
Abstract
Mammalian reoviruses are promising oncolytic agents, but most preclinical and clinical work has focused on the type 3 Dearing (T3D) prototype, potentially underestimating the therapeutic relevance of broader reovirus genetic diversity. Because reoviruses possess a segmented double-stranded RNA genome, reassortment can generate progeny with novel combinations of traits influencing infectivity, replication, and cytotoxicity. Here, we evaluated a panel of previously generated T1L × T3D reassortants and recombinant reoviruses across three epithelial tumor models: A549 lung adenocarcinoma and the oral squamous carcinoma cell lines OECM-1 and CAL-27. Across all three models, the tested viruses displayed marked cell line-dependent heterogeneity in both cytotoxicity and infectivity. Several reassortants reduced viability more effectively than the parental T1L and T3D strains in one or more cell lines, and DB62 emerged as the most broadly active candidate across the panel. Infectivity and cytotoxicity overlapped only partially, indicating that efficient infection alone does not fully predict oncolytic potency. Together, these findings show that reassortment can generate reoviruses with enhanced or selective activity across epithelial tumor contexts and support future studies examining how segment-dependent differences in interferon antagonism, entry, and cell death shape oncolytic potency.
Introduction
Solid tumors of epithelial origin, including lung adenocarcinoma and oral squamous cell carcinoma, remain difficult to treat when disease recurs, metastasizes, or becomes refractory to standard therapies. These clinical challenges continue to drive interest in oncolytic virotherapy, a treatment strategy that uses replication-competent viruses to preferentially infect and kill malignant cells (Lin et al., 2023). Mammalian reoviruses (family Spinareoviridae) are especially attractive in this context because they are generally nonpathogenic in humans and can replicate efficiently in transformed cells with dysregulated oncogenic signaling and altered antiviral defenses (Müller et al., 2020). Among the prototype strains, type 3 Dearing (T3D), clinically developed as pelareorep, has been the most extensively studied in preclinical and clinical settings (Chakrabarty et al., 2015; Müller et al., 2020). However, the broader therapeutic potential of reovirus remains limited by marked differences in tumor permissiveness, viral spread, and virus-induced cell death across cancer types and even among individual cell lines.
Despite sustained interest in reovirus-based oncolysis, most studies have focused heavily on T3D, with comparatively few systematically evaluating alternative strains, laboratory variants, or reassortant viruses (Kim et al., 2011; Rodríguez Stewart et al., 2019; van den Wollenberg et al., 2012; Yip et al., 2021). Because reoviruses possess a segmented double-stranded RNA (dsRNA) genome, coinfection can generate reassortant progeny that rapidly acquire new combinations of traits affecting attachment, replication, spread, and cytotoxicity. Classical studies using T1L × T3D reassortants established mammalian reovirus as a tractable system for linking strain-specific phenotypes to individual genome segments (Doyle et al., 2015; Sherry et al., 1996; Sherry and Fields, 1989; Sherry et al., 1998), including the demonstration that the nonparental myocarditic phenotype of clone 8B mapped primarily to the M1 gene and, in later work, to additional core gene contributions (Sherry and Fields, 1989; Sherry et al., 1989; Sherry et al., 1998). Notably, although both T1L and T3D engage JAM-A as a protein receptor through the S1-encoded attachment protein σ1, they differ in σ1-mediated glycan binding, with T1L recognizing GM2 gangliosides and T3D preferentially binding a variety of sialylated glycans (Dermody and Sutherland, 2025). These differences are thought to contribute to strain-specific attachment and downstream cell tropism, although other viral proteins engage with receptors in a serotype-independent manner as well (Dermody and Sutherland, 2025). Related reassortant analyses further showed that T3D-like viruses induce stronger type I interferon (IFN-α/β) responses and remain more sensitive to IFN-mediated restriction, whereas T1L-like viruses are relatively interferon resistant, a difference later linked mechanistically to strain-specific functions of the M1-encoded µ2 protein as an IFN-α/β antagonist (Irvin et al., 2012; Rivera-Serrano et al., 2017; Sherry and Fields, 1989; Zurney et al., 2009).
