
Abstract
A previously healthy 10-year-old girl presented with subacute onset of ataxia and acute-onset cardiac and pulmonary failure. Magnetic resonance imaging (MRI) of the brain showed symmetric T2 fluid-attenuated inversion recovery hyperintensities in the dorsal pons, medulla, and inferior cerebellar peduncles; nerve conduction velocities and electromyography demonstrated a sensorimotor axonal neuropathy consistent with Friedreich ataxia. Within 12 months, the patient fully recovered and molecular testing of the frataxin gene was unremarkable. Two years later, the patient returned with acute neurologic decompensation and died one month later from progressive demyelination of the brainstem. Mitochondrial DNA sequencing revealed a mutation at 8344A>G in transfer RNA lysine with heteroplasmy at 98% consistent with a diagnosis of a primary mitochondrial disorder.
Case Report
A previously healthy 10-year-old girl presented to the hospital after a choking spell. Within 24 hours she developed cardiac and respiratory failure. Review of systems was remarkable for a 1-year history of progressive generalized weakness, ataxia, dysphagia, dysmetria, dysarthria, ophthalmoplegia, nyctalopia, hoarseness, and bilateral pes cavus.
Magnetic resonance imaging (MRI) of her brain demonstrated symmetric T2 fluid-attenuated inversion recovery hyperintensities in the dorsal pons, medulla, and inferior cerebellar peduncles (Figure 1A). Spine imaging was unremarkable. Nerve conduction velocities and electromyography were compatible with a sensorimotor axonal neuropathy. Vitamin E deficiency, β-lipoproteinemia and other infantile metabolic disorders, as well as spinal muscular atrophy were included in the initial