
Abstract
Pompe disease is an autosomal recessive disorder caused by lysosomal acid α-glucosidase deficiency. Infantile-onset Pompe disease presents with cardiomyopathy and hypotonia, leading to premature death. This article describes 7 infantile Pompe disease cases and provides their molecular bases and clinical outcomes after enzyme replacement therapy for the first time in Korea. Molecular genetic analyses revealed the presence of 9 different mutations, including 5 novel mutations (c.2171C>A, c.2774C>T, c.1582_3de12, c.1261_1263Tms, and c.1322_1326+9de114). The most common mutation in these 7 patients was c.1316T>A (28%). Four patients received intravenous recombinant human acid α-glucosidase therapy for 2 years, on average, without significant side effects during the treatment course. They all exhibited increased muscle power, with considerable improvement in cardiac function. Pompe disease is heterogeneous regarding both clinical features and molecular characteristics. Early identification of Pompe disease is very important, considering that enzyme replacement therapy is a safe and effective treatment for early-onset patients.
Keywords
Pompe disease is an autosomal recessive lysosomal storage disorder caused by acid α-glucosidase deficiency caused by mutations in the acid α-glucosidase gene. This enzyme defect leads to the accumulation of lysosomal glycogen in multiple tissues, which results in various clinical features. 1 The classic infantile form of Pompe disease (infantile glycogen storage disease type ІІ) is a rapidly progressive disease with hypotonia, generalized muscle weakness, and hypertrophic cardiomyopathy, usually leading to death from cardiorespiratory failure or respiratory infection in the first year of life. 2–4 The late-onset type (or atypical form) shows less progressive clinical characteristics and absence of severe cardiomyopathy; these phenotypical differences are related to residual enzyme activity. 5,6 The acid α-glucosidase gene is located on human chromosome 17q25.2-25.3 and more than 200 different sequence variations have been characterized to date (www.pompecenter.nl). 7 Recent clinical applications of enzyme replacement therapy raise the possibility of its application to Pompe disease. 8
In this report, we described the clinical features and the molecular bases of 7 cases of infantile Pompe disease for the first time in Korea (3 classic and 4 nonclassic infantile forms), together with a clinical experience of enzyme replacement therapy over an average follow-up period of 2 years.
Patients and Methods
Patients
Seven Korean patients (3 males and 4 females) diagnosed with Pompe disease between 2001 and 2008 in 2 hospitals (Seoul National University Children’s Hospital and Samsung Medical Center) were included in this study. The patients were unrelated, with the exception of a pair of siblings (patients 2 and 3). The diagnosis of 3 patients (patients 5-7) was confirmed by enzymatic determination of acid α-glucosidase activity in peripheral blood leukocytes. In the remaining 4 patients (patients 1-4), the disease was first suspected after histologic examination of their muscles and was later confirmed via genetic analysis of the acid α-glucosidase gene. Intravenous recombinant human alglucosidase alpha (Myozyme; Genzyme Co., Framingham, Massachusetts) was administered to 4 patients (patients 4-7) every alternate week at a dose of 20 mg/kg. We did not administer the enzyme to all patients because Myozyme was available in our country only at the end of 2005. Informed parental consent was obtained for the collection of clinical data and extraction of DNA to perform mutation analysis.
Molecular Genetic Analysis
Genomic DNA was extracted from peripheral blood leukocytes or skeletal muscle tissues using the Puregene DNA isolation kit (Gentra Systems, Inc., Minneapolis, Minnesota). Polymerase chain reaction was performed to amplify the entire coding region of exons 2 to 20 and the surrounding intronic sequences of the acid α-glucosidase gene, as described previously by Ko et al. 9 We screened 100 alleles in normal subjects to determine the significance of novel variations.
