
Abstract
The aim of the study was to evaluate interictal electroencephalogram features in 22 patients with Dravet syndrome from the onset of the disease through the next 5 years. Electroencephalogram was abnormal in 5 patients (22.7%) at onset, and in 17 (77.3%) at the end of the study. Epileptiform abnormalities (focal, multifocal, or generalized) were seen in 6 patients at the onset and in 14 (27% vs 64%) at the end of the study. Photoparoxysmal response was present in 41% of patients at the end of follow-up. No statistical differences were found between mutated and nonmutated groups regarding evolution of background activity, interictal abnormalities, and presence of photoparoxysmal response. Electroencephalogram findings seemed to be age dependent, variable among different patients, and not influenced by the presence of sodium channel, voltage-gated, type I, alpha subunit (SCN1A) mutation. The lack of specific epileptiform abnormalities contributes to the difficulty of patients’ management in Dravet syndrome.
Keywords
Dravet syndrome, first reported as severe myoclonic epilepsy in infancy, was described by Dravet in 1978. 1 In the last Report of the International League Against Epilepsy Commission on Classification and Terminology, 2 it was suggested that Dravet syndrome should be considered as “genetic epilepsy.”
The syndrome is characterized by the occurrence of generalized seizures during the first year of life often triggered by fever, and unilateral clonic seizures in otherwise normal children. Myoclonia, atypical absences, and focal seizures appear soon after the onset. 3 Psychomotor retardation becomes evident usually during the second year of life. 4,5 Moreover, progressive neurologic deficits such as ataxia and pyramidal signs subsequently can develop. 6
Sodium channel, voltage-gated, type I, alpha subunit (SCN1A) mutations have been identified in 30% to 80% of patients with Dravet syndrome. 7–12 A small proportion (10%-25%) of negative cases were found to have pathogenic deletions or duplications of SCN1A. 13–15
The electroencephalogram (EEG) pattern in Dravet syndrome is characterized by the association of generalized, focal, and multifocal abnormalities. 16,17 Usually background activity is slow and is associated with bilateral frontocentral theta activity while the patient is awake. 18 Paroxysmal abnormalities can be absent especially in the early stages. If present, they consist of generalized or focal spikes, spikes and waves, synchronous and/or asynchronous, isolated, or in brief bursts. Usually sleep EEG is normal. A lack of specific pattern has been underlined. 16 Photosensitivity is reported in a large proportion of cases (>40%), although it is not a constant feature over time. 19
Moreover, limited data are available documenting the progression of EEG features during the disease. Therefore, we wish to contribute to the understanding of the dimensions of EEG changes over time in patients with Dravet syndrome by reporting the EEG findings through retrospective examination of all traces from the onset of the disease up to 5 years of follow-up.
Methods
This is a retrospective study on EEG features of patients affected by Dravet syndrome who were referred to the Bambino Gesù Children’s Hospital in Rome and Epilepsy Centre University Federico II in Naples from 1991 to 2005. The diagnosis of Dravet syndrome was based on accepted clinical and genetic criteria. 16 All included patients had been followed since the onset of epilepsy and for at least 5 years. Children were evaluated during follow-up every 3 to 6 months.
Patient Population and Assessments
Twenty-two patients were included in the study. Epileptic seizures were classified according to the International League Against Epilepsy criteria. 20,21
SCN1A point mutations were tested by denaturing high-performance liquid chromatography and direct sequencing; genomic deletions by fluorescence in situ hybridization or multiplex ligation-dependent probe amplification. EEGs were performed using the bipolar and monopolar 10-20 International system. Intermittent photic stimulation was performed following international protocols. 22 The frequency of each stimulation was progressively increased up to 30 Hz, or up to 60 Hz when intermittent photic stimulation did not elicit any response. Examiners of the EEGs were blind to the patients’ clinical and genetic characteristics.