Given the utility of T1L × T3D reassortants for linking viral gene segments to biologically meaningful phenotypes, we asked whether previously generated reassortants and recombinant reoviruses would display distinct infectivity and cytotoxicity profiles across three epithelial tumor models, and whether these phenotypes might reveal segment-dependent patterns not apparent in a single cell type. Specifically, A549 cells provide a well-established lung adenocarcinoma model, whereas OECM-1 and CAL-27 represent oral squamous cell carcinoma models derived from the gingiva and tongue, respectively, commonly used to study epithelial tumor biology and therapeutic responses. Our results show that previously generated T1L × T3D reassortants and recombinant reoviruses exhibit distinct, cell line-dependent infectivity and cytotoxicity profiles across these models, identifying candidate strains with enhanced oncolytic activity relative to the parental viruses.
Materials and Methods
Cell culture
Human A549 (CCL-185), OECM-1 (CVCL-6782), and CAL-27 (CRL-2095) cells were cultured in high-glucose Dulbecco’s modified Eagle medium (DMEM; Thermo Fisher Scientific) supplemented with 10% fetal calf serum (Atlas Biologicals), 1% sodium pyruvate, 100 U/mL penicillin, and 100 µg/mL streptomycin. Cells were maintained under standard culture conditions at 37°C in a humidified 5% CO2 atmosphere and were used at a low passage number for all experiments. For infection assays, cells were detached with trypsin, seeded in 96-well plates at a concentration of 1 × 104 cells/well (A549) or 5 × 104 cells/well (OECM-1 and CAL-27), and allowed to attach overnight before infection.
Viruses
All viruses were low-passage-number stocks originating from plaques of either cultured virus or virus generated by reverse genetics (Kobayashi et al., 2007) and kindly provided by Dr. Barbara Sherry (North Carolina State University). Parental T1L and T3D viruses (Irvin et al., 2012; Rivera-Serrano et al., 2017; Zurney et al., 2009), type 3 Abney (T3A) (Wilson et al., 1994), T3SA− and T3SA+ (Barton et al., 2001), KC150 and 1HA3 (Connolly et al., 2001), 8B (Sherry et al., 1989), and P208S (Irvin et al., 2012; Rivera-Serrano et al., 2017) have been previously described. The origin and genomic characterization of EB series (Haller et al., 1995; Hermann and Coombs, 2004), the EW series, and the DB series (Sherry et al., 1989; Sherry et al., 1998; van den Wollenberg et al., 2012) of reassortants have been previously described. The genomic architecture of each reassortant is listed in Figure 1. All viruses were titrated by plaque assay in L929 cells as previously described (Irvin et al., 2012; Zurney et al., 2009). Multiplicity of infection (MOI) values were calculated from plaque titers obtained in L929 cells and therefore represent L929-derived PFU per target cell. Because permissiveness and cytopathic readouts differ among cell types, the same nominal MOI may not correspond to equivalent productive infection or cytotoxicity in other cell types.