differential diagnosis. Cerebral spinal fluid cell count, protein, and glucose levels were normal; serum vitamin E and B12 levels were normal; α-fetoprotein level was normal; and lactic and pyruvic acid levels were normal. A diagnosis of Friedreich ataxia was considered given her clinical, radiologic, and electrophysiologic findings. Subsequent molecular analysis of the frataxin gene was normal, prompting a change in her diagnosis to Friedreich-like ataxia. Within 12 months, the patient fully recovered, ultimately achieving her pre-hospitalization baseline. Repeat nerve conduction velocities and brain imaging showed complete resolution of her previously noted abnormalities (Figure 1B).
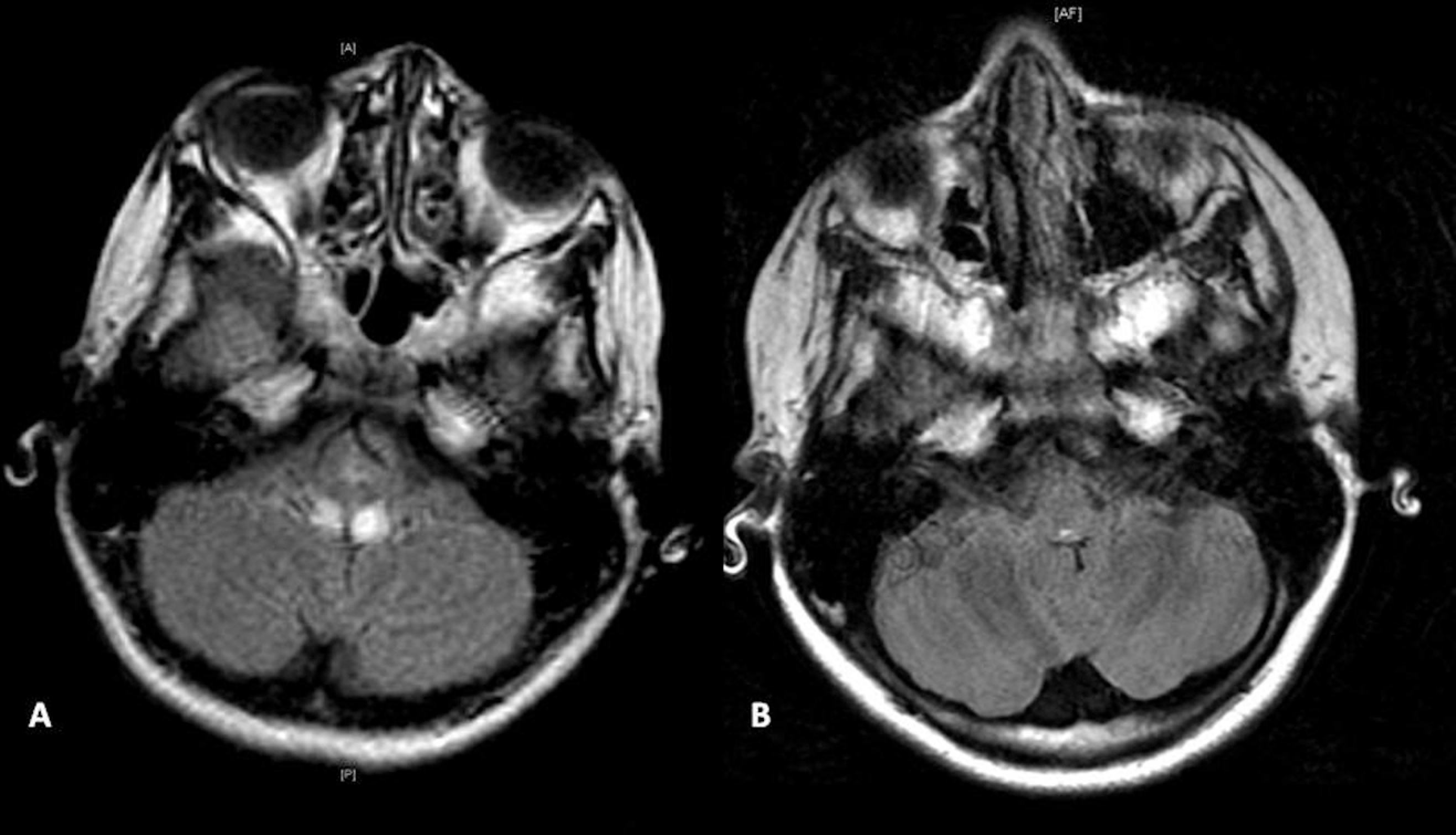
A, Axial MRI of the brain on initial presentation shows symmetric T2 fluid-attenuated inversion recovery hyperintensity of the posterior pons, posterior medulla, and inferior cerebellar peduncles. B, Axial MRI of the brain 12 months later shows full resolution of the previously noted abnormalities.
Two years later, she had acute neurologic decompensation and presented with acute ataxia, upward gaze palsy, seizures, and tremors. Magnetic resonance imaging of the brain showed symmetric T2 fluid-attenuated inversion recovery hyperintensities in the dorsal pons, medulla, and midbrain extending upward to the left thalamus (Figure 2). Nerve conduction velocities demonstrated sensorimotor axonal neuropathy. Blood lactic acid level was elevated at 7.8 mmol/L (reference 0.5-2.2 mmol/L). Mitochondrial DNA sequencing demonstrated a mutation at 8344A>G in transfer RNA lysine with heteroplasmy at 98%. One month later, the patient developed a fever of 104° F, became unresponsive, and died.