Primary and Secondary End Points
The primary end point of the study was the evaluation of EEG features from the onset of the disease through the next 5 years. We analyzed all the EEG traces from the baseline (time 0) and every 6 months (±1 month; time 1, 2, 3, etc.). If patients had intermediate EEGs, they were not considered. Patients were excluded if EEGs were not performed in that particular time interval or if EEG recordings did not fit with the above indications. The following parameters were considered for each evaluation: background activity, interictal abnormalities in terms of type and localization, distinguishing slow waves (theta and/or delta rhythm), epileptiform abnormalities (spikes, spikes and waves, and poly-spikes), and presence of photoparoxysmal response. To differentiate interictal from ictal paroxysms, technicians witnessed all the examinations and pointed out all clinical events. Although there is evidence that focal or multifocal EEG abnormalities can only appear during sleep in the present study, sleep EEG was not evaluated because it was not available in all cases.
The secondary endpoint was the comparison of EEG findings between patients with and without SCN1A mutation. The analysis of the data was focused on the results at the beginning and end of the study period—time trend analysis was complex, and 22 patients was not a great enough number for anything more than an “impression.”
Statistical Analysis
Continuous data were tested for normality, and differences between means were tested with Student t test.
Results
Study Sample
Demographic and clinical characteristics of the 22 patients with Dravet syndrome are shown in Table 1 . Mean age at onset was 4.7 ± 2.6 months (range 1.5-11). Familial history for epilepsy and/or febrile seizures was reported in 5 patients (22.7%). Two patients were siblings.
Demographic and Clinical Characteristics
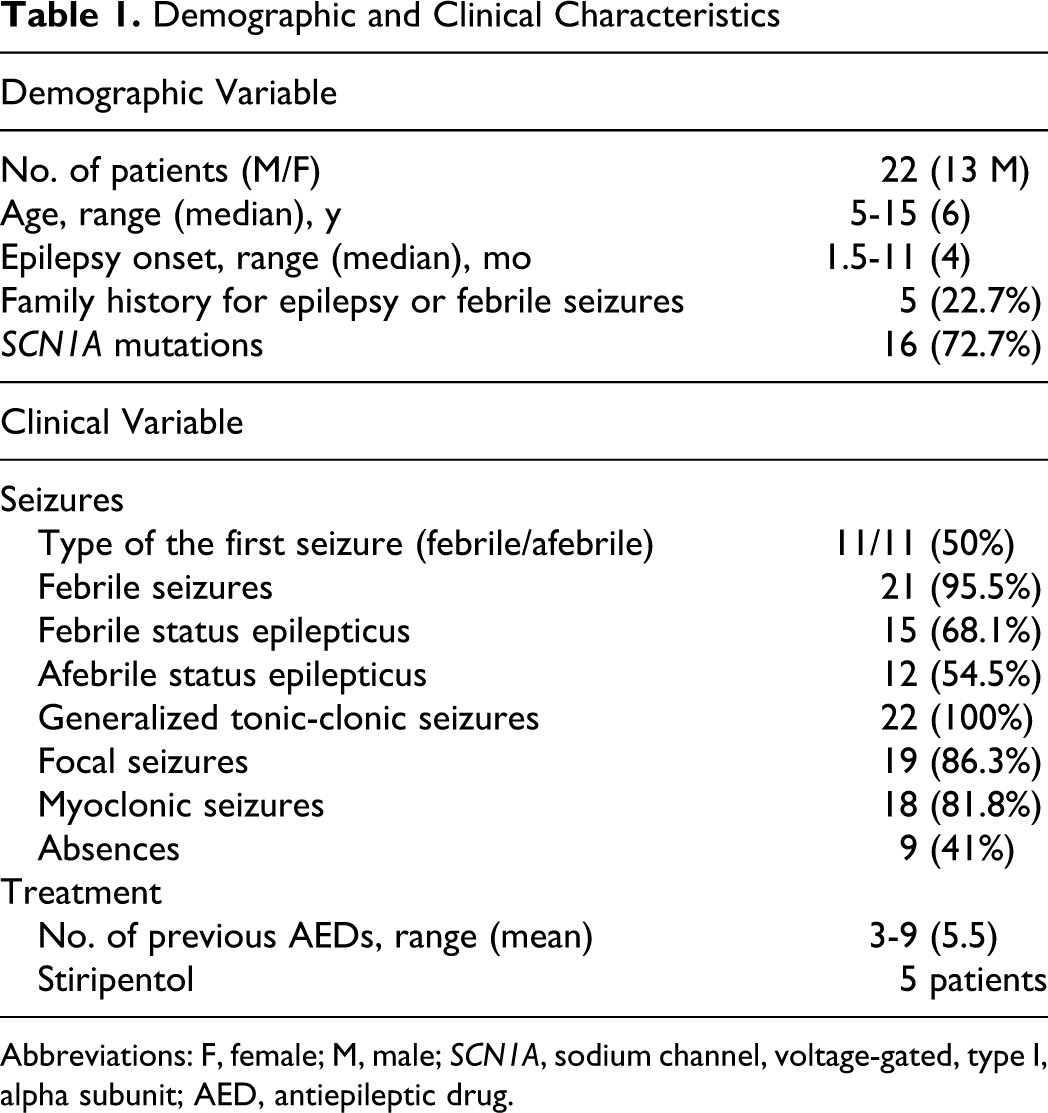
Abbreviations: F, female; M, male; SCN1A, sodium channel, voltage-gated, type I, alpha subunit; AED, antiepileptic drug.