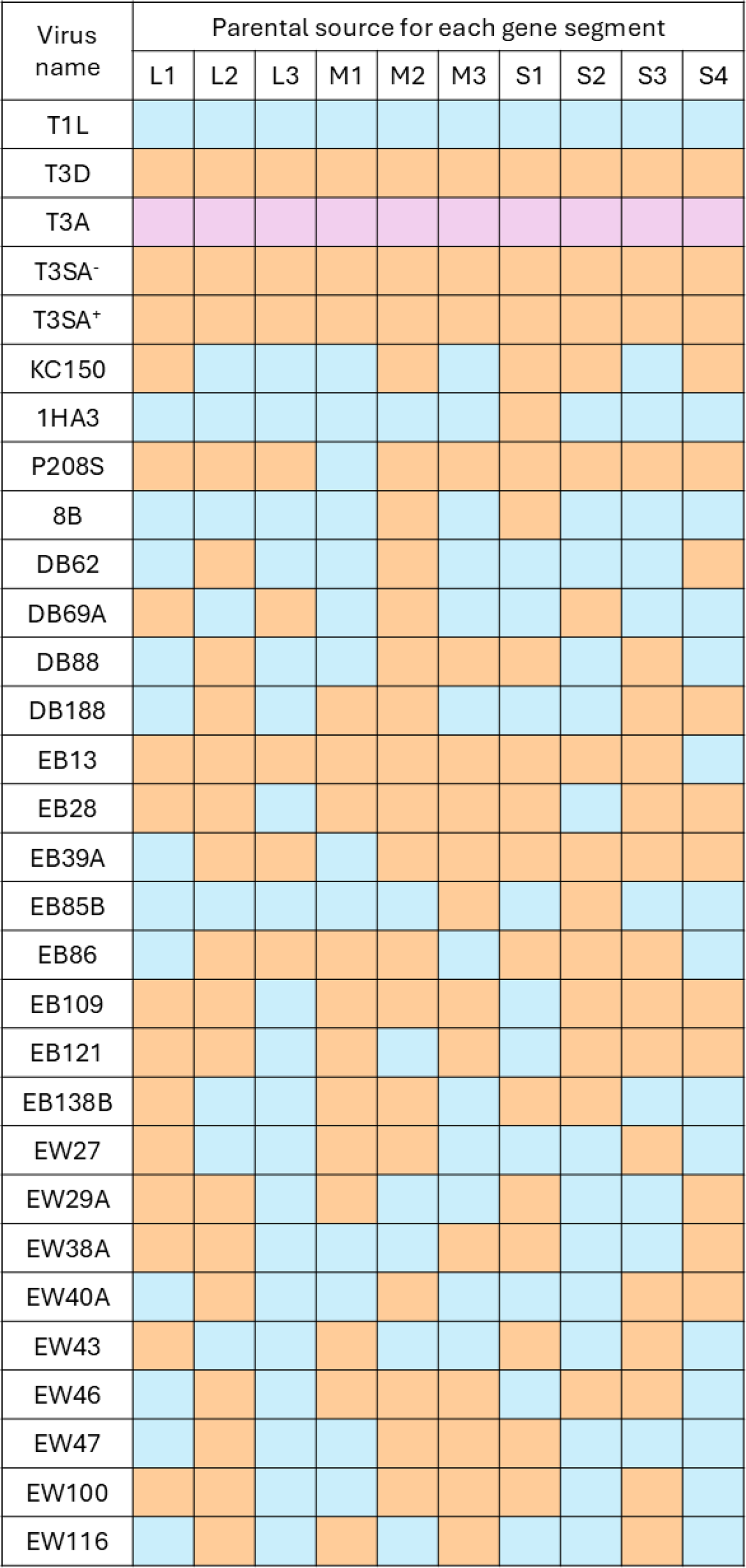
Reovirus panel analyzed for oncolytic activity in the A549, OECM-1, and CAL-27 cells. Mammalian reoviruses contain 10 gene segments: three large (L), three medium (M), and four small (S). Gene segments derived from the T1L serotype are shown in orange, whereas those derived from T3D are shown in blue. T3A is a type 3 strain distinct from both T1L and T3D. Additional information regarding the origin and initial characterization of these viruses is provided in the “Materials and Methods” section. T3A, type 3 Abney; T3D, type 3 Dearing.
Cell viability assay
At 48 h post-infection, or 96 h post-infection for A549 cells, 20 µL of a 6 mg/mL solution of membrane-permeable MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] prepared in complete DMEM was added to each 200-µL culture well. Plates were incubated for 4 h at 37°C in a humidified 5% CO2 atmosphere. Supernatants were then aspirated, and 100 µL of 0.04 N HCl in 2-propanol was added to each well to solubilize the formazan product. After 15 min at room temperature, 100 µL of H2O was added to each well, and optical density was measured at 570 nm with background subtraction at 630 nm using an automated microplate reader. Normalized absorbance values were expressed as percent cell viability relative to mock-infected controls for each experiment. Statistical analysis was performed in GraphPad Prism using one-way ANOVA followed by Dunnett’s multiple-comparison test, with each virus-infected condition compared with the corresponding mock-infected control within the same cell line. p < 0.01 was considered statistically significant.
