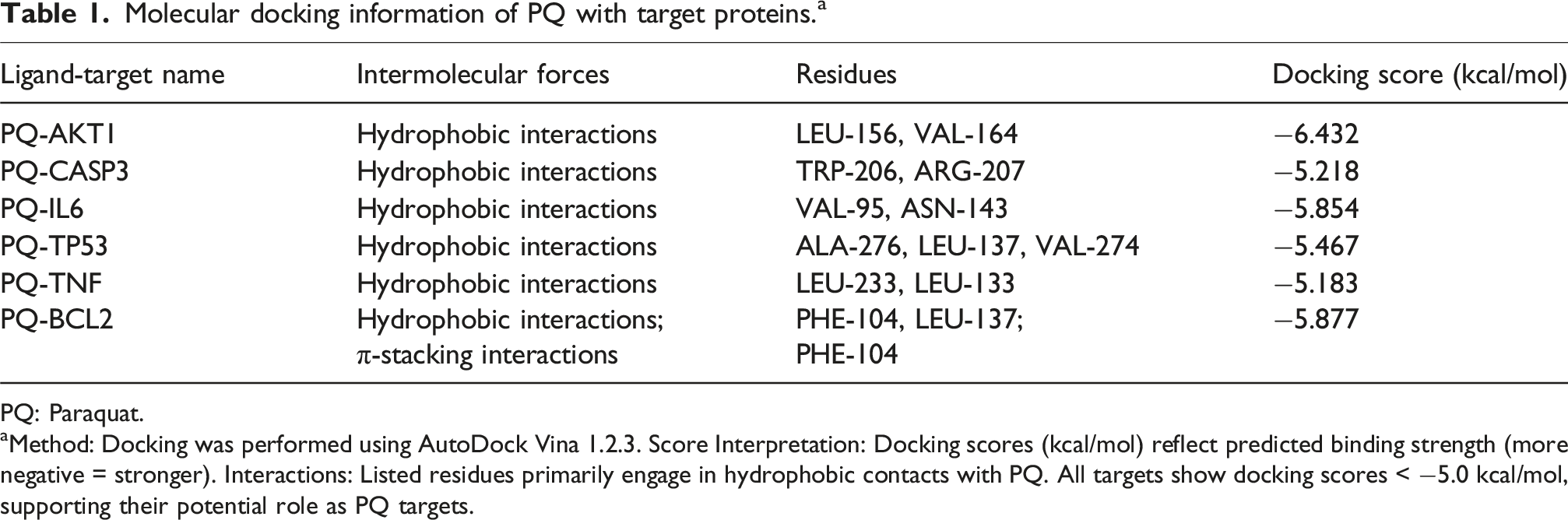
PQ: Paraquat.
aMethod: Docking was performed using AutoDock Vina 1.2.3. Score Interpretation: Docking scores (kcal/mol) reflect predicted binding strength (more negative = stronger). Interactions: Listed residues primarily engage in hydrophobic contacts with PQ. All targets show docking scores < −5.0 kcal/mol, supporting their potential role as PQ targets.
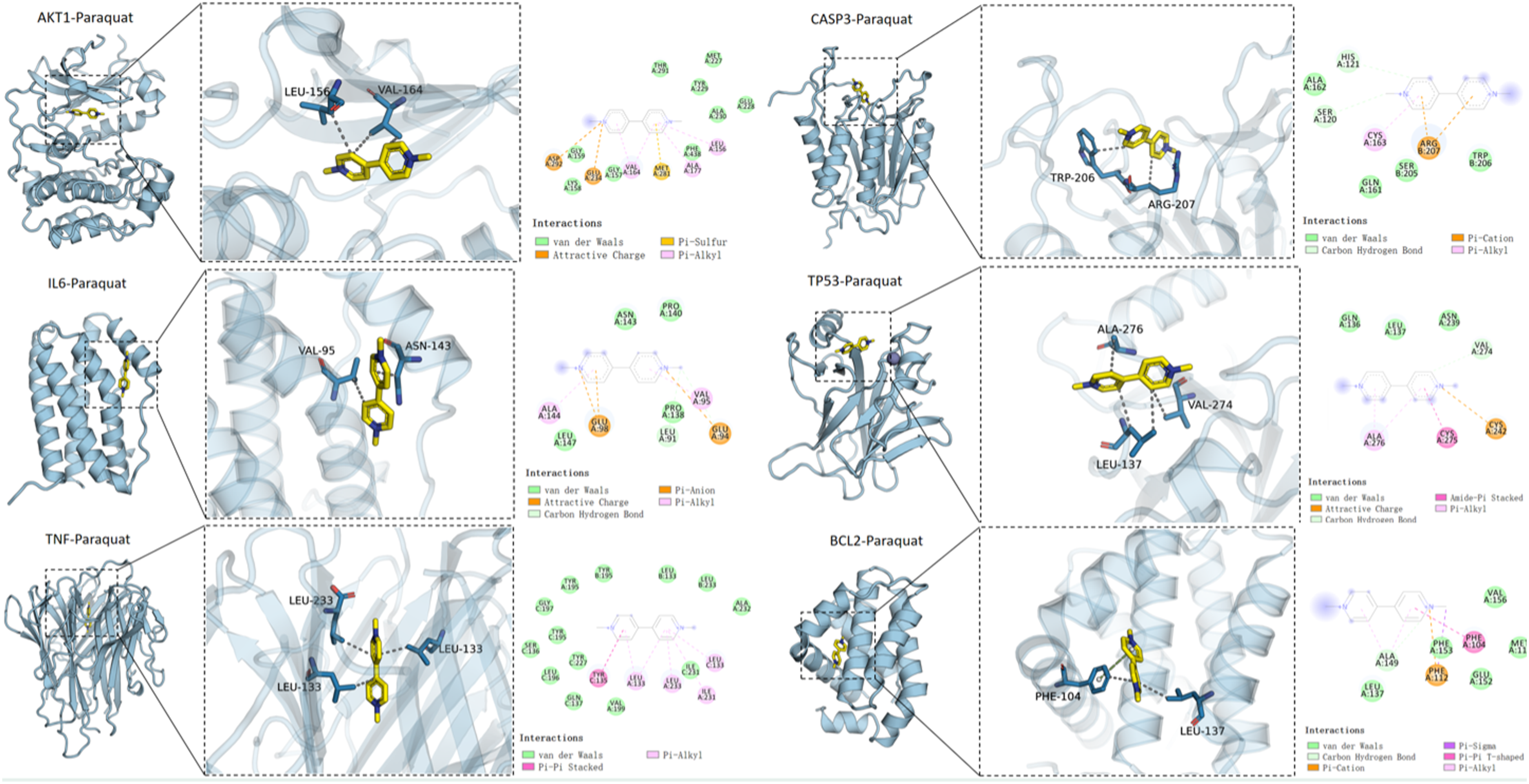
The molecular docking results of PQ with AKT1, IL6, TP53, CASP3, TNF, and BCL2 proteins. The left panel presents an overall view of the complex; the middle panel shows a close-up view of the binding site; and the right panel displays a 2D ligand-protein interaction diagram (Discovery Studio, 2019). In the images, the PQ molecule was depicted as yellow sticks, and the protein structure was represented by a light blue cartoon. Molecular interactions were indicated as follows: hydrogen bonds by blue lines, hydrophobic interactions by gray dashed lines, cation-π interactions by orange dashed lines, and π-π stacking interactions by green dashed lines.