Results
Clinical Features
Initial symptoms manifested from birth to the age of 15 months and diagnoses were established at an average age of 13 months (range, 2-66 months). The patients presented with hypotonia, delayed motor development, cardiomegaly, hepatomegaly, and/or recurrent pulmonary infections, with wide variation in severity. Two patients (patients 1 and 7) received respiratory support using home ventilators, and patient 1 died of respiratory infection at the age of 7 years. Creatine kinase and liver transaminase levels were elevated in all patients. The echocardiographic findings of all patients revealed the presence of hypertrophic cardiomyopathy with or without ventricular dysfunction. One of the patients (patient 6) presented with medically intractable paroxysmal supraventricular tachycardia from the age of 7 months; she was diagnosed with Pompe disease with Wolff-Parkinson-White syndrome. The clinical and laboratory findings of the 7 patients are summarized in Table 1 .
Characteristics of the 7 Korean Patients With Pompe Disease

Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; CK, creatine kinase; GAA, acid α-glucosidase; LVEF, left ventricular ejection fraction; LVMI, left ventricular mass index; PSVT, paroxysmal supraventricular tachycardia; WPW, Wolff-Parkinson-White syndrome.
Note: Patients 2 and 3 are siblings. GAA activity was the leukocyte α-
Mutation Analysis
Nine different pathogenic mutations, including 5 novel mutations, were detected in all patients. Table 1 summarizes the genotypes of the 7 patients. The 4 mutations reported previously were c.1309C>T, c.1822C>T, c.1316T>A, and c.875A>G. The remaining 5 mutations were novel: c.2171C>A, c.2774C>T, c.1582_3de12, c.1261_1263Tms, and c.1322_1326+9de114. The most common mutation was c.1316T>A, as it accounted for 29% of the total number of alleles. The locations of the acid α-glucosidase gene mutations identified in our 7 patients are presented in Figure 1.
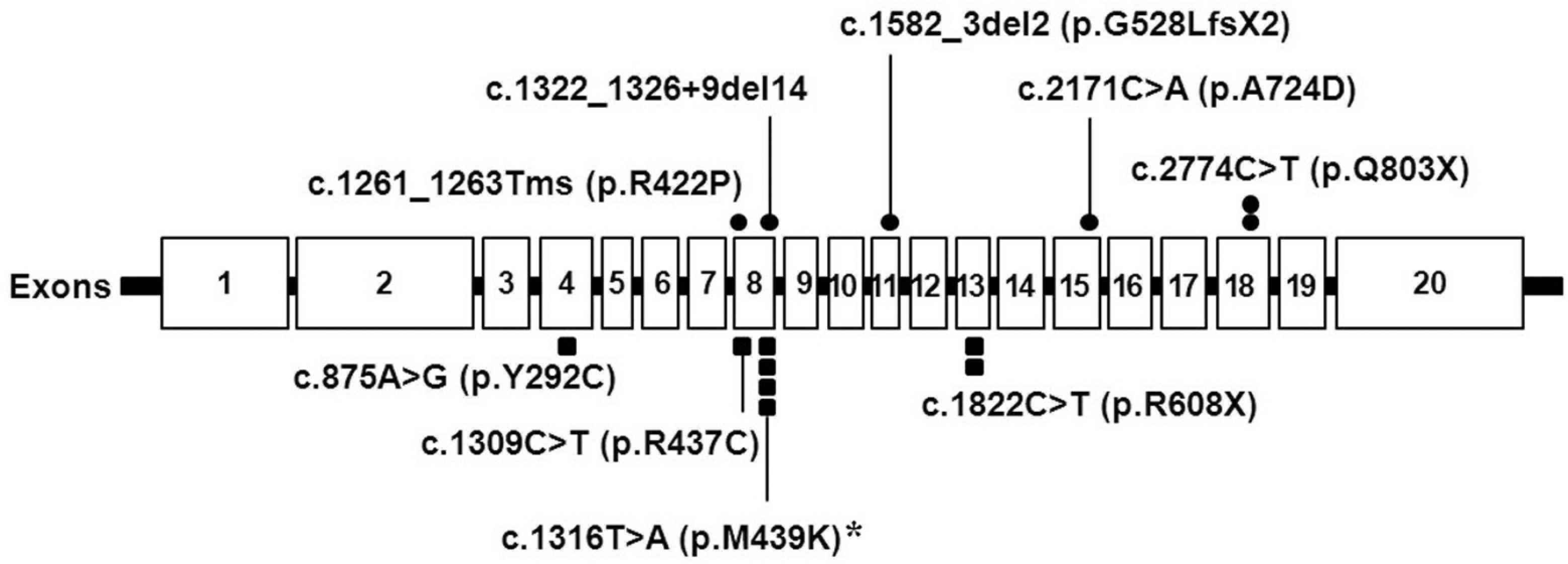
Location of GAA mutations identified in the 7 Korean patients with Pompe disease. Exons are shown as rectangles. The circles above the gene represent novel mutations, and the squares below the gene represent known mutations. The asterisk indicates the most common mutation in the 7 patients.