Indirect immunofluorescence and quantification of viral infectivity
Cells were fixed in 2% paraformaldehyde (Electron Microscopy Sciences) in phosphate-buffered saline (PBS) and permeabilized with 0.25% Triton X-100 (Millipore-Sigma) for 7–10 min. Samples were then blocked with 2% normal goat serum (Millipore-Sigma) for 1 h at room temperature and incubated for 1 h at room temperature with a 1:1 mixture of rabbit antisera raised against reovirus T1L and T3D (Zurney et al., 2009), each diluted 1:1,000 in 0.1% IgG-free bovine serum albumin (Jackson ImmunoResearch). After rinses with PBS, cells were incubated for 60 min at room temperature with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Thermo Fisher Scientific) diluted 1:1,000. Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole), and F-actin was labeled with CruzFluor™ 594-conjugated phalloidin (Santa Cruz Biotechnology) diluted 1:1,000. Cells were imaged on an EVOS Cell Imaging System (Thermo Fisher Scientific) using a 20 × objective, and representative images were assembled for presentation in Photoshop CS4.
For quantification of viral infectivity, three to five micrographs were analyzed per virus condition using Fiji, an open-source distribution of the ImageJ2 scientific image-processing platform (Schindelin et al., 2012). Image analysis was automated using Apple Automator to execute a shell script linked to a custom Fiji macro developed for this application. For each image, the pipeline sequentially converted the image to 8-bit format, applied an automatic threshold, performed binary watershed segmentation, and conducted particle analysis with outline generation. This workflow was used to quantify the total number of nuclei in each field and was applied uniformly to all images within the designated folder, with output data compiled into a single file. Reovirus antigen-positive cells were scored manually for each micrograph, and infectivity was calculated as the number of reovirus antigen-positive cells divided by the total number of nuclei. For each cell type, the highest infectivity value observed among the virus panel was set to 100%, and all other strains were normalized relative to that value. Infectivity data were used as descriptive quantitative measures of antigen-positive infection and were not subjected to inferential statistical testing.
Results and Discussion
To place these experiments in context, we examined a panel of previously characterized reoviruses spanning parental strains, isogenic variants, reassortants, and selected viruses generated by reverse genetics (Fig. 1). We infected A549, OECM-1, and CAL-27 cells and quantified cell viability by MTT assay at the indicated endpoints to assess the oncolytic properties of each virus across these epithelial tumor models (Fig. 2). T1L and T3D served as genetically and phenotypically distinct parental reference strains, with T3D representing the clinically developed prototype strain described above. In addition, T3SA− and T3SA+ are isogenic viruses that differ by a single point mutation in S1 that determines the capacity to bind sialic acid, a property previously linked to enhanced apoptosis and NF-κB activation (Barton et al., 2001; Connolly et al., 2001). Additionally, we included T3A as an additional type 3 strain distinct from T3D and the reassortants KC150 and 1HA3, which were previously linked to apoptosis through activation of c-Jun N-terminal kinase in cultured cells and therefore provided useful comparators for evaluating whether earlier virus-specific phenotypes extend to epithelial tumor models (Clarke et al., 2001; Connolly et al., 2001). Comparing their cytotoxic effects across our cell lines, we observed clear cell type-specific responses, with viruses such as KC150 producing marked cytopathic effects in A549 cells (Fig. 2A) but not in OECM-1 or CAL-27 cells (Fig. 2B,C). These early comparisons prompted us to examine the broader reassortant panel more systematically to determine whether segment exchange could uncover additional patterns of selective or enhanced oncolytic activity across epithelial tumor contexts.