Molecular dynamics simulation results
Through 100 ns molecular dynamics simulations of the AKT1/PQ, CASP3/PQ, IL6/PQ, and TP53/PQ complexes, the root mean square deviation (RMSD) of PQ was analyzed. The results showed that the RMSD values of PQ remained stable within all four protein binding sites, fluctuating within a narrow range of less than 1 Å, indicating stable binding between PQ and each protein (Figure 4(a)). The root mean square fluctuation (RMSF), calculated from the simulation trajectories, reflects the flexibility of protein residues during the dynamics. After binding to PQ, all proteins exhibited reduced RMSF values, particularly in their core regions, suggesting that ligand binding decreased protein flexibility and enhanced local structural stability (Figure 4(b)). Molecular dynamics simulation results. (a) The changes in the RMSD of the ligand in the protein over time during the molecular dynamics simulation. (b) RMSF calculated based on molecular dynamics simulation trajectories. (c) The RoG obtained from the molecular dynamics simulation varies with simulation time.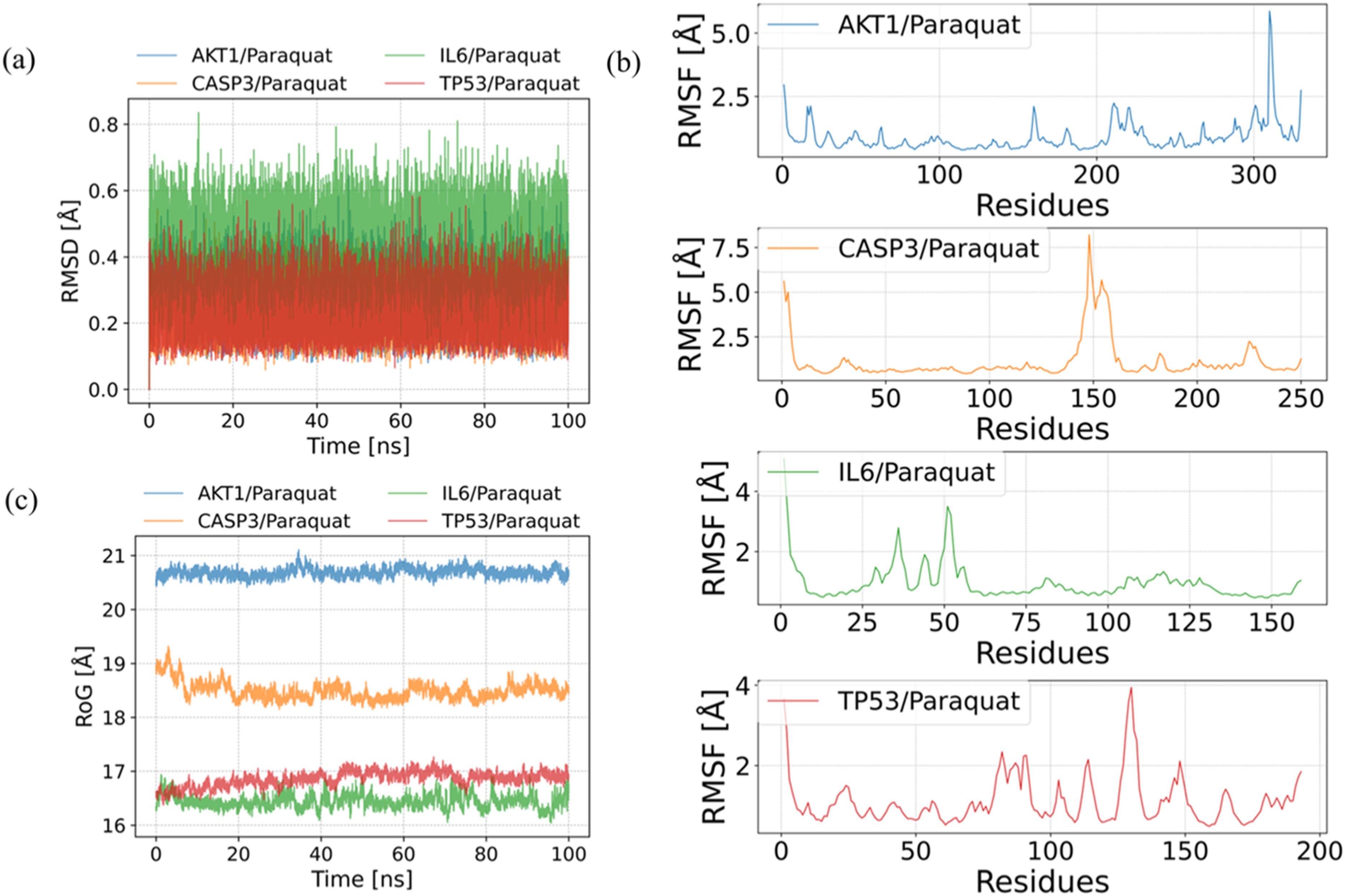
The radius of gyration (RoG) serves as an indicator of structural compactness. We compared the RoG values of the four complexes—AKT1/PQ, CASP3/PQ, IL6/PQ, and TP53/PQ—over a 100 ns molecular dynamics simulation to evaluate their conformational stability and compactness. The results indicated that the AKT1/PQ complex exhibited the highest RoG data, averaging approximately 20.7 Å, suggesting a relatively loose yet stable overall structure. The RoG of CASP3/PQ averaged around 18.3 Å, showing an initial contraction followed by stabilization. In contrast, TP53/PQ and IL6/PQ displayed the lowest RoG values, maintaining averages of about 16.9 Å and 16.5 Å, respectively, which reflects a more compact conformational state throughout the simulation (Figure 4(c)).