Enzyme Replacement Therapy
Four patients (patients 4-7) were under enzyme replacement therapy and exhibited clinical improvement of varying degrees. In patient 4, motor power and cardiac ventricular function did not worsen for more than 3 years after the initiation of enzyme replacement therapy. Patient 5 showed marked improvement in heart function with enzyme replacement (Figure 2 ). Patient 6 started enzyme replacement therapy at the age of 12 months, and her recurrent paroxysmal supraventricular tachycardia attacks disappeared after the second dose of enzyme replacement therapy, despite the presence of persistent delta waves on electrocardiogram. Unfortunately, at the age of 15 months, enzyme replacement therapy was discontinued temporarily because of inadequate supply from the manufacturer and her paroxysmal supraventricular tachycardia attacks returned 1 month after the cessation of enzyme replacement therapy. However, soon after resuming enzyme replacement therapy, her paroxysmal supraventricular tachycardia disappeared once again and has not reoccurred as of the last follow-up. Patient 7 remains under respiratory support using a home ventilator, but his heart function has improved considerably and he currently has grossly normal left ventricular contractility (left ventricular ejection fraction, 67.3%). Creatine kinase and liver transaminase levels in all patients have remained unchanged for an average of 49 months (range, 19-114 months) after enzyme replacement therapy. The clinical and laboratory results after enzyme replacement therapy for the 4 patients that received this treatment are summarized in Table 2 . No significant side effects of enzyme replacement therapy were observed during treatment in these 4 patients.

Chest radiography in patient 5 and M-mode view of 2-dimensional echocardiography in patient 6. (
Results of Enzyme Replacement Therapy
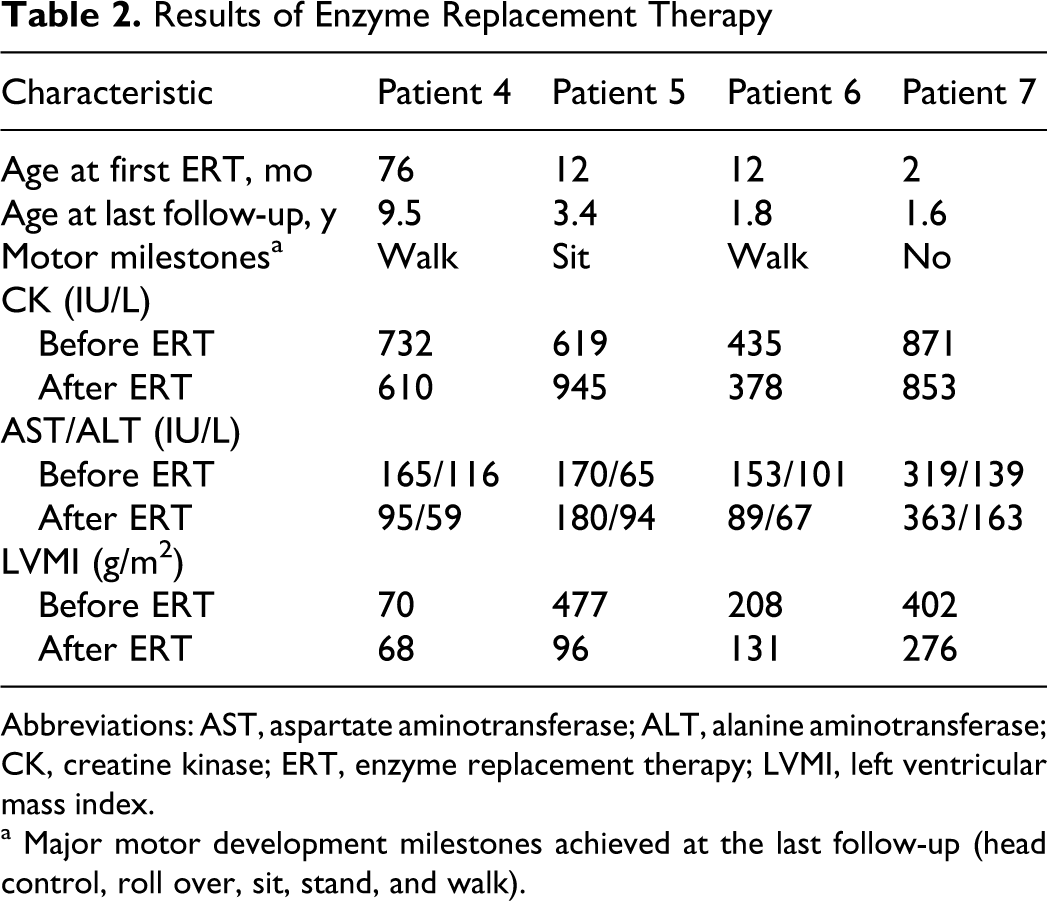
Abbreviations: AST, aspartate aminotransferase; ALT, alanine aminotransferase; CK, creatine kinase; ERT, enzyme replacement therapy; LVMI, left ventricular mass index.
a Major motor development milestones achieved at the last follow-up (head control, roll over, sit, stand, and walk).
Discussion
Pediatric Pompe disease can present with neurologic, gastrointestinal, pulmonary, and/or cardiac manifestations, with a wide spectrum of severity according to residual enzyme activity, as described for our 7 patients. As the clinical presentation is heterogeneous and can greatly vary in terms of severity, early diagnosis of the disease remains challenging for pediatricians. 