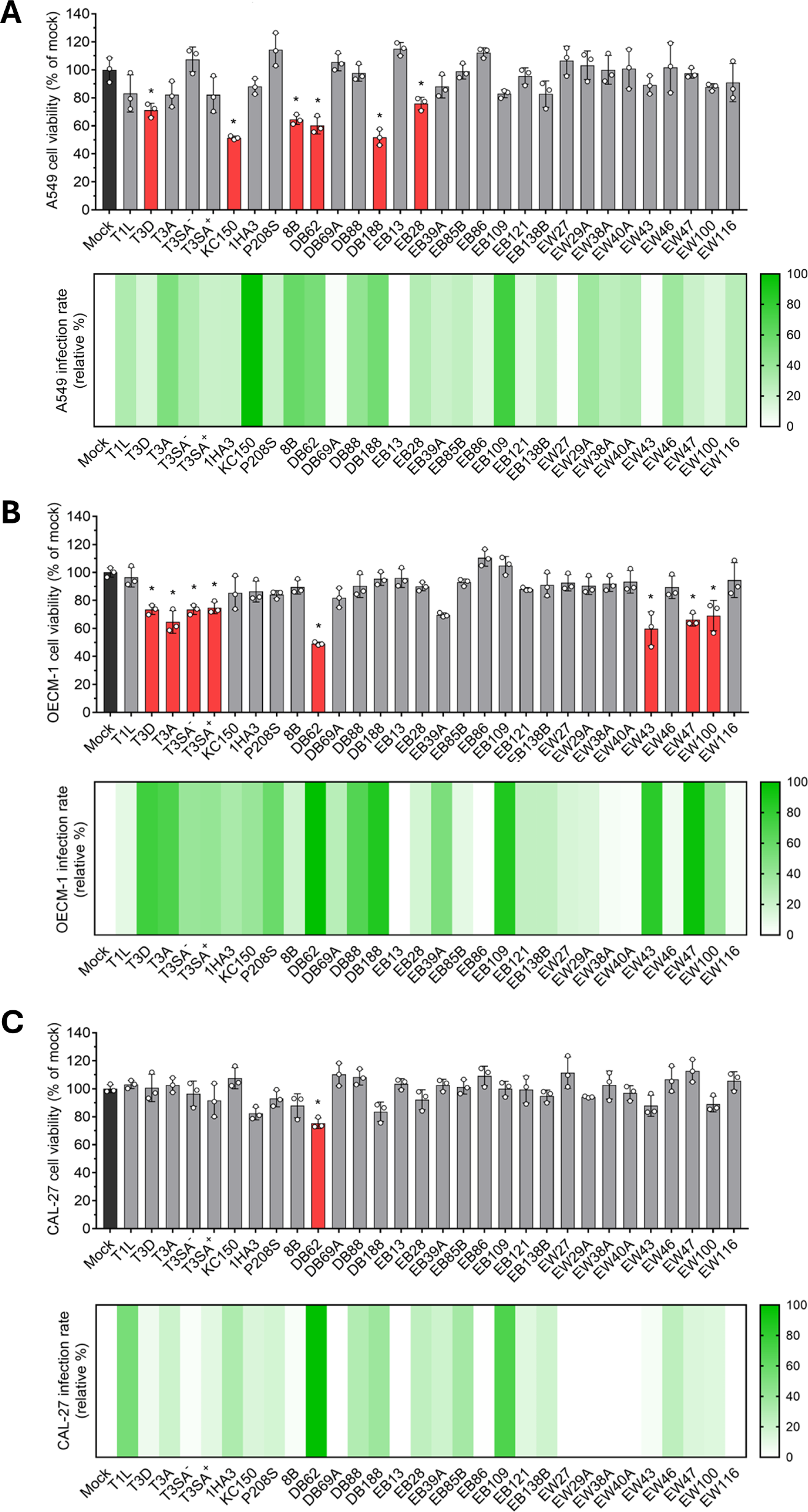
Reovirus reassortants exhibit cell line-dependent infectivity and cytotoxicity across epithelial cancer models. A549
Across the three models, the tested viruses exhibited marked, cell line-dependent heterogeneity in cytotoxicity. Several reassortants reduced viability more effectively than the parental T1L and T3D strains in one or more cell lines, indicating that reassortment can generate segment constellations with enhanced or selective oncolytic activity. These findings extend prior work centered on the T3D prototype by showing that broader reovirus genetic diversity can uncover phenotypes not readily apparent from either parental strain alone. The differences observed among A549, OECM-1, and CAL-27 cells further underscore that tumor cell susceptibility to reovirus is strongly context-dependent and likely reflects variation in host cell entry, intracellular antiviral defenses, and other host determinants that influence productive infection and virus-induced cell death. Receptor availability is one possible contributor to these cell-line-specific patterns. JAM-A is the canonical protein receptor engaged by the reovirus σ1 attachment protein, but JAM-A abundance, receptor accessibility at cell junctions, and σ1-dependent glycan engagement can differ among epithelial tumor contexts. While we did not quantify basal JAM-A expression side-by-side in the present study and therefore do not ascribe the observed phenotypes solely to receptor abundance, preliminary antibody-blocking experiments with σ1-directed monoclonal antibodies reduced infection by selected viruses in these cells (not shown), supporting a role for canonical σ1-mediated entry while leaving open the possibility that downstream replication, innate immune responses, and cell-death pathways also shape the observed outcomes.
In addition, we sought to determine whether differences in cytotoxicity were accompanied by differences in viral infectivity. To address this, we quantified infection in parallel experiments by indirect immunofluorescence using a mixture of reovirus antisera that recognize T1L- and T3D-derived viral antigens across reassortants (Fig. 2). Infectivity profiles overlapped only partially with the viability data, and the two readouts were not uniformly coupled across the tested cell lines. In some contexts, relatively high levels of antigen-positive infection were not matched by proportional losses in viability (e.g., EB109), whereas in others, cytotoxicity appeared more pronounced. This pattern suggests that efficient entry or establishment of antigen-positive