Hydrogen bonds represent one of the strongest forms of non-covalent interactions, and their abundance generally correlates with stronger binding affinity. Throughout the simulation, the number of hydrogen bonds in the four complexes—AKT1/PQ, CASP3/PQ, IL6/PQ, and TP53/PQ—was monitored over time. All complexes exhibited a relatively low number of hydrogen bonds, suggesting that hydrophobic interactions play a dominant role in stabilizing these complexes (Figure 5). Changes in the number of hydrogen bonds between PQ and proteins during molecular dynamics simulations.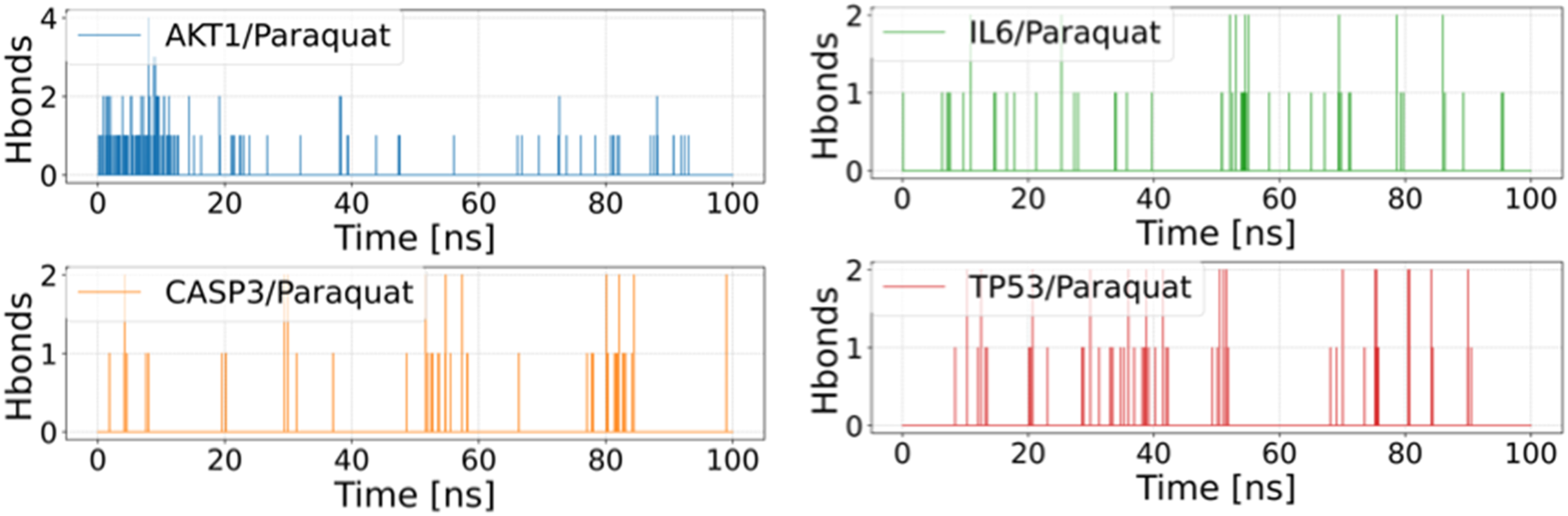
Binding free energies and energy components predicted by MM/GBSA (kcal/mol). a
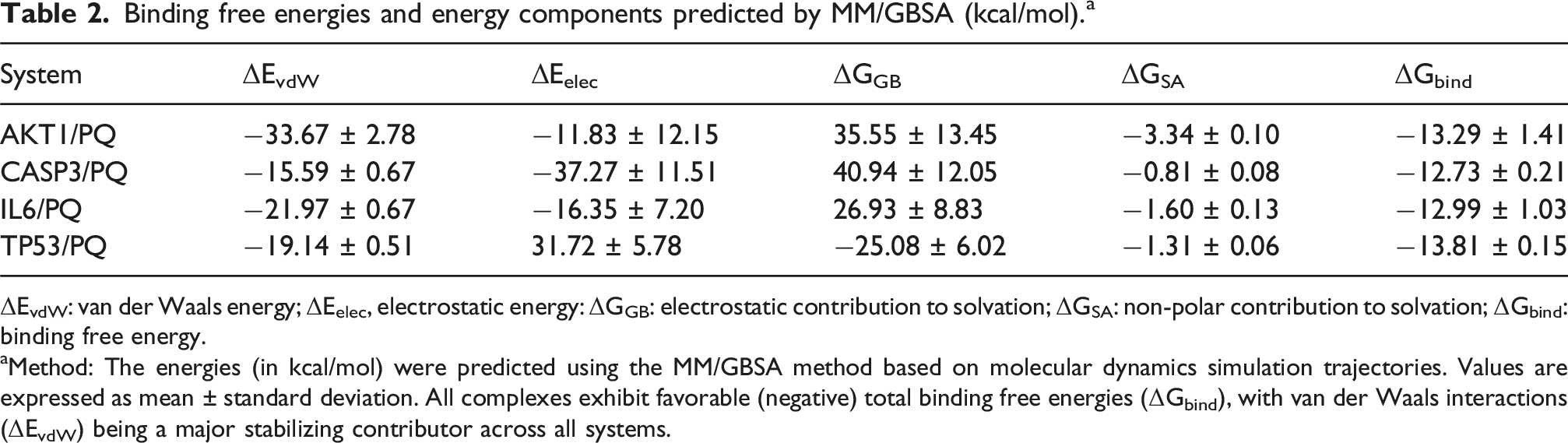
ΔEvdW: van der Waals energy; ΔEelec, electrostatic energy: ΔGGB: electrostatic contribution to solvation; ΔGSA: non-polar contribution to solvation; ΔGbind: binding free energy.
