
Abstract
The rapid evolution of viral pathogens presents significant challenges for global health, as traditional methods for virus detection often fail to identify novel or genetically diverse viruses. The emergence and reemergence of viral pathogens necessitate more advanced and inclusive diagnostic approaches. This review aims to explore the role of metagenomics in overcoming the limitations of traditional viral detection methods and to assess its impact on the discovery, characterization, and surveillance of viral pathogens. A comprehensive review of recent studies employing metagenomic approaches to viral detection was conducted. High-throughput sequencing technologies and bioinformatics tools were highlighted as key components in enabling broad-spectrum viral identification and characterization. Metagenomic approaches have successfully identified novel pathogens, including new arboviruses and reemerging strains of known viruses. These techniques provide a more complete understanding of viral diversity and dynamics, surpassing the limitations of targeted assays and culturing methods. Key findings emphasize the capability of metagenomics to detect viruses previously undetected by conventional methods, improving the scope of surveillance. Metagenomics offers transformative advantages for viral surveillance and outbreak management. It enhances early detection, allows for better-informed responses to viral threats, and contributes to more effective strategies for managing emerging and reemerging viral pathogens. Integration of metagenomic techniques into public health practices is crucial for combating the evolving landscape of viral diseases.
Introduction
The global health landscape is increasingly challenged by the emergence and reemergence of viral pathogens. The rapid evolution and adaptability of viruses have led to significant public health threats, as evidenced by recent outbreaks of diseases such as COVID-19 (World Health Organization, 2020), Ebola (World Health Organization, 2014), and various arboviral infections (Fauci and Morens, 2012). These pathogens not only pose immediate health risks but also strain health care systems, disrupt economies, and complicate disease control efforts worldwide (Morse and Schluederberg, 1990).
Traditional methods for detecting and monitoring viral pathogens, such as polymerase chain reaction (PCR) assays (Mackay et al., 2002) and viral culturing techniques (Chiu and Miller, 2019), have been pivotal in managing well-characterized outbreaks and identifying known viruses. However, these approaches have notable limitations. They are heavily dependent on prior knowledge of the pathogen, which constrains their ability to detect novel or genetically diverse viruses (Allander et al., 2005). Furthermore, many viruses cannot be easily cultured in laboratory settings, complicating their study and surveillance (Hay et al., 2006). As a result, these conventional methods often fall short in the rapid identification and understanding of new or emerging viral threats.
In response to these limitations, metagenomic approaches have emerged as a transformative solution (Chiu and Miller, 2019). Metagenomics involves the comprehensive analysis of genetic material from environmental or clinical samples using high-throughput sequencing technologies (Mardis, 2008). This method allows for the detection and characterization of a broad spectrum of viral pathogens without requiring prior knowledge of their presence (Delwart, 2007). By utilizing advanced sequencing and bioinformatics tools, metagenomics provides an unbiased and detailed view of viral diversity, enabling the discovery of previously unknown pathogens and a deeper understanding of viral dynamics (Naccache et al., 2014).
This review aims to explore how metagenomics is revolutionizing the identification and management of viral threats. We will examine how metagenomic techniques address the limitations of traditional methods, highlight key discoveries facilitated by these approaches, and discuss their implications for enhancing viral surveillance and outbreak response. Through this exploration, we seek to demonstrate the transformative potential of metagenomics in improving our ability to combat both emerging and reemerging viral pathogens.
Historical background and genome characteristics of emerging and Reemerging pathogen human viruses
This document offers a detailed examination of the historical context and genomic features of 20 emerging and reemerging human viral pathogens. The study of these viruses, which have significantly influenced public health, has been greatly advanced by genomic research (Table 1).
Characteristics of Emerging Pathogen Human Viruses
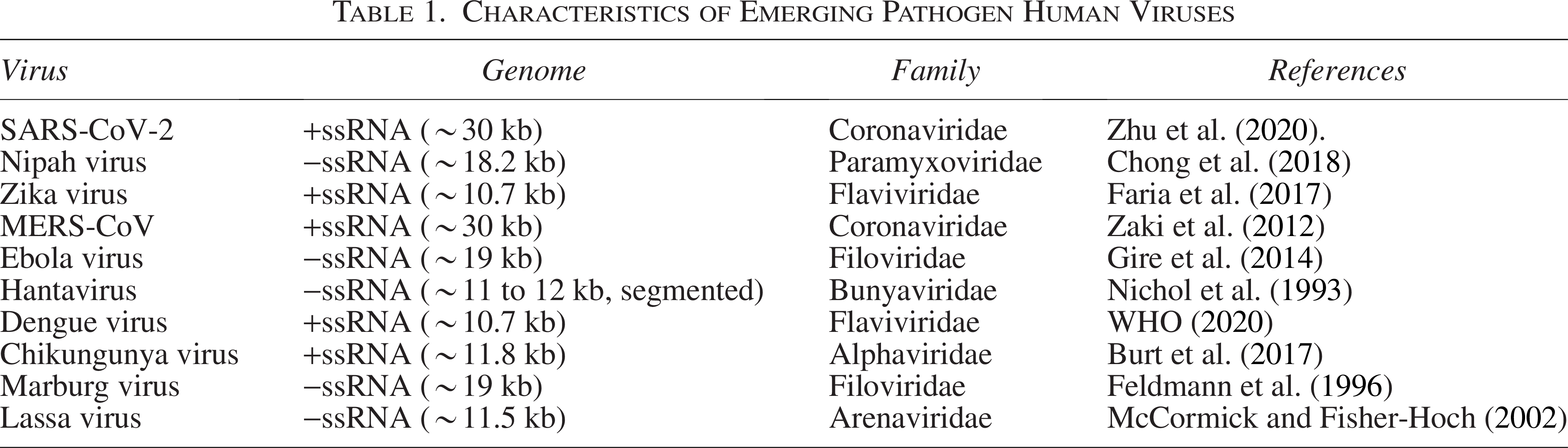
Emerging pathogen human viruses
SARS-CoV-2, the virus responsible for COVID-19, was identified in December 2019 in Wuhan, China, leading to a global pandemic (Zhu et al., 2020). It has a positive-sense single-stranded RNA genome of approximately 30 kb and belongs to the coronavirus family (Table 1). The virus is noted for its significant mutations, resulting in various variants of concern (Gordon et al., 2020).
Nipah virus, first recognized during an outbreak in Malaysia in 1998, causes severe encephalitis and respiratory illness (Chong et al., 2018). This pathogen features a negative-sense single-stranded RNA genome of about 18.2 kb (Table 1). It is categorized within the Paramyxoviridae family and exhibits a high mutation rate, which contributes to its virulence (Bossart et al., 2009).
