
Abstract
Atypical teratoid/rhabdoid tumor is a rare, highly malignant central nervous system tumor most commonly occurring in very young children. Atypical teratoid/rhabdoid tumor most often presents as an expanding mass with symptoms consistent with the location of the tumor and may present with metastatic leptomeningeal disease. The authors describe 2 cases of rapidly progressive, diffuse leptomeningeal atypical teratoid/rhabdoid tumor without a solid primary mass. These cases demonstrate a clinical picture that can easily be confused with a basilar meningitis, encephalomyelitis, or vasculitis.
Case Reports
Case 1
A 16-month-old, previously healthy boy presented to an outside hospital with a 10-day history of right ptosis, gait instability, and nausea. He was considered to have acute demyelinating encephalomyelitis following lumbar puncture and magnetic resonance imaging (MRI); (results unavailable). He was then treated with steroids and antibiotics and discharged from the hospital on a steroid taper. Over the following several weeks, he presented to our institution with progressive right-sided third nerve palsy, torticollis, opisthotonos, and increased irritability. A computed tomography (CT) scan revealed acute hydrocephalus; the patient was electively intubated, and an external ventricular drain placed. Initial lumbar puncture revealed normal protein and glucose, negative cultures, and no cerebrospinal fluid leukocytosis. The patient developed fever on hospital day 2. Repeat cerebrospinal fluid testing revealed leukocytosis (2237 red blood cells/μL and 140 white blood cells/μL with 52% neutrophils), elevated protein (51 mg/dL), and negative cultures. Magnetic resonance imaging of the brain revealed increased T2 signal intensity in an expanded medulla at the cervicomedullary junction and enhancement of the third cranial nerve (Figure 1). A small soft-tissue nodule extended from the medulla near midline and continued to the fourth ventricle in the region of the foramen of Magendie. Subarachnoid and dural enhancement was present in the basilar regions and in the spinal canal. Magnetic resonance angiography revealed right-sided narrowing of the vertebral artery, M1 and M2 segments of the middle cerebral artery, and A1 segment of the anterior cerebral artery, consistent with a vasculopathy. The patient underwent a course of intravenous immunoglobulin for possible central nervous system vasculitis, without clinical improvement.
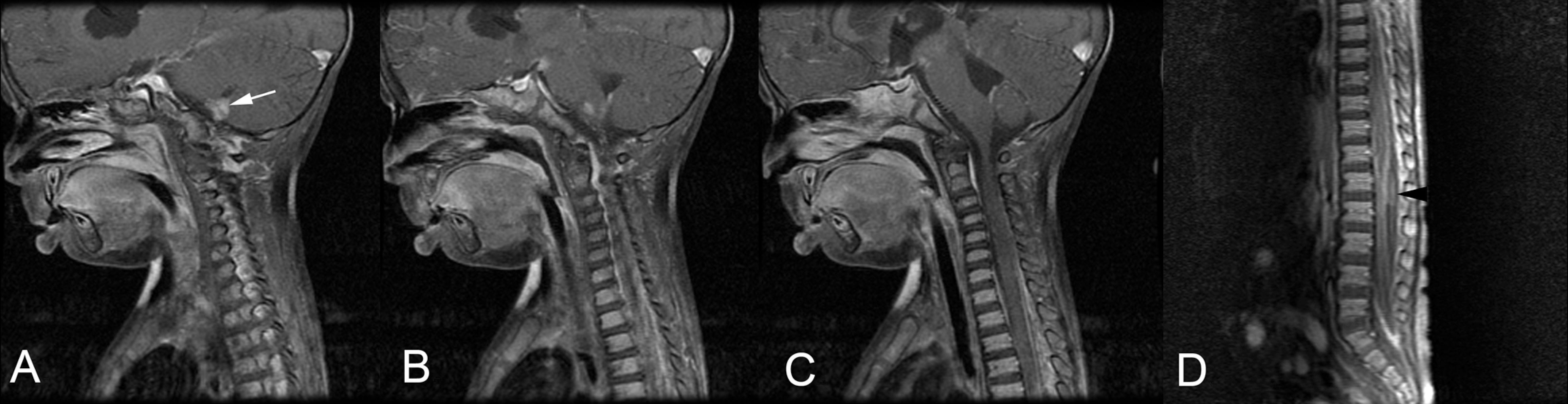
(A-C) Magnetic resonance image (MRI) of the brain of patient 1 on hospital day 2 revealed increased T2 signal intensity in an expanded medulla at the cervicomedullary junction and enhancement of the third cranial nerve. A small soft-tissue nodule (white arrow) extended from the medulla near midline and continued to the fourth ventricle in the region of the Magendie. (D) Spine MRI on hospital day 4 revealed subarachnoid and dural enhancement in the basilar regions and in the spinal canal and an intradural extramedullary mass at L1-L2 (black arrowhead).