aMethod: The energies (in kcal/mol) were predicted using the MM/GBSA method based on molecular dynamics simulation trajectories. Values are expressed as mean ± standard deviation. All complexes exhibit favorable (negative) total binding free energies (ΔGbind), with van der Waals interactions (ΔEvdW) being a major stabilizing contributor across all systems.
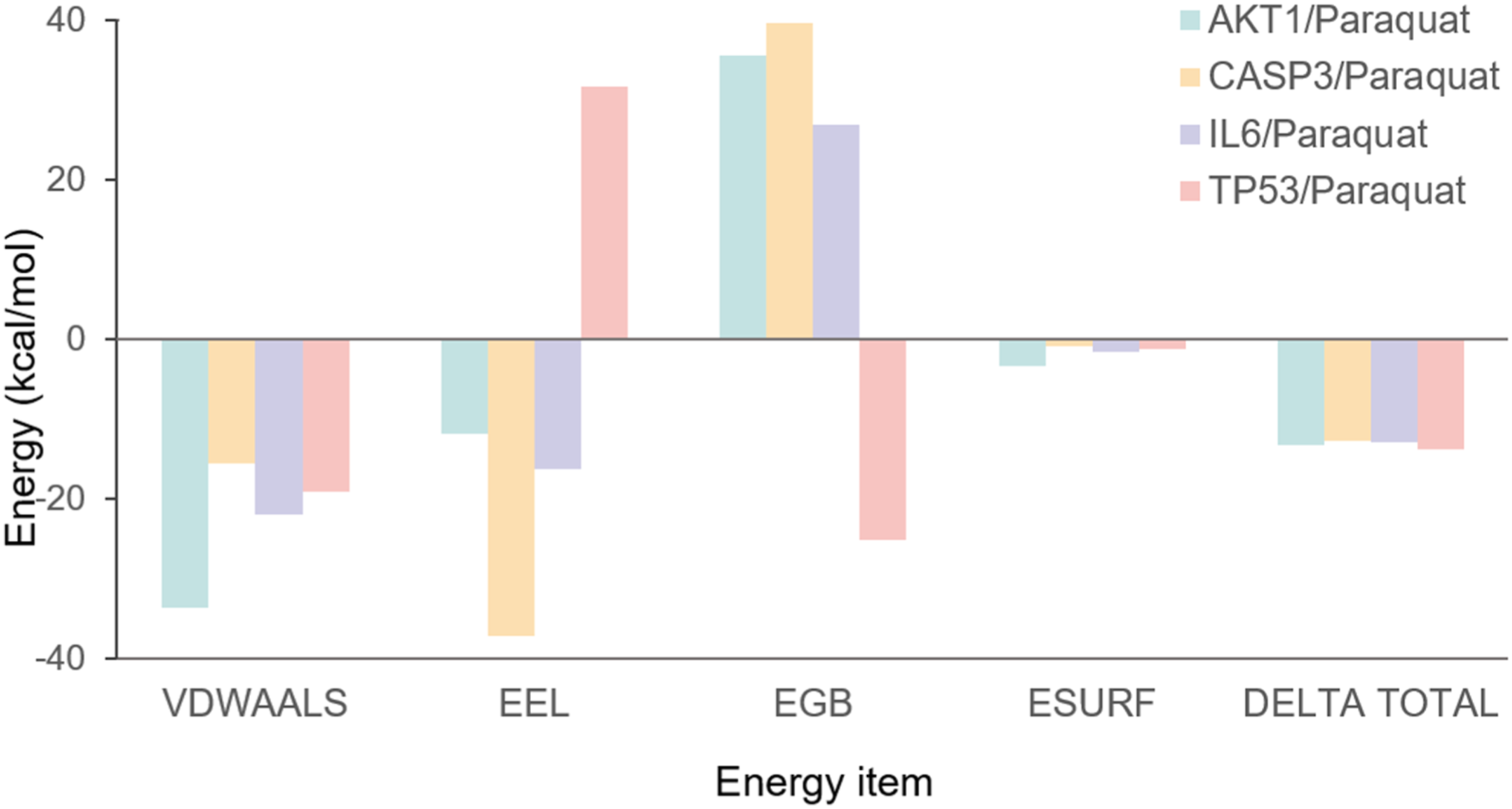
MM/GBSA binding energy and energy decomposition. All energy values are reported in kcal·mol-1 and represent the average calculated from the final 20 ns of triplicate simulations. Abbreviations: VDWAALS, van der Waals energy; EEL, Electrostatic energy; EGB, Polar solvation energy; ESURF, Non-polar solvation energy; DELTA TOTAL, VDWAALS + EEL + EGB + ESURF.