Zika virus was identified in 1947 and garnered global attention during the 2015 to 2016 outbreak due to its link to birth defects (Faria et al., 2017). It has a positive-sense single-stranded RNA genome of approximately 10.7 kb (Table 1). Zika virus is part of the Flaviviridae family and is known for its genetic variability (Faria et al., 2017).
Middle East Respiratory Syndrome Coronavirus was discovered in 2012 in Saudi Arabia and is known for causing severe respiratory illness (Zaki et al., 2012). The virus possesses a positive-sense single-stranded RNA genome of about 30 kb (Table 1). It belongs to the coronavirus family and has been studied extensively for its spike protein, which is critical for viral entry into host cells (Al-Tawfiq and Memish, 2014).
Ebola Virus was first identified in 1976 in Sudan and Zaire and caused a major outbreak in West Africa in 2014 (Gire et al., 2014). This virus features a negative-sense single-stranded RNA genome of approximately 19 kb and is a member of the Filoviridae family (Table 1). It displays significant genetic diversity among its various species (Marston et al., 2017).
Hantavirus, recognized in 1993 in the southwestern United States, is associated with hemorrhagic fever and renal syndrome (Nichol et al., 1993). The virus has a negative-sense single-stranded RNA genome segmented into three parts (S, M, and L), totaling about 11 to 12 kb in length (Nichol et al., 1993).
Dengue virus has been known since the 1950s and is endemic in tropical and subtropical regions, where it causes dengue fever and dengue hemorrhagic fever (WHO, 2020). It possesses a positive-sense single-stranded RNA genome of approximately 10.7 kb and is classified in the Flaviviridae family (Table 1). Dengue virus exists in four distinct serotypes (Guzman and Harris, 2015).
Chikungunya virus was first identified in Tanzania in 1952 and has caused outbreaks in various regions, including the Indian Ocean area during 2005 to 2006 (Burt et al., 2017). The virus has a positive-sense single-stranded RNA genome of approximately 11.8 kb and belongs to the Alphaviridae family (Schmaljohn and Hjelle, 1997).
Marburg virus, first discovered in 1967 in Marburg, Germany, causes severe hemorrhagic fever and is closely related to Ebola virus (Feldmann et al., 1996). It has a negative-sense single-stranded RNA genome of about 19 kb and is classified within the Filoviridae family (Guglielmi et al., 2021).
Lassa virus was identified in 1969 in Nigeria and causes Lassa fever, a type of viral hemorrhagic fever (McCormick and Fisher-Hoch, 2002). The virus features a negative-sense single-stranded RNA genome of about 11.5 kb and belongs to the Arenaviridae family (Patterson et al., 2021).
Reemerging pathogen human viruses
Influenza virus has been responsible for pandemics throughout the 20th and 21st centuries, including the 1918 Spanish flu and the 2009 H1N1 pandemic (Smith et al., 2004; Taubenberger and Morens, 2006). It possesses a segmented negative-sense single-stranded RNA genome, consisting of eight segments with a total length of about 13.5 kb (Table 2) (Webster et al., 1992).
Characteristics of Reemerging Human Pathogen Viruses

Varicella-Zoster Virus (VZV) causes chickenpox and shingles, with a resurgence of shingles linked to reduced population immunity (Arvin, 1996). VZV has a double-stranded DNA genome of approximately 125 kb (Table 2) and is classified in the Herpesviridae family (Krause et al., 2014).
Hepatitis B Virus has been known for decades, with notable advancements in vaccines and treatments. However, issues such as drug resistance have emerged (Scholz et al., 2021). The virus features a partially double-stranded circular DNA genome of about 3.2 kb (Table 2) and belongs to the Hepadnaviridae family (Seeger and Mason, 2015).
Herpes Simplex virus (HSV) causes oral and genital herpes, with reemergence observed in immunocompromised individuals. Recent advances in genomic research have provided insights into resistance mechanisms (Kolb et al., 2015). HSV has a double-stranded DNA genome of approximately 152 kb and is classified in the Herpesviridae family (Roizman and Whitley, 2013) (Table 2).
Human Papillomavirus (HPV) is associated with cervical cancer and other malignancies. The emergence of new strains has raised concerns about increased cancer risk (de Villiers et al., 2004). HPV features a circular double-stranded DNA genome of about 8 kb and belongs to the Papillomaviridae family (Schiffman et al., 2016) (Table 2).
Measles virus was largely controlled through vaccination efforts, but recent outbreaks have occurred due to vaccine hesitancy (Rota et al., 2016). It has a negative-sense single-stranded RNA genome of approximately 15.9 kb and is part of the Paramyxoviridae family (Rota et al., 2016).
Yellow fever virus, primarily spread by mosquitoes, has reemerged in Africa and South America (Monath and Vasconcelos, 2015). The virus has a positive-sense single-stranded RNA genome of about 10.7 kb and belongs to the Flaviviridae family (Gubler, 2012) (Table 2).
Smallpox virus, which was eradicated in 1980 through vaccination, still raises concerns about potential reemergence for bioterrorism (Guzman and Harris, 2015). It features a double-stranded DNA genome of approximately 186 kb and is classified in the Poxviridae family (Guzman and Harris, 2015).
Cytomegalovirus (CMV) remains a significant pathogen, particularly among immunocompromised individuals and transplant recipients (Britt, 2018). CMV has a double-stranded DNA genome of approximately 230 kb (Table 2).
Factors contributing to emerging and reemerging human viruses
Emerging and reemerging human viruses are those that have recently appeared or reappeared in human populations, posing significant challenges to public health. Understanding the factors contributing to their emergence and reemergence is crucial for effective prevention and control. These factors encompass ecological and environmental changes, human behavior, genetic mutations, global travel, and socioeconomic conditions.
Ecological and Environmental Changes: Ecological shifts, such as deforestation, urbanization, and climate change, significantly contribute to the emergence and reemergence of viral pathogens. Deforestation and habitat destruction force wildlife into closer contact with human populations, increasing the likelihood of zoonotic spillover. For example, the deforestation of tropical rainforests has been linked to the emergence of viruses such as Ebola, as it disrupts natural ecosystems and brings humans into proximity with wildlife reservoirs (Wolfe et al., 2007). In addition, climate change can alter the distribution of vector-borne diseases. Warmer temperatures and altered precipitation patterns can expand the range of vectors such as mosquitoes, thereby facilitating the spread of diseases such as dengue and malaria to new regions (Patz et al., 1996).
Human Behavior and Societal Factors: Changes in human behavior and societal dynamics also play a significant role in the emergence and reemergence of viral pathogens. High population density, increased urbanization, and global travel contribute to the rapid spread of viruses. Overcrowded urban areas, often with poor sanitation, can act as hotspots for the transmission of infectious diseases. For instance, the rapid urbanization and population density in cities have been linked to the spread of SARS-CoV-2 during the COVID-19 pandemic (Zhu et al., 2020). Furthermore, global travel facilitates the rapid dissemination of viruses across international borders, turning localized outbreaks into global pandemics (Gordon et al., 2020).