A spine MRI on hospital day 4 revealed an intradural extramedullary mass at the level of L1-L2 just below the conus. The mass was resected and cultured. Pathological testing revealed fibrotic leptomeninges, macrophages, and degenerated material consistent with a hematoma with no evidence of a neoplasm. By hospital day 5, the patient had developed flaccid quadriplegia, areflexia, absent corneal reflexes, hypotension, and ventilator dependence. During his hospital course, he was treated with a prolonged course of steroids and antibiotics without clinical improvement. A repeat brain MRI on hospital day 20 revealed multiple diffuse cerebral infarcts, a stable small medullary nodule, and no new nodules or enhancing masses. Six weeks following hospitalization, the patient lacked any signs of improvement. An electroencephalograph (EEG) was consistent with global encephalopathy without electrocerebral silence. Several days following, the patient failed serial apnea challenge tests, and support was withdrawn. An autopsy was performed and pathological testing revealed an atypical teratoid/rhabdoid tumor, World Health Organization (WHO) grade IV, with diffuse involvement of the brain stem, spinal cord, cerebellum, temporal lobe, occipital lobe, basal ganglia, thalamus, and ventricles (Figure 2). Multiple acute and chronic infarcts were found.
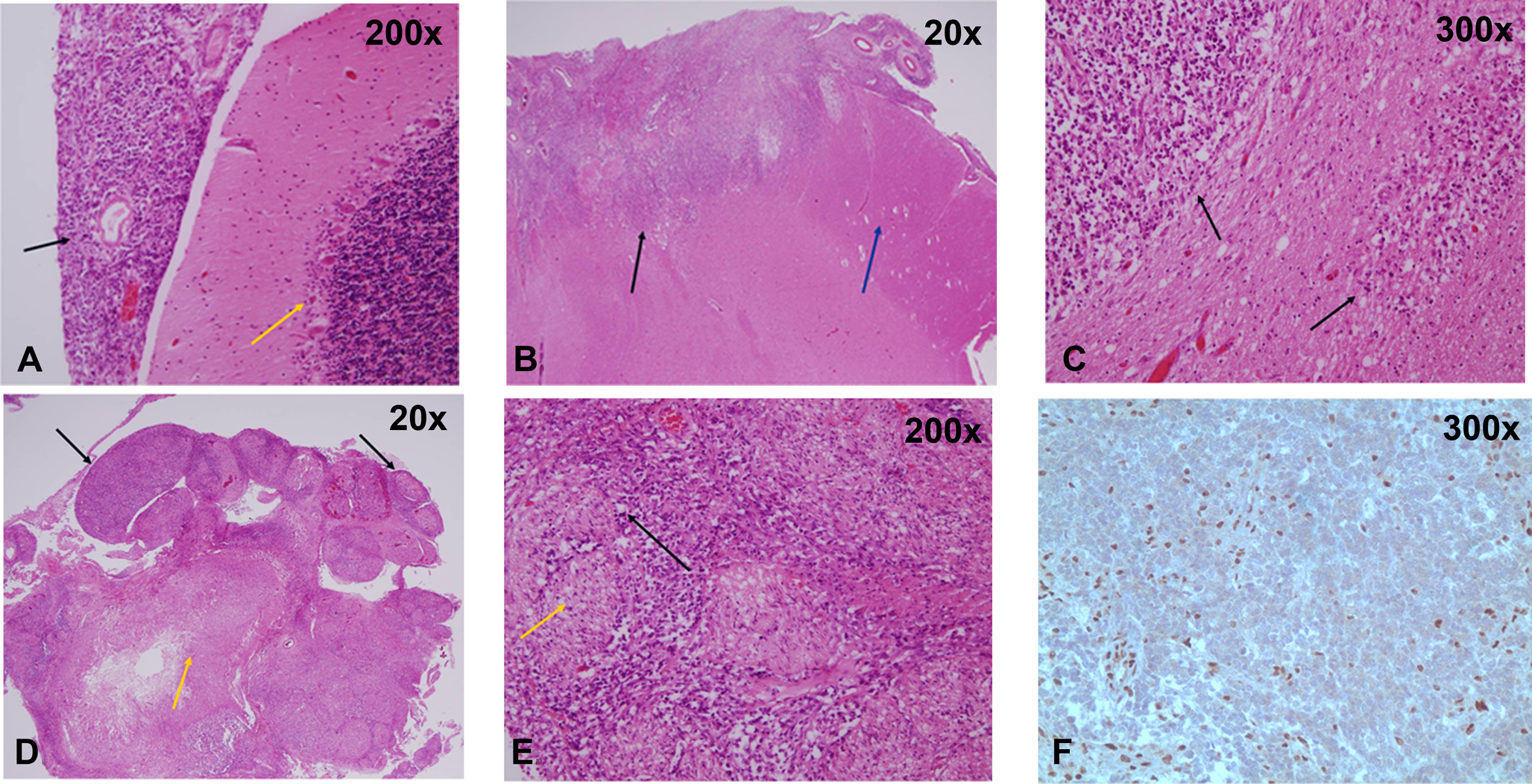
Autopsy sections of the brain of patient 1 show a primitive population of tumor cells with variable histological features. Some tumor cells are spindled and bizarre-appearing, with moderate numbers of mitotic figures. Some of these nonrhabdoid cells show areas of palisading necrosis. Other regions show tumor cells with a more classic rhabdoid appearance, characterized by large, eccentric round nuclei with a densely eosinophilic cytoplasm resembling inclusions. Immunohistochemically, the tumor stains strongly for vimentin, actin, and epithelial membrane antigen (EMA) and is focally positive for desmin, neuron specific enolase (NSE), and AE1/AE3. The tumor cells are negative for CD117, neurofilament, glial fibrillary acidic protein (GFAP), and synaptophysin. The rhabdoid component of the tumor shows negative staining for integrase interactor-1 (INI-1) marker. (A) High-power view of the normal cerebellum (yellow arrow) with associated leptomeninges with atypical teratoid/rhabdoid tumor infiltration (black arrow). (B) This section shows infiltration of the midbrain by atypical teratoid/rhabdoid tumor (black arrow) and the mostly uninvolved cerebral peduncle (blue arrow). (C) Higher power view of the midbrain being infiltrated by atypical teratoid/rhabdoid tumor (black arrows) characterized by primitive blue cells and scattered rhabdoid cells. (D) Spinal cord (yellow arrow) with diffuse infiltration of atypical teratoid/rhabdoid tumor around the spinal nerves (black arrow). (E) The spinal nerve roots (yellow arrow) are surrounded by atypical teratoid/rhabdoid tumor (black arrow). (F) The immunohistochemical stain INI-1 is negative in the atypical teratoid/rhabdoid tumor cells with background staining of normal brain parenchyma.
Case 2
A 28-month-old infant with a history of congenital cytomegalovirus infection and with resultant conductive deafness and global developmental delay presented with progressive neurological changes. Over a span of 2 weeks he developed new-onset tonic seizures, staring-off spells, language regression, ataxia, dysphagia, and dysconjugate gaze. On examination, he had right cranial nerve VI and VII palsies. Head CT showed increased ventriculomegaly, and a ventriculoperitoneal shunt was placed. Cerebrospinal fluid showed a mild leukocytosis with 11 white blood cells/μL and normal protein at 43 mg/dL. Electroencephalograph showed diffuse slowing but no epileptiform activity. New findings on brain MRI included thickening of the quadrigeminal plate with narrowing of the aqueduct and pial enhancement of the brain, brain stem, third ventricle, and vermis (Figure 3A). No enhancement of the brain parenchyma was seen. Magnetic resonance angiography was normal. The MRI findings were thought to be secondary to the acute hydrocephalus. The patient was discharged 6 days following admission with clinical improvement.