infection alone is insufficient to predict the full oncolytic phenotype and instead supports a model in which downstream events, including replication efficiency, particle disassembly or stability, induction of cell death pathways, and antagonism of antiviral IFN-α/β responses, also contribute to strain-specific outcomes. Thus, the present data support the idea that infectivity and tumor cell killing are related but mechanistically distinct phenotypes.
The observation that antigen-positive infection and short-term cytotoxicity did not fully overlap is also important for interpreting the high-MOI infection design. All inocula were standardized using L929-derived PFU, and the 48–96 h endpoints were selected to compare early cytotoxic responses across epithelial tumor models rather than to measure maximal virus spread or endpoint killing. Thus, high input infection by parental T1L or T3D does not necessarily result in rapid loss of cellular metabolic activity in every tumor cell line. Reovirus cytopathicity can be strongly cell-type and kinetics dependent, reflecting differences in productive replication, antiviral signaling, death pathway engagement, and lytic versus nonlytic egress (Fernández de Castro et al., 2020; Smith et al., 2024). Accordingly, the limited early cytotoxicity of some highly infectious conditions should be interpreted as evidence that entry/antigen expression and tumor cell killing are related but separable phenotypes.
Importantly, our interpretation considers the parental origin of each genome segment rather than uncharacterized point mutations within individual isolates. This distinction is relevant because reovirus phenotypes are not always fully predicted by segment provenance alone, as illustrated by the historically important 8B myocarditic variant, whose phenotype was initially associated with M1 and later shown to involve contributions from multiple viral core genes (Sherry and Fields, 1989; Sherry et al., 1989). These studies provide an important cautionary framework for the present work, because segment-of-origin logic alone may not fully explain phenotype and additional genetic factors, including point mutations, may also contribute. Indeed, even a single amino acid polymorphism between reovirus strains can produce drastically different phenotypes (Erickson et al., 2025; Irvin et al., 2012; Parker et al., 2002; Rivera-Serrano et al., 2017). DB62 is particularly informative because its broad activity coincides with a mixed segment constellation that includes T3D-derived L2, M2, and S4 segments in an otherwise T1L × T3D reassortant background.