Genetic Mutations and Viral Evolution: Genetic mutations in viruses can lead to the emergence of new viral strains with enhanced transmission or virulence. Viral evolution is driven by high mutation rates and selective pressures, which can result in the development of new variants. For example, the influenza virus undergoes frequent genetic changes through antigenic drift and shift, leading to seasonal flu epidemics and occasional pandemics (Neumann and Kawaoka, 2015). Similarly, the emergence of new variants of SARS-CoV-2 with mutations in the spike protein has impacted the virus’s transmissibility and vaccine efficacy (Gordon et al., 2020).
Global Travel and Trade: The modern world’s extensive travel and trade networks contribute significantly to the spread of emerging and reemerging viruses. International travel facilitates the rapid movement of pathogens between countries, while global trade, particularly in animals and animal products, can introduce new pathogens to different regions. For example, the outbreak of H5N1 avian influenza in poultry spread to humans and affected various countries due to international trade and travel (Neumann and Kawaoka, 2015). Similarly, the spread of the Zika virus across the Americas was facilitated by international travel and trade (Faria et al., 2017).
Socioeconomic Factors: Socioeconomic conditions influence the emergence and reemergence of viral pathogens. Populations with limited access to health care, poor living conditions, and inadequate public health infrastructure are more vulnerable to viral infections. Disparities in health care access can lead to delayed detection and response to outbreaks. For instance, impoverished regions may lack the resources for effective disease surveillance and control, contributing to the persistence and spread of diseases such as Lassa fever (McCormick and Fisher-Hoch, 2002). Moreover, socioeconomic factors can affect behaviors such as vaccination uptake, influencing the reemergence of vaccine-preventable diseases.
Antibiotic Use and Resistance: Although antibiotics are not used to treat viral infections directly, their misuse and overuse can have indirect effects on viral diseases. Overuse of antibiotics can lead to resistant bacterial strains, complicating the treatment of secondary bacterial infections in patients with viral diseases. This can impact the management of viral outbreaks and contribute to the overall disease burden (Ventola, 2015). In addition, disruptions to microbial communities caused by inappropriate antibiotic use can create opportunities for opportunistic viral pathogens to emerge or spread.
In conclusion, the factors contributing to the emergence and reemergence of human viruses are multifaceted and interrelated. Addressing these factors requires comprehensive strategies that include monitoring environmental changes, enhancing public health infrastructure, and improving global cooperation to manage and prevent viral outbreaks effectively.
Traditional and PCR methods for detecting human viral pathogens
Traditional methods for detecting and monitoring viral pathogens have long been crucial for diagnosing infections and managing disease outbreaks. These techniques include viral culture, serology, electron microscopy, and PCR. Each method has specific applications and limitations that influence its use in different contexts (Table 3).
Traditional and PCR Methods for Detecting Human Viral Pathogens

Viral culture has historically been a cornerstone in virology for isolating and identifying viruses. This method involves growing the virus in a controlled laboratory setting using cell lines that are susceptible to the virus. Once the virus is cultured, it can be further characterized through various assays or observed directly. The primary advantage of viral culture is that it allows researchers to study the live virus, including its behavior, replication, and response to antiviral agents. This is crucial for understanding the virus’s properties and for drug development. However, the limitations of viral culture are significant. The process is time-consuming, often requiring several days to weeks to yield results. It also demands specialized laboratory facilities and skilled personnel. Moreover, some viruses are challenging to culture or may not grow in standard cell lines, potentially leading to false negatives and limiting the method’s effectiveness for all pathogens (Knipe and Howley, 2013).
Serology involves detecting viral antibodies or antigens in a patient’s blood and is a widely used diagnostic tool. Common serological assays include enzyme-linked immunosorbent assays, immunofluorescence assays, and western blotting. These tests are advantageous because they can identify both acute and past infections by measuring the immune response to the virus. They are also useful in epidemiological studies to assess population-level exposure and immunity (Sabath et al., 2012). Despite these benefits, serological tests have limitations. They can suffer from cross-reactivity, where antibodies bind to antigens of similar viruses, leading to false positives. In addition, serology does not detect the virus itself, which means it may not identify infections during the early stages when antibodies are not yet present (Lazear and Diamond, 2016).
Electron microscopy offers a direct method for visualizing viral particles through high-resolution imaging using electron beams. This technique provides detailed images of viral morphology, which can be critical for identifying and studying the structure of unknown or newly discovered viruses. Electron microscopy is particularly useful for observing viral particles in clinical samples and understanding their structural features. However, the method is limited by its high cost and complexity. The equipment required is expensive and requires specialized training to operate. In addition, while electron microscopy provides valuable morphological information, it does not offer insights into the viral genetic material or specific strains (Murray et al., 2015).
PCR is a molecular technique that has revolutionized viral detection. PCR amplifies specific segments of viral DNA or RNA, making it possible to detect even minute amounts of viral genetic material. This method is highly sensitive and can provide results quickly, often within a few hours. PCR is invaluable for diagnosing infections in their early stages, monitoring viral load, and identifying specific viral strains. Despite its advantages, PCR has limitations, including the need for precise primers and probes, which can be challenging to design for all viruses. In addition, PCR requires sophisticated equipment and can be susceptible to contamination, which might lead to false positives (Mullis and Faloona, 1987).
In summary, traditional methods such as viral culture, serology, electron microscopy, and PCR each play a crucial role in the detection and monitoring of viral pathogens. However, their limitations often necessitate complementary approaches or advancements in technology to address gaps in accuracy, speed, and comprehensive viral characterization.
Viral metagenomics for detecting and monitoring human viral pathogens
Metagenomics has revolutionized viral research by providing powerful tools for discovering novel viruses, enhancing surveillance, and characterizing viral diversity. This approach leverages high-throughput sequencing technologies to analyze complex mixtures of genetic material directly from environmental or clinical samples, bypassing the need for prior isolation and cultivation of viruses. Below is a detailed exploration of its applications in viral research, supported by case studies, recent findings, and comparisons with traditional diagnostic methods.
Discovery of novel viruses
Metagenomics has been instrumental in uncovering previously unknown viruses, expanding our understanding of viral diversity. A landmark example is the discovery of the SARS-CoV-2 virus responsible for COVID-19. Early in 2019, metagenomic sequencing of patient samples in Wuhan, China, identified a novel coronavirus that was not previously known (Zhu et al., 2020). This identification was crucial for the rapid development of diagnostic tests, vaccines, and treatments.
Another pivotal case is the real-time identification of a novel arenavirus (Lujo virus) during an outbreak of hemorrhagic fever in southern Africa. Metagenomic next-generation sequencing (NGS) enabled the characterization of the virus from clinical samples within days, which significantly accelerated outbreak containment efforts (Briese et al., 2009).