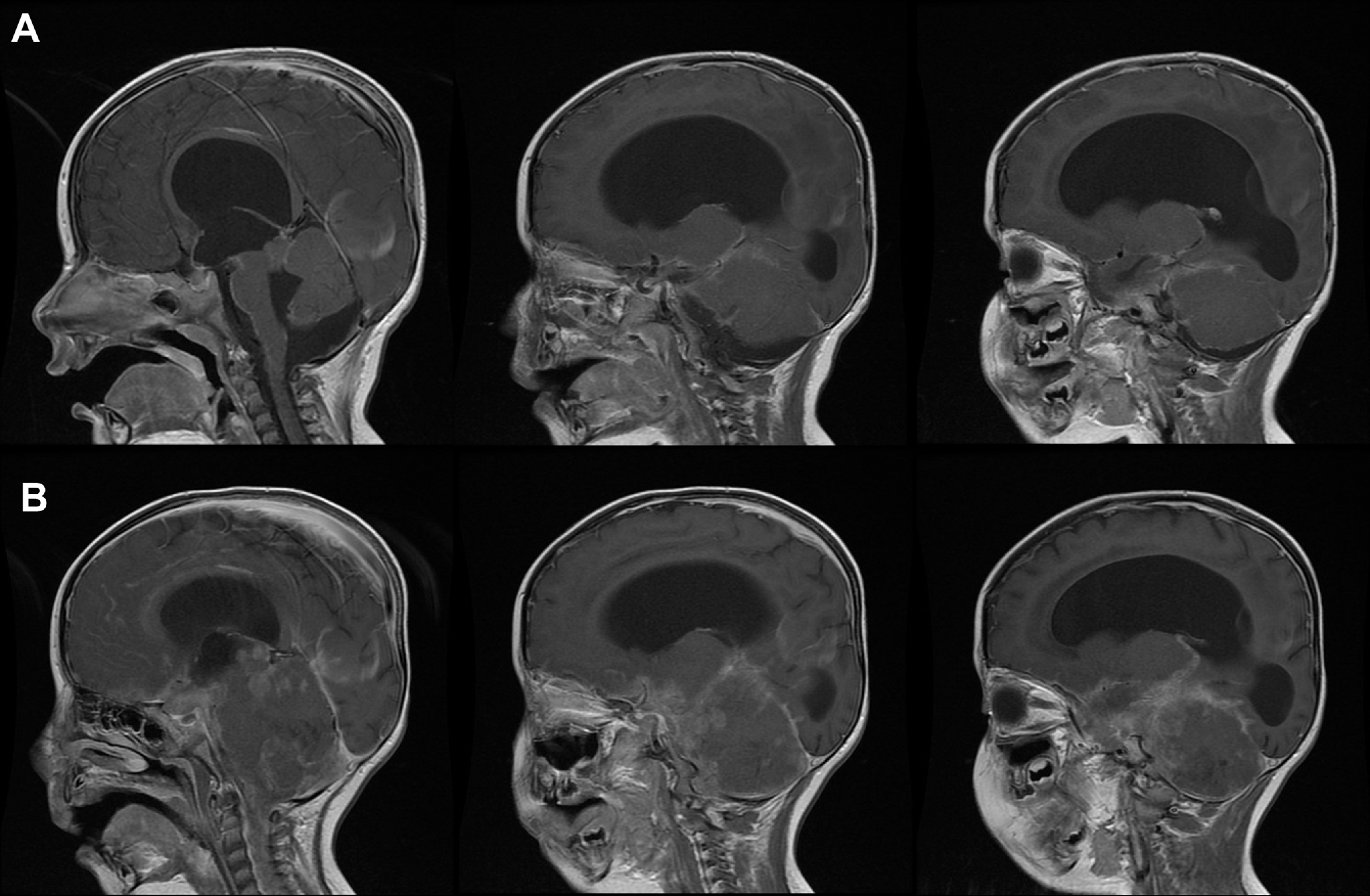
(A) Consecutive slices from the sagittal T1 gadolinium-enhanced magnetic resonance image (MRI) during initial hospitalization of patient 2 revealed acute hydrocephalus, thickening of the quadrigeminal plate, and pial enhancement of the brain, brain stem, third ventricle, and vermis. (B) Corresponding slices from the sagittal T1 gadolinium-enhanced brain MRI 6 weeks following previous MRI showed diffuse bulky meningeal enhancement.