Sixteen patients had a point mutation or a deletion of SCN1A gene (72.7%). In addition, first seizure was febrile in 11 patients (50%). Twenty-one patients (95.5%) subsequently had at least 1 seizure during fever. All patients presented at least 1 status epilepticus during follow-up. Fifteen of 22 (68.1%) patients had a febrile status epilepticus and 12 (54.5%) had an afebrile status epilepticus during the disease course. In addition, the patients also experienced generalized tonic-clonic (100%), focal (86.3%), myoclonic (81.8%), and absence (41%) seizures.
All patients were treated with 3 to 9 different antiepileptic drugs in their past (mean 5.5), and 5 of them were also treated with stiripentol. The mean age at evaluation was 8 ± 3.8 years (range 5-15 years). Mean follow-up period was 8.7 ± 3.9 years.
EEG Findings
Figure 1 and Table 2 show EEG findings from the onset through the study period. All patients had an EEG every 6 months. EEG was abnormal at onset in 5 patients (22.7%), and in 17 patients (77.3%) at the end of the study. Background activity was normal in all patients at onset. After 6 months, 6 of 22 patients (27%) presented a slowing of background activity. This finding tended to be constant during the duration of the study, and it was not different between mutated and nonmutated patient groups (18.6% vs 14.6%; P = .4).
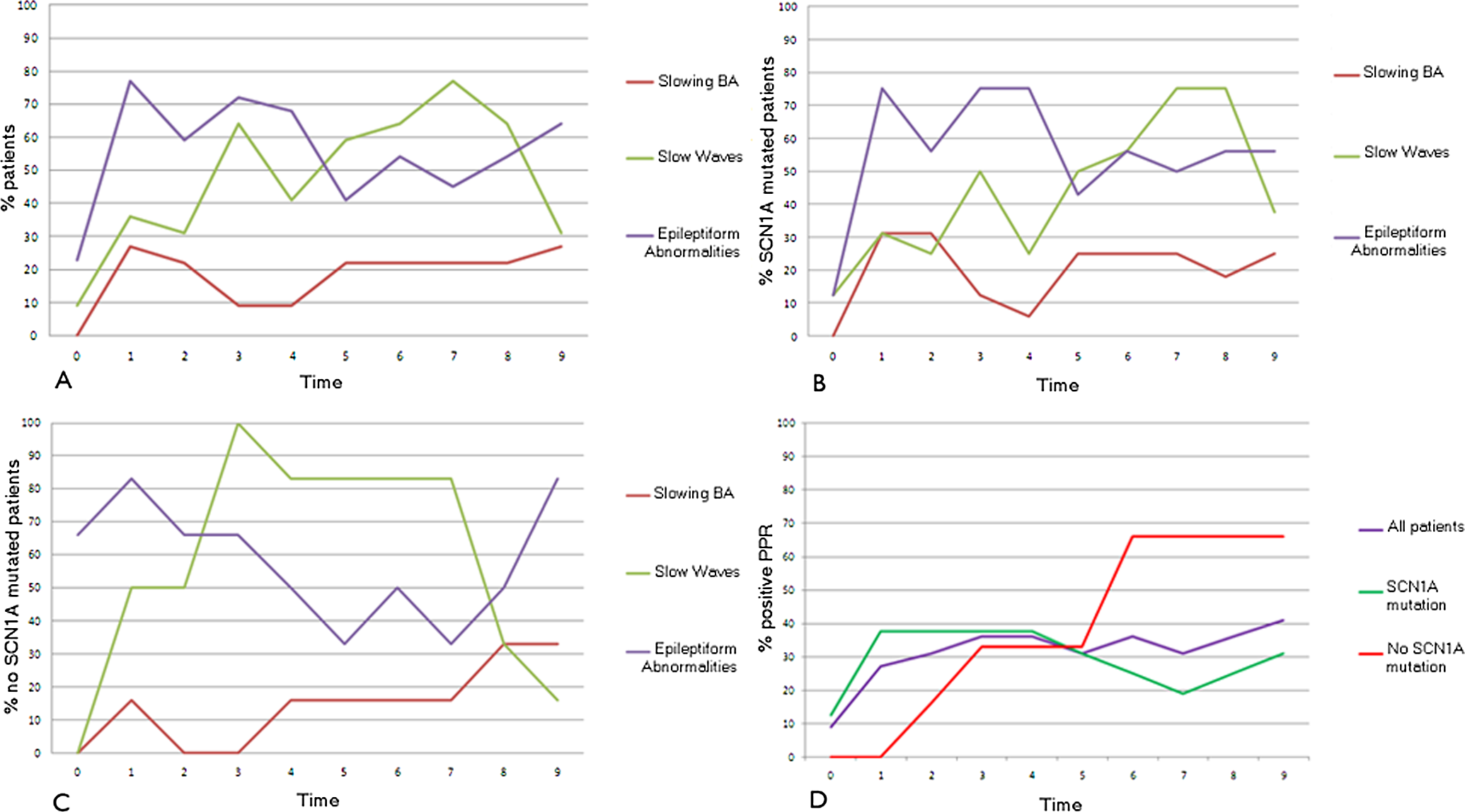
Electroencephalogram (EEG) findings from the onset through the study period.
Electroencephalogram (EEG) Findings and Presence of Photoparoxysmal Response (PPR) in All Patients, and in SCN1A Mutated and Non-mutated Groups of patients

Abbreviation: SCN1A, socium channel, voltage-gated, type I, alpha subunit. Values within parentheses are percentages.
Interictal abnormalities