Another significant case is the discovery of the Mosaic virus, identified in bats from the Congo Basin. Metagenomic sequencing revealed this novel virus, which shares genetic features with known filoviruses but represents a new branch within this family (Goldstein et al., 2018). Such discoveries illustrate how metagenomics can uncover hidden viral diversity that traditional methods might miss.
More recently, metagenomic studies have uncovered numerous novel viruses in humans, animals, and environmental reservoirs. For instance, Shi et al. (2018) used metagenomics to identify over 200 new RNA viruses in invertebrates, highlighting the vast and underexplored viral biodiversity. These findings have significant implications for zoonotic spillover prediction.
Enhancing viral surveillance
Metagenomics enhances viral surveillance by enabling the detection of a broad range of pathogens in various environments. For instance, environmental surveillance of water and soil samples using metagenomic techniques has identified viral contaminants that were previously undetected by conventional methods. A study examining wastewater samples from various cities found previously unrecognized viruses, providing insights into the viral landscape and potential public health risks (Bibby and Peccia, 2013; Kong et al., 2020).
A more recent example is the Global Sewage Surveillance Project, which utilized metagenomics to track enteric and respiratory viruses across 79 countries, revealing patterns in global viral transmission and antimicrobial resistance (Hendriksen et al., 2019). Such large-scale efforts demonstrate the scalability and global relevance of metagenomic surveillance.
In clinical settings, metagenomics allows for comprehensive pathogen detection from patient samples. For example, a study on respiratory infections demonstrated that metagenomic sequencing of nasopharyngeal swabs identified a wide range of viruses, including rare and emerging ones that were not detected by traditional assays (Quick et al., 2016). This broad detection capability is crucial for tracking viral outbreaks and understanding their epidemiology.
In addition, a landmark case occurred in California in 2014, where metagenomic sequencing identified Leptospira in a child with meningoencephalitis—a diagnosis missed by standard microbiological tests—highlighting its diagnostic value in clinical practice (Wilson et al., 2014).
Understanding viral diversity and evolution
Metagenomics significantly contributes to understanding the genetic diversity and evolution of viral populations. By sequencing the genetic material from viral communities, researchers can analyze variations in viral genomes and track evolutionary changes over time.
A notable example is the study of the human virome. Metagenomic analysis of human gut samples has revealed extensive viral diversity, including phages and eukaryotic viruses, and provided insights into their roles in health and disease (Norman et al., 2014). Recent updates to this work have shown that the gut virome is highly personalized and stable over time, potentially influencing immune development and susceptibility to disease (Cinek et al., 2021).
In addition, metagenomic sequencing has been used to study the evolution of influenza viruses (Krishnamurthy and Wang, 2017). By analyzing viral sequences from diverse geographical regions, researchers have mapped the genetic changes that drive seasonal epidemics and pandemic events (Baillie et al., 2012). This information is crucial for predicting future outbreaks and informing vaccine development strategies.
A 2021 study by Jenkyn-Bedford et al. (2021) used metagenomic datasets combined with phylodynamics to track the evolutionary trajectory of SARS-CoV-2 variants in real time. Their work helped forecast variant emergence and informed public health interventions more rapidly than conventional genomic surveillance methods.
Basic Approaches to Human Viral Metagenomics
Sample collection, transportation, and storage
Human sample collection, transportation, and storage are critical steps in human viral metagenomics, a field focused on analyzing viral communities within human samples for diagnostic, epidemiological, and research purposes. Each phase collection, transportation, and storage require meticulous handling to ensure the integrity and reliability of the metagenomic data (Fig. 1).
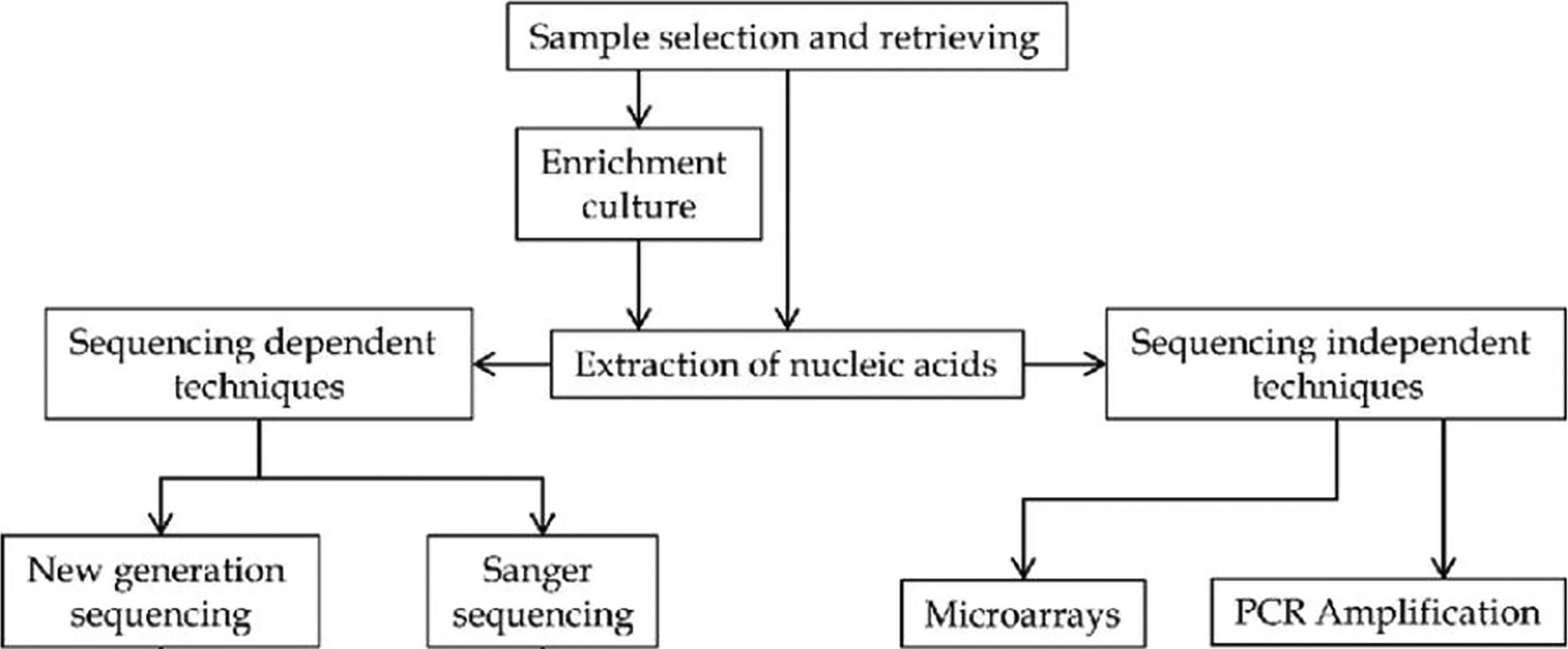
Schematic overview of sample preparation and nucleic acid extraction in viral metagenomics.