With those caveats in mind, the historical use of T1L × T3D reassortants as genetic tools provides a useful framework for interpreting the present findings. Although this study was not designed to assign causality to individual gene segments, the segment composition of the more active viruses highlights several plausible contributors, including M1, M2, L2, L3, and S4. Prior work has linked M1, which encodes the multifunctional protein µ2, to strain-specific differences in interferon sensitivity and antagonism of IFN-α/β signaling (Irvin et al., 2012; Lemay and Boudreault, 2025; Rivera-Serrano et al., 2017; Zurney et al., 2009), making this segment an attractive candidate for shaping the context-dependent differences observed across epithelial tumor lines. The T3D M2 gene segment encodes µ1, an outer-capsid protein that mediates membrane penetration during entry and has been linked to apoptosis and myocarditic phenotypes in multiple systems (Danthi et al., 2008; DeAntoneo et al., 2022; Tyler et al., 1996). Likewise, the L3-encoded λ1 protein may influence capsid properties relevant to particle stability and postentry events (Gummersheimer and Danthi, 2020). The T3D S4 segment encodes σ3, a dsRNA-binding outer-capsid protein with roles in regulating host stress and antiviral responses, including stress granule biology and PKR/RNase L-associated pathways (Guo et al., 2021), while the L2-encoded λ2 turret protein may also influence postentry transcription, capping, or early replication events that affect innate immune detection.
In this context, the reassortants tested here, particularly DB62, may combine T3D-derived entry/cell-death or host-stress determinants with T1L-derived segments such as M1, which have been associated with IFN-α/β antagonism. Because this screen was not designed for single-segment causal assignments, these interpretations remain hypothesis-generating, but they provide a clear rationale for prioritizing DB62 in future mechanistic studies. Together, these findings support the idea that reassortment can generate viral constellations with improved or selective activity across distinct epithelial tumor contexts and provide a foundation for future studies aimed at defining how individual segments, or segment combinations, regulate infectivity, interferon responses, and cytotoxicity.
Conclusions
Our findings show that previously generated T1L × T3D reassortant reoviruses exhibit distinct, cell line-dependent infectivity and cytotoxicity profiles across epithelial tumor models, providing evidence that segment composition contributes to context-specific oncolytic phenotypes. Several reassortants displayed enhanced activity relative to the parental strains in one or more cell lines, with some candidates demonstrating broader activity across the tested models. These results reinforce the value of reassortant-based approaches for identifying viral determinants of tumor cell susceptibility and for expanding the range of reovirus strains considered for oncolytic development beyond the extensively studied T3D prototype. Although the present study was designed as a comparative screen to identify reassortants with enhanced activity across epithelial tumor models, the strong performance of DB62 now justifies focused follow-up experiments, including single-step and multi-step growth curves, virus yield measurements in each tumor line, assessment of IFN and interferon-stimulated gene involvement, death pathway characterization, and full-genome sequencing to distinguish segment-of-origin effects from isolate-specific mutations. Collectively, this work provides a foundation for future studies aimed at defining segment-specific mechanisms underlying these phenotypes, including potential contributions of gene segments associated with interferon antagonism and cell death, and for evaluating the most promising candidates in more complex preclinical models.
Authors’ Contributions
E.E.R.-S., C.V.D., and J.R.H. conceptualized the study and designed experiments. E.E.R.-S. and J.R.H. performed experiments. E.E.R.-S., J.R.H., and O.M.C. analyzed data. E.E.R.-S. wrote the original draft. E.E.R.-S., J.R.H., O.M.C., and C.V.D. reviewed and edited the article.
Footnotes
Acknowledgments
The authors thank Dr. Barbara Sherry (North Carolina State University) for kindly providing A549 cells, purified reoviruses, and reovirus antisera. They thank Dr. Victoria Moore (High Point University) for kindly providing the OECM-1 and CAL-27 cell lines.
Author Disclosure Statement
All authors declare that no conflicts of interest exist.
Funding Information
This research was supported with the institutional faculty support of