We considered epileptiform and slow waves. Epileptiform abnormalities (focal, multifocal, or generalized) were seen in 6 patients at the onset of the disease and in 14 (27% vs 64%) at the end of the study. All patients at least in one observation presented epileptiform abnormalities during the study period. Slow abnormalities (theta and delta rhythms) were evident at the onset in 2 patients and in 7 (9% vs 31%) at the end of study. No differences were found comparing mutated and nonmutated patients regarding the percentage of patients with interictal abnormalities during the study period (79.8% vs 83.3%; P = .7). Overall within 22 patients, at 12, 24, and 36 months we had the chance to record sleep EEG in respectively 16, 12, and 10 patients. The analysis of EEG trace revealed that all patients who presented epileptiform abnormalities when awake tended to increase during sleep. Only in 4 patients who had a normal awake EEG, did the epileptiform abnormalities appear during sleep.
Photoparoxysmal response
At onset, 2 of 22 (9%) patients were photosensitive. This finding progressively increased during the study period: at the end, it was present in 9 of 22 (41%) patients. No statistical differences were found between mutated and nonmutated groups regarding the presence of photoparoxysmal response during the study period (29% vs 37.9%; P = .4).
Localization
Figure 2 and Supplemental Table 1 and 2 (tables available online at jcn.sagepub.com) show types and localization of epileptiform abnormalities from the onset through the study period. At onset, epileptiform abnormalities were generalized in 3 of 6 patients (50%) and mainly over central-posterior areas in the other 3 (50%). At the end of the study period, 2 of 14 patients (14%) presented generalized, 8 (57%) multifocal, and 4 (28.5%) focal abnormalities (2 anterior, 1 central, and 1 posterior regions).