Human sample collection
The process of collecting human samples is foundational for effective viral metagenomics. Various types of samples are used depending on the target virus and the study objectives. For instance, blood samples are frequently collected to assess systemic viral infections and are drawn using venipuncture. These samples are typically collected in sterile tubes with anticoagulants to prevent clotting and are often processed by separating plasma or serum through centrifugation (Simmonds et al., 2017). Saliva and nasal swabs offer less invasive methods for detecting respiratory viruses, with nasal swabs particularly useful for collecting samples from the nasopharyngeal region. The swabs used are usually made of synthetic materials such as nylon or Dacron to avoid introducing inhibitors that could affect PCR results (Spencer et al., 2013). Stool samples are essential for studying enteric viruses and must be collected in sterile containers and kept cool to prevent degradation (Barrell et al., 1976). Tissue biopsies, collected during surgical procedures or autopsies, require preservation using formalin or other fixatives to maintain both cellular and viral integrity (Simmonds et al., 2017).
Sample transportation
Proper transportation of human samples is crucial for preserving the integrity of viral nucleic acids and preventing contamination. Temperature control is vital, as many viral nucleic acids are sensitive to temperature changes. Samples are generally transported on ice or in refrigerated containers to maintain low temperatures and prevent degradation. In cases where long-term transport is necessary, samples may need to be kept at temperatures below −80°C to ensure their stability (Chiu and Miller, 2019). Packaging is also a key consideration; samples should be placed in leak-proof containers and surrounded by absorbent materials to prevent leakage and contamination. Compliance with biohazard regulations is essential to ensure safety during transportation (CDC, 2017). Minimizing the time between sample collection and processing is critical to reduce the risk of degradation, though if delays occur, samples should be stored under conditions that limit nucleic acid breakdown (Nolan et al., 2006).
Sample storage
The storage of human samples is essential for maintaining the quality of viral nucleic acids and ensuring accurate metagenomic analyses. For short-term storage, samples are typically kept at 4°C, which is suitable for preserving viral integrity for a few days. However, for long-term preservation, samples should be stored at ultralow temperatures, such as −80°C, or in liquid nitrogen at −196°C. This helps prevent nucleic acid degradation and preserves virus viability for extended periods (Mraz et al., 2009). In addition, to avoid the negative effects of freeze-thaw cycles on nucleic acids, it is advisable to aliquot samples into small portions and minimize the number of freeze-thaw events (Feldmann et al., 1996).
These approaches ensure that human viral samples are handled properly throughout their lifecycle, from collection to analysis, thus enhancing the reliability and accuracy of viral metagenomic studies.
Human sample preparation for nucleic acid extraction in viral metagenomics
Human sample preparation for nucleic acid extraction in viral metagenomics is a crucial process that involves several methods to ensure the purity and integrity of nucleic acids. The removal of contaminants is essential to obtain accurate and reliable results. Various techniques are employed to achieve this, including density centrifugation, filtration, nuclease treatments, and non-nuclease approaches. Each of these methods plays a distinct role in contaminant removal and nucleic acid purification.
Density Centrifugation
Density centrifugation is a widely used technique to separate viral particles from cellular debris and other contaminants based on their density. This method involves the use of a gradient medium, such as sucrose or cesium chloride, which creates a density gradient in a centrifuge tube. When the sample is subjected to high-speed centrifugation, viral particles, which have a specific density, migrate to distinct layers in the gradient. This allows for the separation of viruses from proteins, nucleic acids, and other cellular components (Sambrook and Russell, 2001). Density centrifugation is particularly effective in isolating viruses from complex biological fluids, such as blood or plasma, where the viral particles are mixed with a large amount of host material. However, this method can be time-consuming and may require multiple centrifugation steps to achieve a high degree of purity (Chomczynski and Sacchi, 1987).
Filtration
Filtration is another critical method for removing contaminants from human samples before nucleic acid extraction. This process uses membrane filters with specific pore sizes to physically separate viral particles from larger debris, such as cells and cell fragments. Filtration can be performed using various types of filters, including membrane filters and ultrafiltration devices, which are selected based on the size of the target viruses and the nature of the sample (Walker, 2002). For instance, ultrafiltration is effective for concentrating viral particles by retaining them on a filter while allowing smaller contaminants and excess fluids to pass through. Filtration is advantageous for its simplicity and speed, but it may require careful optimization to avoid loss of viral particles or clogging of the filter membranes (Bustin et al., 2009).
Nuclease treatments
Nuclease treatments are employed to degrade nucleic acids that are not of interest, such as host DNA or RNA, which can interfere with the analysis of viral nucleic acids. This approach involves the use of specific enzymes, such as DNases or RNases, to selectively degrade the unwanted nucleic acids (Chomczynski and Sacchi, 1987). DNase treatment is used to remove contaminating genomic DNA from samples containing RNA viruses, while RNase treatment is used to degrade RNA in samples containing DNA viruses. Nuclease treatments must be carefully controlled to prevent degradation of viral nucleic acids, and thorough removal of the enzymes is necessary to avoid any potential interference in downstream applications (Mullis and Faloona, 1987). Although nuclease treatments can significantly improve nucleic acid purity, they may require additional purification steps to ensure complete removal of enzyme residues.
Non-Nuclease approaches
Non-nuclease approaches involve alternative methods for contaminant removal that do not rely on enzymatic degradation of nucleic acids. These approaches include chemical methods, such as the use of organic solvents, which can selectively precipitate proteins and other contaminants while leaving nucleic acids in solution (Sambrook and Russell, 2001). Another non-nuclease method involves the use of affinity-based purification, where specific binding agents or materials are used to capture and remove contaminants based on their interactions with the viral particles or nucleic acids (Chomczynski and Sacchi, 1987). These methods can complement nuclease treatments by providing additional layers of purification, thus ensuring higher purity and yield of the target nucleic acids. However, they may require optimization to achieve the desired level of contamination control without adversely affecting the recovery of viral nucleic acids (Chomczynski and Sacchi, 1987).
Viral Nucleic Acid Extraction and Amplification from Human Samples
Viral nucleic acid extraction
The extraction and amplification of viral nucleic acids from human samples are critical steps in viral metagenomics and diagnostics. These processes are essential for identifying and characterizing viral pathogens, but they require meticulous techniques to ensure the quality and accuracy of the results (Fig. 1).
The extraction of viral nucleic acids begins with the careful processing of human samples, which may include blood, plasma, serum, or tissue biopsies. The primary goal is to isolate viral RNA or DNA from a complex mixture of host cells, proteins, and other contaminants. One common method for nucleic acid extraction is the use of commercial kits designed specifically for viral RNA or DNA. These kits typically employ silica-based columns or magnetic bead-based technology to bind nucleic acids, followed by washing and elution steps to obtain pure nucleic acid extracts (Chomczynski and Sacchi, 1987). The efficiency of these kits is often high, but the process requires strict adherence to protocols to avoid contamination and ensure consistent results.