AlphaFold2 predicts proteins that competitively bind to PQ
AlphaFold2 was used to predict the proteins that interact with PQ. It showed that three proteins (1owyq, ioz0h0, qxknu) exhibit strong binding affinity with PQ, surpassing that of core targets such as AKT1 (Figures 7 and 8). It indicates that these three proteins can competitively bind to PQ. AlphaFold2 predicts the proteins interacted with PQ. Three proteins including 1owyq, ioz0h0, and qxknu were observed to strongly bind with PQ, along with their binding energies and modes. Sequence information for AlphaFold2-designed proteins.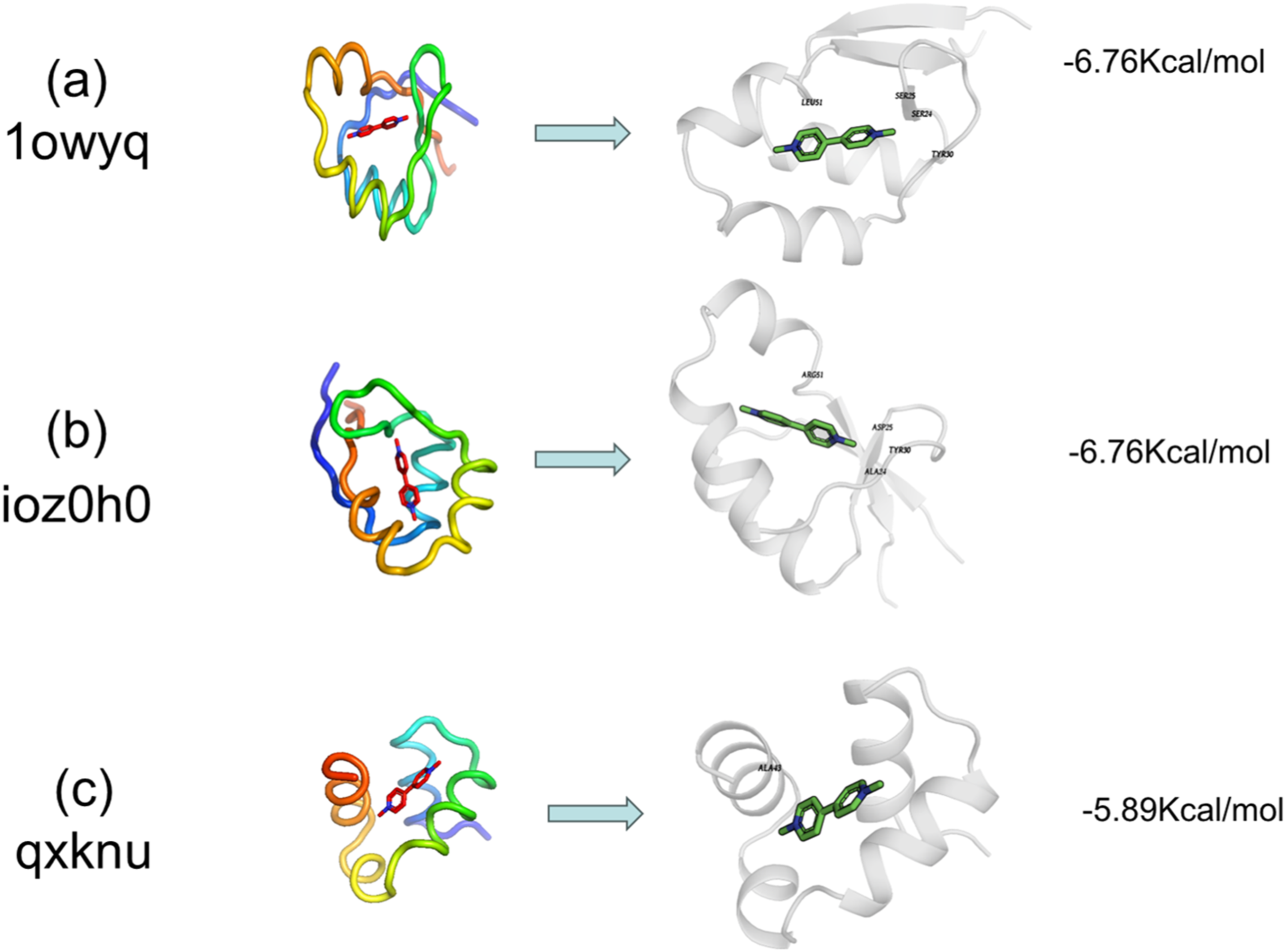
Discussion
PQ can enter the human body through various routes, then accumulate in the lungs and cause damage (Zhang et al., 2024). Once pulmonary fibrosis develops, effective clinical treatments are lacking. The mechanism of PQ-induced pulmonary fibrosis is complex and interconnected, presenting challenges for diagnosis and therapy. This study elucidates the potential molecular interplay between PQ and core cellular targets in pulmonary fibrosis. By integrating network toxicology, molecular docking, dynamics simulations, and de novo protein design, we shift focus toward a less explored aspect: PQ’s capacity for direct and stable interactions with key regulatory proteins, which may serve as an upstream event triggering downstream cascades of oxidative stress, inflammation, and apoptosis.
Our network toxicology analysis identified 111 common targets shared between PQ toxicity and pulmonary fibrosis, with AKT1, TP53, IL6, and CASP3 emerging as central hubs. AKT1, a key node in the PI3K/AKT pathway, regulates cell survival, proliferation, and metabolism, and its dysregulation is implicated in fibrotic diseases (Nie et al., 2019). It promotes pulmonary fibrosis by inducing mitochondrial ROS and mitophagy in macrophages, leading to TGF-β1 activation (Larson-Casey et al., 2016). Furthermore, the natural compound daphnetin attenuates silica-induced lung fibrosis by targeting the PI3K/AKT1 pathway, underscoring the therapeutic relevance of this axis (Yang et al., 2024).