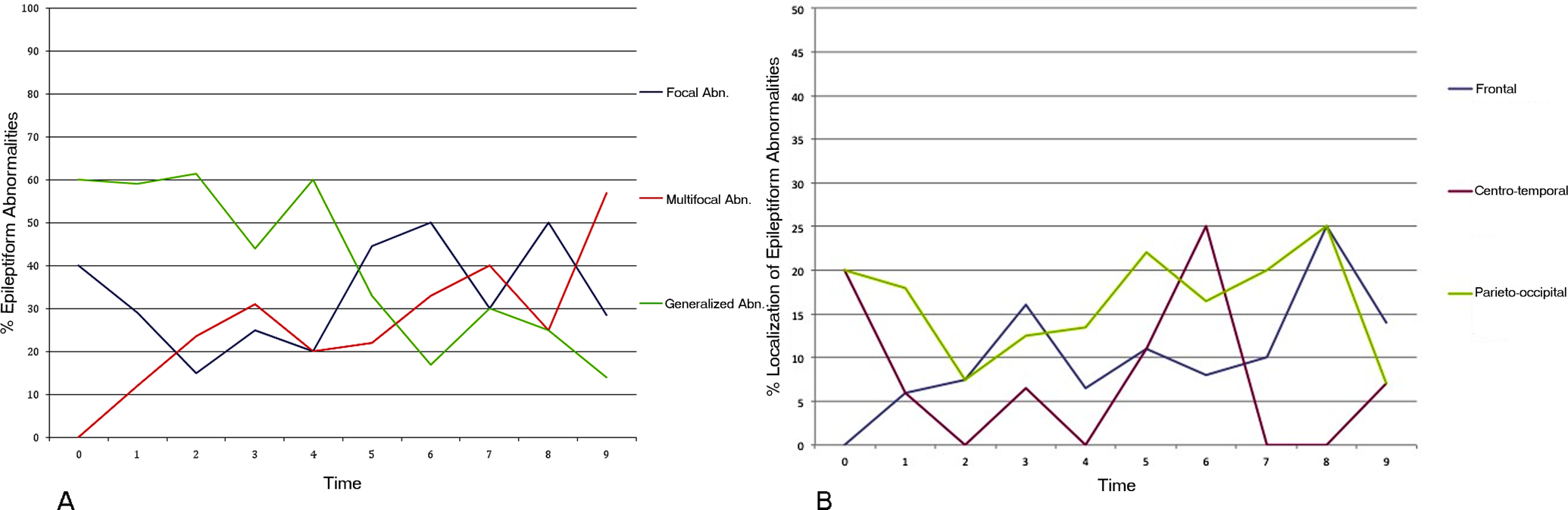
Types and localization of epileptiform abnormalities and slow waves from the onset through the study period.
Slow waves at onset were seen mainly over central regions, while at the end of follow-up they were mainly diffuse (5 of 7, 71.5%) or over the anterior areas (2 of 7, 28.5%). We did not consider the differences between mutated and nonmutated groups because of the small number of patients.
Discussion
To date, few data are available on EEG features in Dravet syndrome and on their modifications thorough the disease course. 5,23–26 Three stages of the disease have been recognized, and EEG features might change over time. 17,27 It is known that EEG at onset might be normal; epileptiform and slow abnormalities tend to appear during the second year, associated with a slowing down of background activity. 16 A systematic study on the evolution of EEG abnormalities since the onset of the disease is not yet available. Our study shows the progression of EEG features over a 5-year period of observation.
EEG at onset could be normal in a large number of patients, 16,23,28 and we confirm that in 77% of our patients EEG was normal at onset. In early stages, paroxysmal discharges are not evident; later on, especially during the second year of life, generalized spikes and waves and focal or multifocal abnormalities appear. 16,23,24 They tended to be stable throughout the study period, while background activity deteriorated over time. We looked at differences between SCN1A mutated and nonmutated groups of patients, and no statistical differences were found, showing that SCN1A mutation does not affect the EEG evolution any more than other causes of Dravet syndrome.
Epileptiform abnormalities (generalized, focal, or multifocal) tended to increase through the years. This was confirmed for both the mutated and nonmutated groups. Epileptiform focal abnormalities appeared quite soon (within the first year) in our patients, and remained stable through the follow-up. Focal abnormalities have been previously reported to occur later in life, 29 and an individual variability cannot be excluded.