Another approach involves the use of organic solvents, such as phenol–chloroform, which can separate nucleic acids from proteins and other cellular debris. This method is effective but labor-intensive and requires careful handling to avoid cross-contamination and degradation of the nucleic acids (Sambrook and Russell, 2001). In addition, specialized techniques such as density gradient centrifugation can be used to isolate viral particles before nucleic acid extraction, providing a more targeted approach (Chomczynski and Sacchi, 1987).
Viral Nucleic Acid Amplification Strategies for Human Viral Metagenomics
Once the nucleic acids are extracted, amplification is required to detect and quantify the viral genomes. The most commonly used method for amplification is PCR, which allows for the specific amplification of viral DNA or complementary DNA (cDNA) from RNA viruses. PCR involves the use of specific primers that bind to the viral nucleic acids, along with a DNA polymerase enzyme that extends these primers to amplify the target sequences (Mullis and Faloona, 1987). This technique is highly sensitive and specific, making it ideal for detecting low-abundance viral nucleic acids in human samples.
For RNA viruses, the extracted RNA must first be converted into cDNA using reverse transcription. This process employs the enzyme reverse transcriptase to synthesize cDNA from RNA templates. The cDNA can then be used as a template for subsequent PCR amplification (Sambrook and Russell, 2001). Alternatively, quantitative PCR (qPCR) can be employed to simultaneously amplify and quantify the viral nucleic acids in real-time, providing a quantitative measure of viral load (Bustin et al., 2009).
Multiple displacement amplification (MDA) is a technique designed for amplifying small quantities of DNA using a unique DNA polymerase that operates at a constant temperature. MDA utilizes the high-fidelity Phi29 DNA polymerase, which is capable of generating large amounts of DNA from minute initial quantities. This method involves the use of random primers to initiate the amplification process, allowing for the displacement and replication of DNA strands without the need for thermal cycling (Dean et al., 2002). MDA is highly effective for generating sufficient quantities of DNA for subsequent analysis, especially when dealing with limited or degraded samples. However, a notable limitation is the potential for amplification bias, which can lead to uneven representation of certain regions of the genome (Dean et al., 2002). In addition, the technique may be sensitive to contaminating DNA, which requires stringent contamination control measures.
Sequence-independent single-primer amplification (SISPA) is a method designed to amplify a broad spectrum of nucleic acids without prior knowledge of the sequences involved. SISPA uses a single primer that binds to a conserved region or random sequence in the nucleic acids. This approach allows for the amplification of a diverse range of viral genomes, including those from novel or poorly characterized viruses (Allander et al., 2001). The method is particularly useful in metagenomics and environmental virology for identifying unknown viral species. However, SISPA can suffer from biases related to the efficiency of primer binding and amplification, which may affect the representation of less abundant viral genomes (Willner et al., 2009). The method also requires careful optimization of the primer and amplification conditions to minimize nonspecific amplification.
Single-primer isothermal amplification (SPIA) is an amplification technique that enables the generation of large quantities of nucleic acids at a constant temperature using a single primer. SPIA employs a single, long primer that binds to the target DNA or cDNA and initiates continuous amplification via a combination of DNA polymerase activity and strand displacement (Dean et al., 2001). This method is advantageous for its simplicity and efficiency, as it eliminates the need for thermal cycling equipment. SPIA is particularly beneficial in settings where conventional PCR may be challenging due to equipment constraints. Despite its advantages, SPIA can exhibit limitations in terms of amplification bias and the potential for nonspecific amplification, which necessitates careful protocol optimization to achieve reliable results (Gordon et al., 2020).
Random PCR, also known as random amplification of polymorphic DNA, is a technique that employs arbitrary primers to amplify various DNA segments randomly throughout the genome. This method is particularly useful for generating a wide array of amplification products from viral DNA, allowing for the detection and characterization of diverse viral genomes (Williams et al., 1990). Random PCR is valued for its simplicity and the ability to amplify unknown or variable sequences without prior knowledge of the target sequences. However, it is limited by issues such as primer-dimer formation and nonspecific amplification, which can complicate the interpretation of results (Welsh and McClelland, 1990). In addition, the technique may not be as reproducible or sensitive as other amplification methods, particularly when dealing with low-abundance viral genomes.
Sequencing Technologies in Human Viral Metagenomics
First-generation sequencing, also known as Sanger sequencing, represents the pioneering technology for DNA sequencing and was developed in the 1970s by Frederick Sanger and his colleagues. This method relies on chain termination during DNA synthesis, using fluorescently labeled dideoxynucleotides to terminate the growing DNA strand at each base position. The resulting fragments are then separated by capillary electrophoresis, and the sequence is read based on the fluorescent signals (Sanger et al., 1977). Although Sanger sequencing was revolutionary for its time, it is limited by its relatively low throughput and higher cost per base compared with newer technologies. Its main strength lies in its accuracy and reliability for sequencing shorter DNA fragments, making it suitable for targeted sequencing applications but less practical for large-scale viral metagenomic studies (Mardis, 2008) (Table 4).
Sequencing Technologies

Second-generation sequencing (NGS), or high-throughput sequencing, represents a major advancement in sequencing technology, enabling massively parallel sequencing of millions of DNA fragments simultaneously (Table 4). Technologies such as Illumina (Solexa), Roche 454, and ABI SOLiD fall under this category. Illumina sequencing, for example, involves the bridge amplification of DNA fragments on a flow cell, followed by sequencing-by-synthesis, where fluorescently labeled nucleotides are incorporated one at a time, and the emitted signals are detected to determine the sequence (Mardis, 2008). This approach provides high throughput and low cost per base, making it ideal for comprehensive viral metagenomic studies that require extensive data generation. However, it also has limitations, including relatively short read lengths and difficulties in accurately assembling sequences from repetitive regions (Mardis, 2008; Metzker, 2010).
Third-generation sequencing technologies, including Single-Molecule Real-Time (SMRT) sequencing by Pacific Biosciences and nanopore sequencing by Oxford Nanopore Technologies, represent the latest advancements in sequencing technology (Table 4). These methods offer significant improvements over previous generations, particularly in read length and real-time sequencing capabilities. SMRT sequencing, for instance, uses zero-mode waveguide technology to observe DNA polymerase activity in real-time, allowing for the sequencing of long DNA molecules with high accuracy and reduced error rates in repetitive regions (Eid et al., 2009). Nanopore sequencing involves passing DNA through a nanopore and measuring changes in electrical current to determine the sequence, enabling ultra-long reads and direct sequencing of RNA molecules (Mikheyev and Tin, 2014). While these technologies provide valuable insights into complex viral genomes and metagenomic diversity, they face challenges such as higher error rates and higher costs, though ongoing advancements continue to enhance their accuracy and accessibility (Oxford Nanopore Technologies, 2021).