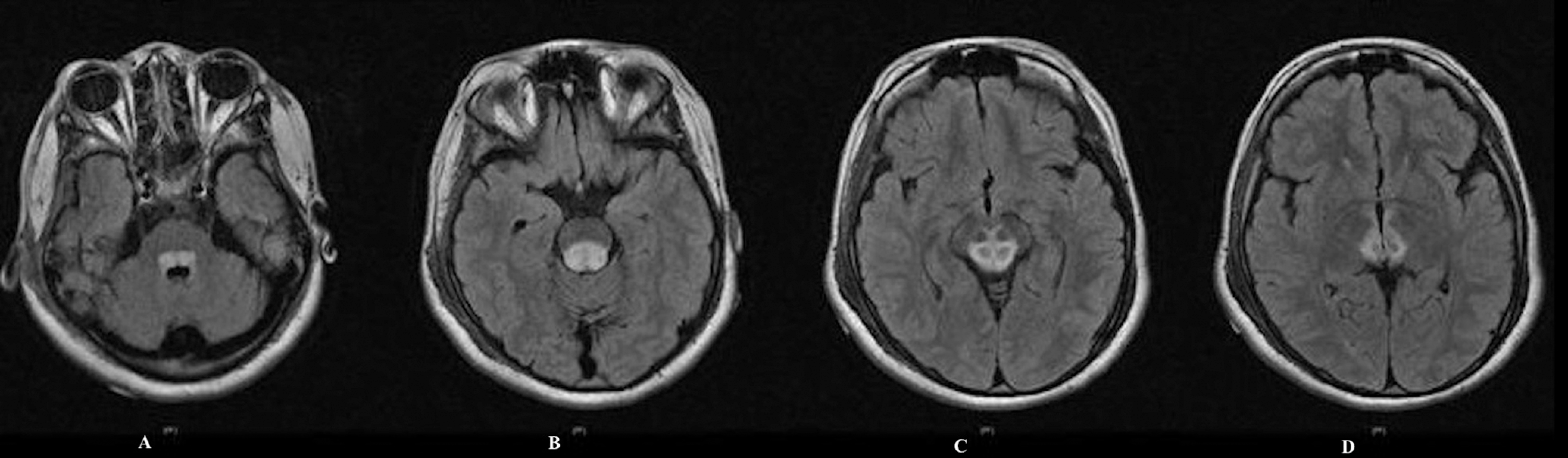
Axial MRI of the brain on recurrence shows significantly increased T2 fluid-attenuated inversion recovery signal with areas of hyperintensity in the dorsal medulla and inferior cerebellar peduncles.
Discussion
Friedreich ataxia most commonly results from hyperexpansion of triplet GAA repeats in the first intron of the frataxin gene and although molecular analysis is the gold standard, there are patients with “Friedreich-like ataxia” who have a typical Friedreich phenotype but no identifiable triplet repeats and no mutations in the frataxin gene. 1,2 Our patient’s presentation and lack of molecular diagnosis placed her in this category. The consideration of a mitochondrial disease was only raised following the complete amelioration of her condition with an abrupt recurrence several years later as is characteristic in many primary disorders of mitochondrial metabolism.
Heteroplasmic mitochondrial DNA mutations at 8344A>G present most commonly as myoclonic epilepsy associated with ragged-red fibers (MERRF). 3 Myoclonic epilepsy associated with ragged-red fibers was first reported as a new nosological entity belonging to mitochondrial encephalomyopathies in 1982 in the San Remo symposium on mitochondrial pathology. Classically, patients develop progressive myoclonus, epileptic seizures, cerebellar ataxia, dementia, sensorineural hearing loss, optic atrophy, muscular wasting, and foot deformities. Cardiac involvement is frequent in patients with these mtDNA mutations, with a high prevalence of ventricular dysfunction and Wolf-Parkinson-White syndrome. 4 Other clinical features include spasmodic dysphonia, 5 seizures, 6 sudden respiratory failure, 7 central and peripheral nervous system demyelination, 8 external ophthalmoplegia, and retinopathy. 9 As with many mitochondrial disorders, clinical expression depends to some degree on the level of heteroplasmy. 3
The clinical heterogeneity of mitochondrial disease in children presents a new challenge for modern pediatric neurology. The purpose of this report is to consider the inclusion of mitochondrial disease in the differential diagnosis of a patient with Friedriech-like ataxia, particularly should molecular studies fail to confirm the diagnosis.
Footnotes
Acknowledgments
We thank Dr Ian Butler for his assistance in manuscript revisions.
Author Contributions
JAC wrote the first draft of this manuscript. MKK assisted JAC in revisions.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
This case report did not require ethical board review.