In a study of 53 patients, EEG findings confirmed that in 79% of cases generalized paroxysmal activity predominated, less frequently associated with focal or multifocal spikes: age of appearance of such abnormalities is not reported. 24 Korff et al 23 reported 16 patients with EEG data during the evolution of the disease in an undetermined time interval. Abnormal EEG was reported in 25% of patients at onset and in 81% on evolution, and generalized spikes and poly-spikes and waves discharges mainly characterized abnormalities. This is in contrast with our results, which show that generalized epileptiform abnormalities tend to decrease within a few years (60% at onset vs 14% at follow-up). Moreover, we found an increase of epileptiform abnormalities comparing awake versus sleep EEG; however, a firm conclusion cannot be drawn because of the limited number of patients.
The percentage of patients with slow abnormalities (theta and delta rhythms) also increased during the disease course— at the onset 9% vs 31% at the end of the study; slow abnormalities had a maximum peak after 1 year since the onset. It is possible that in Dravet syndrome the appearance of slow waves might be associated with the cognitive decline, which usually is evident after the second year of life. 5,30
Few data are reported about localization of abnormalities: focal and slow abnormalities are mainly described on central areas. 16,18 In our patients, these abnormalities were mainly over posterior regions during the first periods of observation, and they moved anteriorly during the follow-up, involving frontal and centrotemporal regions. Photosensitivity is inconsistently found during the disease course. It has been observed in 42% of the patients during follow-up 3,29 and can appear very early. 28,29 In our study, photoparoxysmal response increased from 9% to 41% during the first 2 years of observation; and only in mutated patients was it evident since the onset. Intermittent photic stimulation determined a photoparoxysmal response in 26% of patients in the Caraballo series. 24 A discrepancy between laboratory EEGs’ photostimulation and everyday clinical sensitivity to lights has been reported in Dravet syndrome patients. 31
During adolescence, an unexpected EEG pattern in Dravet syndrome has also been reported 25 : 5 patients presented with bifrontal spikes and slow waves while awake and fast poly-spikes during sleep. Authors discuss its possible relationship to a switch to Lennox-Gastaut syndrome, although they consider it unlikely. A peculiar EEG pattern in adulthood also was reported recently. 30 As expected, we did not find such EEG patterns because of the age of our patients, which was lower. Long-term follow-up, however, confirms that interictal EEG findings are multiple and heterogeneous. 32
Our study suggests that interictal EEG findings cannot be considered as a clue in the diagnosis of Dravet syndrome, which remains clinical and genetic. EEG features are highly variable from the onset and through the disease course. It is only in a few cases that generalized epileptiform abnormalities together with an early presentation of photosensitivity might help for diagnosis purposes.
Although EEG cannot be considered as a diagnostic criterion in Dravet syndrome at onset, it might be useful during the follow-up to differentiate cases with an atypical clinical course with patients affected by Lennox-Gastaut syndrome or myoclonic astatic epilepsy.
This study shows the evolution of EEG features from the onset in Dravet syndrome. EEG findings seemed to be age dependent, variable among different patients, and not influenced by the presence of SCN1A mutation. During the disease course, all our patients experienced different types of seizures, which required several antiepileptic drugs. Although all patients were seen at standard time intervals, possible drug influences on EEG activity cannot be excluded, as well as type and frequency of seizures in each period of observation. We collected and analyzed a discrete number of EEG examinations during the disease course in our series of patients. A possible conclusion of this study is that EEG does not add a great deal to the management and, in routine clinical practice, does not need to be repeated very often. As other authors have suggested, 23 the lack of specific epileptiform abnormalities contributes to the difficulty of patient management in Dravet syndrome.
Footnotes
Acknowledgments
The work was done at Bambino Gesù Children’s Hospital in Rome, and preliminary results were presented at the Dravet Syndrome International Workshop—Severe Myoclonic Epilepsy: 30 Years Later Meeting, which was held in Verona, Italy in October 2009. The authors thank the patients and their families for their participation.
NS wrote the first draft of the manuscript. He made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data. MB, MT, NJ, PS, AC, and SC made substantial contributions to acquisition, and interpretation of data; they revised it critically and gave the final approval of the version to be published. LMS, LF, and FV made substantial contributions to conception and design of the manuscript, revised it critically, and gave the final approval of the version to be published.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.
The Scientific Directorate of the Bambino Gesù Children’s Hospital in Rome, Italy, approved the study.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.