The patient returned within 2 weeks for admission for rehabilitation. He was found to have dysphagia, vomiting, weight loss, lethargy, ataxia, and persistent cranial nerve VI and VII palsies. Head CT and EEG were unchanged. He was found to have low-grade fevers and a urinary tract infection and was treated with antibiotics. He was also hyponatremic secondary to syndrome of inappropriate anti-diuretic hormone secretion, which improved with fluid restriction. During the admission, the patient developed truncal ataxia and intermittent alteration of sensorium, which improved transiently with a decrease in valve pressure. By 2 weeks following admission, he developed left III and bilateral IV and VI cranial nerve palsies, became more lethargic, and developed opisthotonic movements. He also developed fever. Cerebrospinal fluid obtained via ventriculoperitoneal shunt aspiration showed low opening pressure, 32 white blood cells/μL, and elevated protein at 216 mg/dL. An extensive infectious evaluation was obtained, and the patient was started on broad-spectrum antibiotics. Over the following several days, he developed signs of further brain stem involvement that included dilated pupils and absent gag reflex and required intubation. The infectious evaluation failed to reveal an infectious cause. Repeat MRI of the brain 4 weeks following admission showed contrast-enhancing diffuse bulky meningeal enhancement, most prominent in the quadrigeminal plate and posterior fossa (Figure 3B). New enhancement was also present in the spinal cord at the craniocervical junction. A subsequent MRI of the spine revealed foci of nodular dural enhancement throughout the spine, circumferential enhancement in the cervical cord, and bulky enhancement of the lower thoracic spine through the thecal sac. Spinal lumbar puncture was attempted without success. Ventriculoperitoneal shunt cerebrospinal fluid aspiration obtained for cytological analysis showed clumps of abnormal but unidentifiable cells. A T12-L2 laminoplasty with biopsy of spinal cord and nerve root lesion was performed. Pathological testing of the lesion revealed atypical teratoid/rhabdoid tumor. Given the patient’s severe neurological deterioration and poor prognosis, the family elected to withdraw supportive care 6 weeks following admission. The patient died shortly following withdrawal of support.
Discussion
Acute onset of leptomeningeal enhancement is most commonly due to infectious or inflammatory disorder. In the absence of a primary mass, tumor is a less likely consideration. The patients discussed here both presented with a confusing clinical picture of progressive cranial neuropathies, encephalopathy, and slight pleocytosis with imaging suggestive of infectious or inflammatory leptomeningeal processes rather than neoplastic. In both cases, the diagnosis of a diffuse central nervous system primary tumor was elusive and could not be confirmed with cerebrospinal fluid evaluations. The occurrence of complicating cerebral infarctions with nodular disease in the central nervous system further led to concerns about fungal or other infections associated with cerebral vasculitis.
Central nervous system infections can appear on MRI as T1-weighted postcontrast meningeal enhancement with meningitis and T2-weighted and fluid-attenuated inversion recovery changes with meningoencephalitis or vasculitic complications. 1 Diffuse enhancement, nodular lesions, and space-occupying lesions with surrounding edema can be seen with some infections. 1 Vascular complications of severe infections, such as septic vasculitis, can cause rapid clinical deterioration and can be seen with magnetic resonance angiography. 1 Magnetic resonance imaging and magnetic resonance angiography are sensitive, noninvasive tests for evaluating vasculopathies, particularly involving large vessels. 2 Congenital and autoimmune disorders can cause infarctions such as mitochondrial encephalomyelopathy with lactic acidosis. 3 Patient 1 had radiographic evidence of vasculitis, directing the focus on an inflammatory disorder. Although the initial MRI suggested possible tumor, the vasculopathy along with the negative lumbar biopsy was thought to rule out a neoplastic process. The biopsy of the spinal cord lesion revealed a hematoma from a prior lumbar puncture.