Bioinformatics Processes in Viral Metagenomics
Bioinformatics plays a crucial role in viral metagenomics, providing the computational tools and methodologies necessary to analyze and interpret complex metagenomic data (Table 5). This process involves several key stages, including data preprocessing, sequence alignment, taxonomic classification, and functional annotation (Fig. 2).
Bioinformatics Processes in Viral Metagenomics

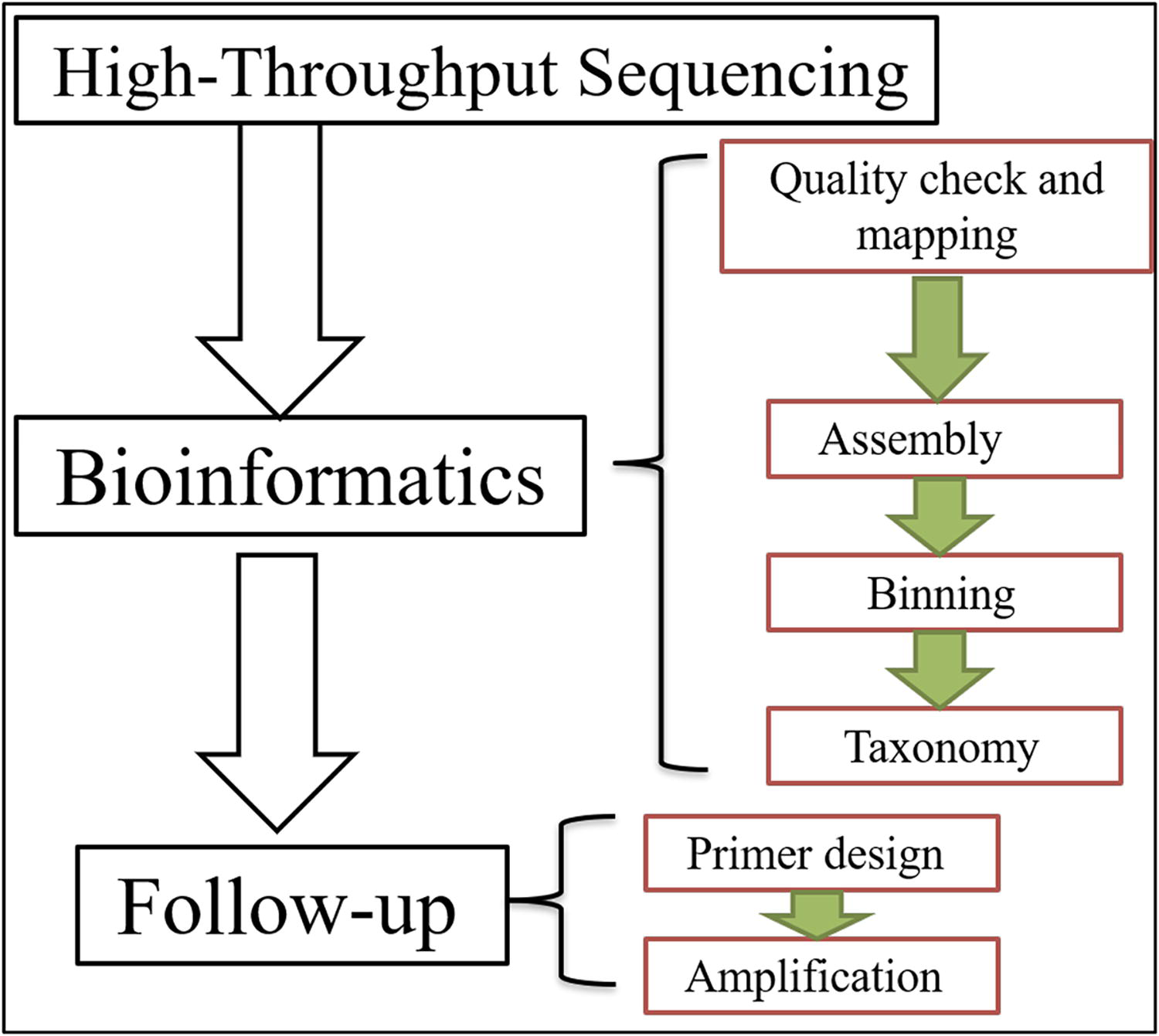
Schematic diagram of bioinformatics and follow-up workflow in viral metagenomics.
Data Preprocessing
The initial step in bioinformatics for viral metagenomics is data preprocessing, which involves quality control and filtering of raw sequencing reads. High-throughput sequencing technologies generate vast amounts of data, and raw reads often contain errors or low-quality sequences that need to be cleaned before further analysis. Tools such as Trimmomatic (Bolger et al., 2014) and Cutadapt (Martin, 2011) are used to trim adapters, remove low-quality reads, and eliminate contaminants. This preprocessing ensures that the dataset is accurate and reliable for subsequent analysis.
Sequence Alignment
After preprocessing, the next critical step is sequence alignment, where the cleaned reads are mapped to reference databases to identify viral sequences. Traditional alignment tools such as BLAST (Altschul et al., 1990) and more recent algorithms such as Bowtie2 (Langmead and Salzberg, 2012) and BWA (Li and Durbin, 2009) are used to align sequencing reads against viral genomic databases. Accurate alignment is essential for identifying the presence of specific viruses within a sample and for quantifying their abundance. High-performance computing resources are often required to handle the large datasets typical in metagenomic studies.
Taxonomic Classification
Following sequence alignment, the taxonomic classification of viral sequences is performed to determine the specific viral species present in the sample. Tools such as Kraken (Wood and Salzberg, 2014) and MetaPhlAn (Truong et al., 2015) use large reference databases to classify sequences based on their similarity to known viral genomes. These tools help in identifying novel or rare viruses that may not be included in existing databases. Accurate taxonomic classification is crucial for understanding the diversity and composition of viral communities in environmental or clinical samples.
Although bioinformatics is acknowledged as a critical component of metagenomics, the review could provide more insights into the specific tools and algorithms currently used in viral analysis. This could help readers appreciate the technological advances necessary for effective metagenomic research.
Follow-Up Processes in Human Viral Metagenomics
Following the initial analysis of viral metagenomic data, several essential follow-up processes are crucial for validating results, interpreting findings, and deriving meaningful insights from the data (Fig. 2). These processes include validation of results, functional analysis, and data integration (Table 5).