10 The most common initial presentation in our patients was cardiomegaly, which was apparent in all but one patient. Among them, 5 patients were managed for cardiomyopathy for a certain period before the correct diagnosis was established. Interestingly, patient 3 was diagnosed with, and managed for, hypertrophic cardiomyopathy of unknown etiology since her early infantile period because she did not show motor weakness. After her younger brother (patient 2) was diagnosed with Pompe disease, she was confirmed as suffering from the same disease. Therefore, we suggest that hypertrophic cardiomyopathy with unknown etiology manifesting in the infantile period should alert pediatricians to the possibility of a Pompe disease diagnosis, even when motor symptoms are not evident.
Patient 5 was initially diagnosed with a congenital cataract, without evidence of congenital infection or related drug exposure. Although there are no previous reports of the coincidence of congenital cataract and Pompe disease, cataracts might be caused by glycogen deposits in the lens of Pompe patients. Massive glycogen deposits in all ocular tissues of a 16-week-old fetus with Pompe disease were examined and reported previously by Pokorny et al, 11 which supports our assumption. Unfortunately, we could not demonstrate our hypothesis in a further study because the cataract lens of our patient was removed by operation before the diagnosis was established.
Molecular analysis of the acid α-glucosidase gene in a specific population allows the characterization of its mutational spectrum within that population. 7,12–16 Genetic analysis performed in our patients revealed that the mutational spectrum of Korean Pompe patients exhibited different features compared with those observed in patients from other countries, including other Asian populations. 15,16 The most frequent mutation in our 7 patients was c.1316T>A (p.439Met>Lys), which is located in exon 8 (29%) and was previously found in 2 Korean patients with a late-onset phenotype. 17 Therefore, we suggest that c.1316T>A is a common recurrent mutation in the Korean population, irrespective of phenotypic expression. As all of our patients with the c.1316T>A mutation had early-onset phenotypes, it is difficult to determine whether this mutation is related with a mild phenotype. Clinical heterogeneity among patients with the same mutation was also described in previous studies. 13,18–20 This supports the theory that other genetic or environmental factors can affect phenotypic variability. As mentioned in many previous studies, it is impossible to predict the clinical course of patients with Pompe disease based solely on genotype.