TP53 orchestrates PQ-induced apoptosis and oxidative damage in lung epithelial cells (Wu et al., 2020). It is transcriptionally activated during alveolar epithelial differentiation, directly regulating the formation of the pre-alveolar type-1 transitional state (PATS). While this TP53-driven transient senescence is essential for normal repair, its aberrant persistence contributes to fibrosis (Kobayashi et al., 2020). Harmine alleviates pulmonary fibrosis by modulating DNA damage response genes, including downregulating TP53, which correlates with improved outcomes in models (Gong et al., 2024).
IL6 is a core pro-inflammatory mediator driving fibrosis. Hesperidin ameliorates fibrosis by inhibiting lung fibroblast senescence, an effect linked to the downregulation of senescence markers and suppression of the IL6/STAT3 pathway (Han et al., 2023). CASP3, as a central executioner of apoptosis, contributes to alveolar epithelial cell death and fibrotic remodeling. A caspase-3-responsive nanotheranostic system (Casp-GNMT) has been developed, which upon activation alleviates oxidative stress and inhibits TGF-β, thereby reducing collagen deposition (Li et al., 2025).
Collectively, these studies underscore that AKT1, TP53, IL6, and CASP3 are pivotal in pulmonary fibrosis. Our molecular docking further revealed strong binding affinities between PQ and these proteins (energies < −5 kcal/mol), suggesting PQ may directly modulate their activity and contribute to pathological signaling.
GO enrichment analysis of the intersecting targets revealed their predominant involvement in inflammatory responses, oxidative stress, and apoptotic regulation—processes central to established PQ toxicology (Wang et al., 2024). Key terms such as “response to lipopolysaccharide” and “ROS metabolic process” highlight the roles of innate immune activation and oxidative damage. This aligns with reports that anti-inflammatory and antioxidant compounds can ameliorate pulmonary fibrosis (Mehdar, 2024). The lung’s susceptibility to ROS-generating pollutants and the consequent oxidative stress and mitochondrial dysfunction underscore the therapeutic potential of targeting ROS-mediated pathways in fibrosis (Sharma et al., 2021).