Validation of results is a critical follow-up process in viral metagenomics to ensure the accuracy and reliability of the identified viral sequences and taxonomic classifications (Table 6). This step often involves cross-referencing the metagenomic data with experimental validation methods, such as PCR or qPCR, which can confirm the presence of specific viruses identified in the metagenomic analysis (Bustin et al., 2009). In addition, the use of independent sequencing runs or replicate samples can help to verify the consistency of the results. Tools such as the Genome Analysis Toolkit are used to assess the quality of variant calls and ensure that the identified sequences are correct and reproducible (McKenna et al., 2010).
Functional analysis is another vital follow-up process, focusing on understanding the biological roles and potential impacts of the identified viral genes and proteins (El-Gebali et al., 2019). This involves using bioinformatics tools to predict the functional capabilities of viral genomes, such as identifying gene ontology terms and potential interactions with host proteins (Eddy, 2011). Functional annotation tools, such as InterProScan and KEGG, help to assign functional categories to viral genes based on known databases, thus providing insights into their roles in viral pathogenesis and their potential effects on the host (Jones et al., 2014; Kanehisa et al., 2017). Understanding these functions can reveal how viral infections influence host biology and contribute to disease processes.
Follow-Up Processes in Human Viral Metagenomics

Data integration involves combining metagenomic data with other types of biological data to provide a more comprehensive view of viral communities and their interactions. This may include integrating metagenomic data with host genomic, transcriptomic, or proteomic data to study virus–host interactions and the impact of viral infections on host gene expression (Wooley et al., 2010). Multi-omics approaches, where data from different sources are combined, can help in constructing detailed models of viral infection dynamics and host response. In addition, integrating metagenomic data with epidemiological and clinical data can enhance the understanding of viral transmission patterns and disease outbreaks (Chiu and Miller, 2019).
Challenges and Limitations of Metagenomic Approaches
While metagenomics offers unmatched capabilities in detecting a wide range of viral pathogens, its adoption in routine clinical and public health workflows is still limited by several significant challenges.
High Cost and Infrastructure Needs
One of the primary limitations of metagenomic sequencing is its relatively high cost, which includes not only the sequencing itself but also reagents, data storage, and computational resources. Unlike traditional diagnostics such as PCR or antigen tests, metagenomics requires access to NGS platforms and high-throughput infrastructure. These requirements can limit its feasibility in low-resource settings (Simner et al., 2018).
Bioinformatics Complexity and Skill Requirements
Metagenomic analysis requires specialized bioinformatics tools and expertise, which are not commonly available in most clinical laboratories. The process of assembling, classifying, and interpreting metagenomic data is technically demanding and computationally intensive. Moreover, accurate pathogen identification depends on the quality and completeness of reference databases, which may be limited for novel or rare viruses (Chiu and Miller, 2019). Misclassification or false negatives can result from poor database representation or sequence divergence.
Clinical Interpretation Challenges
A unique challenge with metagenomic sequencing is determining the clinical relevance of detected organisms. Because the technique is untargeted, it may identify viruses that are not causing disease (e.g., latent infections or commensal virome components), which complicates clinical interpretation. This issue becomes particularly problematic when multiple organisms are present in a sample or when a virus of unclear pathogenicity is detected (Salzberg, 2016; Wilson et al., 2014).
Sample Preparation and Sensitivity Issues
Sensitivity can also be compromised due to sample preparation limitations. Clinical samples often contain a high background of host nucleic acids, which can obscure low-abundance viral sequences. Without adequate enrichment techniques or host depletion methods, viral genomes may remain undetected (Quick et al., 2016). In addition, certain steps such as reverse transcription or amplification can introduce biases, affecting the representativeness of the recovered viral community.
Lack of Standardization and Validation
There is currently no universally accepted standard for metagenomic workflows, leading to variability across laboratories in terms of sample handling, sequencing depth, quality filtering, and analysis pipelines. This lack of standardization limits reproducibility and poses challenges for clinical validation and regulatory approval (Schlaberg et al., 2017). In contrast, traditional diagnostic tests undergo rigorous standardization and validation processes before clinical implementation.
Ethical, Legal, and Privacy Concerns
Metagenomic sequencing may inadvertently capture host genetic information or uncover unexpected pathogens, raising concerns related to patient privacy and ethical reporting. This becomes especially relevant in clinical diagnostics, where returning incidental findings is a complex legal and ethical issue (Quick et al., 2016).
Future Directions in Viral Metagenomics
Future research in viral metagenomics is expected to focus on integrating multi-omics approaches, such as combining metagenomics with transcriptomics, proteomics, and metabolomics, to better understand viral functions and host interactions (Li et al., 2021). In environmental monitoring, portable sequencing technologies and real-time bioinformatics tools are being developed to track viral diversity and detect emerging pathogens in ecosystems impacted by climate change and human activity (Brum and Sullivan, 2015). In clinical settings, viral metagenomics could enhance personalized medicine by enabling rapid, unbiased pathogen detection and by informing tailored therapeutic strategies based on host–virome interactions (Chiu and Miller, 2019). In addition, advances in artificial intelligence and machine learning are expected to improve viral sequence identification, functional annotation, and prediction of virus-host dynamics (Ren et al., 2020). These developments, alongside ethical frameworks for data use and biosafety, will shape the next generation of viral metagenomics applications.
Conclusion
Human viral metagenomics has fundamentally reshaped our understanding of viral infections, enabling the detection and characterization of known and novel viruses with unparalleled accuracy. This approach has significantly advanced the study of viral diversity, pathogenesis, and host–virus interactions, especially in the context of emerging and reemerging pathogens. While challenges such as data interpretation, contamination, and the need for robust computational tools persist, the ongoing evolution of sequencing technologies, AI-driven analysis, and multi-omics approaches promises to overcome these hurdles. Future research should focus on expanding reference databases, developing real-time surveillance tools, and exploring the role of the virome in noninfectious diseases such as cancer and autoimmune disorders. Looking ahead, the integration of viral metagenomics with other cutting-edge technologies and interdisciplinary collaboration will enhance our ability to monitor and combat viral diseases. The application of viral metagenomics in public health holds vast potential, from early outbreak detection and antimicrobial resistance monitoring to informing vaccine design and personalized medicine. To maximize impact, interdisciplinary collaboration is essential, alongside the development of ethical guidelines and infrastructure to support global data sharing. Policymakers, researchers, and public health officials should prioritize investment in metagenomic surveillance systems, training in computational virology, and frameworks for integrating metagenomic data into routine diagnostics and public health strategies. Ultimately, human viral metagenomics will continue to play a pivotal role in shaping the future of virology, public health, and the development of novel diagnostic and therapeutic strategies.
Authors’ Contributions
The author was responsible for all aspects of the research, including conceptualization, methodology, data collection, analysis, and article preparation.
Footnotes
Author Disclosure Statement
No conflicting financial interests exist. In this review, the authors declare no competing interests, indicating they have no affiliations or financial involvement that could potentially sway their findings.
Funding Information
No funding was received for this article.