Enzyme replacement therapy with recombinant human acid α-glucosidase has dramatically improved the life expectancy and clinical features of patients with Pompe disease. 8,21,22 In our study, all patients who received enzyme replacement therapy exhibited clinical improvement, or at least a stable clinical state without progression of the disease. The major effect of enzyme replacement therapy in our patients was the restoration of cardiac function. The most remarkable improvement was observed in patient 5, who was relieved from a life-threatening clinical status and exhibited a notable recovery of cardiac function. Her left ventricular mass index before enzyme replacement therapy was 477 g/m2, which decreased to less than 100 g/m2 after a year of enzyme replacement therapy. Currently, she is also showing considerable improvement in motor development.
The literature reports an incidence of arrhythmias in infants under enzyme replacement therapy of up to 18%. The type of arrhythmia included nonsustained supraventricular tachycardia, ventricular premature beats, atrial bigeminy, ventricular tachycardia, and ventricular fibrillation. 23 Cook et al 24 also reported the results of 24-hour ambulatory electrocardiographic analysis in 12 infants receiving enzyme replacement therapy, of which 2 showed ectopic premature ventricular contractions. However, these reports did not describe any paroxysmal supraventricular tachycardia attacks that necessitated emergency treatment for conversion to normal rhythm. One of our patients (patient 6) was diagnosed with Wolff-Parkinson-White syndrome with recurrent life-threatening paroxysmal supraventricular tachycardia in the infantile period, and the interval of paroxysmal supraventricular tachycardia attacks consistently decreased with increasing age. Remarkably, her paroxysmal supraventricular tachycardia attacks disappeared completely with enzyme replacement therapy, despite persistent delta waves on electrocardiograph.
The etiology of arrhythmia in Pompe disease is unknown; however, an animal model of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy demonstrated that disruption of annulus fibrosis by glycogen-engorged myocytes, rather than distinct bypass tracts, can be responsible for the preexcitation observed in this case. 25 With the advent of enzyme replacement therapy, a remarkable decrease in left ventricular mass index and normalization of ventricular function was demonstrated, 21,22,26 as was the case in our patients. Moreover, conduction abnormalities in patients with infantile Pompe disease can also improve after enzyme replacement therapy, with increase in PR interval and decrease in both QT dispersion and left ventricular voltage. 27 However, there are no reports showing that enzyme replacement therapy itself is useful for the prevention of paroxysmal supraventricular tachycardia attacks, as in patient 6, for whom enzyme replacement therapy was so effective that paroxysmal supraventricular tachycardia terminated and the temporary cessation of enzyme replacement therapy prompted the recurrence of paroxysmal supraventricular tachycardia.
According to our experience and many other published studies, enzyme replacement therapy is a safe and effective therapeutic option for patients with Pompe disease. 8,21,22,26
We presented the clinical features, the molecular bases (including 5 novel mutations), and the outcomes of an enzyme replacement therapy experience in 7 Korean patients with infantile Pompe disease first described in our population. Based on the present knowledge, enzyme replacement therapy can lead to changes in the natural course of Pompe disease. Therefore, the availability of this effective treatment has rendered the establishment of an early diagnosis in affected patients more critical. Our study has improved the understanding of Pompe disease and has emphasized the importance of early diagnosis for early initiation of enzyme replacement therapy.
Footnotes
Twelve authors contributed to this manuscript. AC and SJK collected the data and wrote the paper. BCL, HH, and CSK confirmed the genetic diagnosis. GBK evaluated and managed the cardiologic problems of the patients. JL, JDP, KJK, and YSH evaluated and clinically diagnosed the 7 patients. DKJ directed the enzyme replacement therapy and JHC designed this study and is the corresponding author.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article:
This study was supported by a grant from the Korea Healthcare Technology R&D project, Ministry for Health, Welfare and Family Affairs, Republic of Korea (Grant No. A080588) and a Seoul Broadcasting System Grant-in-Aid for research at the Seoul National University Children’s Hospital (Grant No. 30-2009-0140).
Informed consent forms for the publication of the patients’ medical history and genetic study profile were signed by the patients’ parents.