KEGG pathway analysis further emphasized the involvement of immune-inflammatory and stress-response pathways. The enrichment of “lipid and atherosclerosis” and “AGE-RAGE signaling pathway in diabetic complications” suggests shared mechanisms between metabolic dysregulation, chronic inflammation, and PQ-induced fibrosis, highlighting lipid metabolism as a potential therapeutic target (Chen and Dai, 2023).
Significant enrichment was also observed in classic pathways including the “TNF signaling pathway,” “Apoptosis,” “IL-17 signaling pathway,” and “NOD-like receptor signaling pathway.” This indicates that PQ may exacerbate alveolar injury and fibrosis by activating inflammatory factors, inducing cell death, and engaging immune recognition. Notably, IL-17A plays a central role in driving neutrophilic inflammation in cystic fibrosis, with multiple immune cell types contributing to its production (Hagner et al., 2021; Steele et al., 2025). Furthermore, NOD-like receptors (NLRs), particularly the NLRP3 inflammasome, are key in linking innate immunity to fibrotic remodeling (Zhang et al., 2021).
In summary, KEGG analysis revealed that multiple interwoven inflammatory, apoptotic, and stress-related pathways synergistically mediate PQ-induced pulmonary fibrosis, providing insights for network toxicology and targeted intervention.
Molecular dynamics simulations were conducted to further validate the PQ-protein interactions. Stable RMSD values and reduced RMSF fluctuations indicated that PQ binding enhances the structural stability and restricts the flexibility of the binding sites, potentially impairing functional dynamics. The particularly compact conformations of the IL6/PQ and TP53/PQ complexes, reflected by low radius of gyration values, suggest strong and stable binding.
MM/GBSA calculations confirmed the favorable binding energies, which were primarily driven by van der Waals interactions with minimal contribution from hydrogen bonding, indicating the dominance of hydrophobic effects. This strong and stable binding affinity suggests that PQ can directly disrupt the functions of key proteins involved in apoptosis, inflammation, and proliferation, providing a significant mechanistic basis for its induction of pulmonary fibrosis.
Employing AlphaFold2-based protein design, we identified three novel proteins (1owyq, ioz0h0, qxknu) exhibiting higher binding affinity for PQ than native targets such as AKT1. Based on their structural characteristics, binding to these designed proteins is hypothesized not to significantly interfere with renal PQ excretion, although this requires experimental validation. These proteins may represent potential candidates for sequestering PQ, competitively preventing its interaction with pathological targets and thereby mitigating toxicity. This approach provides new leads for the exploration of protein-based detoxification strategies.
Finally, we acknowledge the inherent limitations of this computational study. The predictions of direct binding and their functional implications remain hypothetical and require rigorous experimental validation through techniques such as surface plasmon resonance, isothermal titration calorimetry, or cellular functional assays following PQ exposure. The network analysis is constrained by the completeness of the underlying databases, and the molecular dynamics simulations, while insightful, are limited in timescale compared to biological processes. Our study did not aim to replace the established model of oxidative stress-driven toxicity but proposed that direct protein binding could be a significant and under-investigated concomitant mechanism. The stability and energy favorability of the predicted complexes suggest that these interactions are likely to occur in vivo and could meaningfully contribute to the overall toxicological profile.
Conclusion
In conclusion, our multi-faceted analysis suggests that PQ induces pulmonary fibrosis through direct interaction with core target proteins such as AKT1, TP53, IL6, and CASP3, leading to dysregulation of oxidative stress, inflammation, and apoptosis pathways. The stable binding of PQ to these targets exacerbates chronic toxicity and fibrotic progression. These insights not only deepen the understanding of PQ’s toxicological mechanisms but also provide a foundation for novel therapeutic strategies, including targeted inhibitors and engineered scavenger proteins.
Footnotes
Author contributions
Guiyan Du: Conceptualization, project administration, and writing—original draft. Shengyao Tang and Tian Shi: Writing—review and editing, supervision, and funding acquisition. Guosheng Liu, Xia Huang, and Jian Chang: Resources and formal analysis.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Hubei Provincial Key Laboratory of Occurrence and Intervention of Rheumatic Diseases Foundation (Grant No. OIR202410Q); Enshi Prefecture Guiding Foundation (Grant No. E20210033); the Hubei Provincial Natural Science Foundation (Grant No. 2025AFD154), and the Minda Hospital of Hubei Minzu University PhD Research Start-up (Funding) Foundation.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All raw data and code are available upon request.