Atypical teratoid/rhabdoid tumor is a rare malignant brain tumor occurring most often in children younger than 3 years, although it can occur in older children. In a meta-analysis review of 134 patients, the mean age was 3.7 years (range, 1 day to 18 years) and median age 2.2 years. There was a predominance of boys (77 boys, 7 unspecified). By location, 51.5% were supratentorial, 41% infratentorial, and 7.5% isolated spinal tumors; 13.6% were metastatic at diagnosis. 4 Presenting signs and symptoms are typically related to the tumor location and age of the patient. In very young children, irritability, vomiting, and lethargy are presenting signs associated with increased intracranial pressure. Cranial nerve deficits and gait instability may also be seen.
Atypical teratoid/rhabdoid tumor is a primitive tumor with a deletion of the tumor suppressor gene, hSNF5/INI1, on chromosome 22q11.2. Atypical teratoid/rhabdoid tumor is distinguished pathologically by characteristic rhabdoid cells and the lack of immunohistochemical staining for INI1. It may be difficult to distinguish pathologically from medulloblastoma or primitive neuroectodermal tumor. In 1 study of 13 patients with intracranial and extracranial rhabdoid tumors, only 5 had classic large areas of rhabdoid cells. 4 One of the 7 central nervous system tumors had a large area of rhabdoid cells. 4 In 2 cases, there were uniform sheets of small tumor cells, typical of primitive neuroectodermal tumor, and 1 had perivascular pseudorosettes characteristic of ependymoma. 4 The histological appearance can frequently make diagnosis difficult. All of the 13 tumors showed absent INI1 staining. 4 Loss of INI1 staining is not specific; however, this is seen in other benign and malignant tumors. 4
Atypical teratoid/rhabdoid tumor was first recognized as a distinct tumor in 1987, after publication of a series of 4 patients under the age of 2 years who were reported to have highly malignant, rapidly fatal tumors with histological features containing neuroepithelial, peripheral epithelial, and mesenchymal elements. 5 Rorke et al 6 subsequently published a report of 52 cases of central nervous system atypical teratoid/rhabdoid tumor. Atypical teratoid/rhabdoid tumor is known to have leptomeningeal dissemination, reported in 15% to 33% of cases at the time of diagnosis.6,7 In Rorke’s study, one-third of the patients evaluated for disseminated disease at diagnosis had leptomeningeal dissemination. 6 The majority of patients who relapsed had leptomeningeal disease at relapse or ultimately developed leptomeningeal spread. 6 Of the patients evaluated postmortem, 10 of 11 had widely spread tumor throughout the subarachnoid space. 6 In a case series of 33 patients, 5 presented with leptomeningeal dissemination; however, all of these patients had primary supratentorial (1), infratentorial (3), or spinal (1) tumors. 7 In a case series of 52 children with atypical teratoid/rhabdoid tumor, 10 of the 11 patients examined postmortem were found to have tumor widespread throughout the central nervous system. 6 Atypical teratoid/rhabdoid tumor has been reported to disseminate along cranial nerves. 8 Extracranial metastases are extremely rare with atypical teratoid/rhabdoid tumor but have been reported. 9
Prior to this report, only 1 case of disseminated atypical teratoid/rhabdoid tumor without a primary tumor mass had been published. 10 El-Nabbout et al 10 published a case report of a 2.5-year-old girl who presented with signs and symptoms of meningitis. Central nervous system imaging revealed hydrocephalus and leptomeningeal enhancement in the perimesencephalic cistern and the cerebral folia. She transiently improved following ventriculoperitoneal shunt placement; however, she developed seizures and rapid clinical deterioration. 10 Repeat MRI several weeks from the initial study showed more diffuse, plaque-like enhancement of the leptomeninges. 10 A leptomeningeal biopsy confirmed atypical teratoid/rhabdoid tumor, and the patient died within 2 weeks. 10
Conclusion
The 2 cases described here exemplify an atypical, confusing presentation of central nervous system atypical teratoid/rhabdoid tumor. They demonstrate that subacute encephalopathy with diffuse leptomeningeal features and cranial neuropathies should prompt an investigation for this tumor as a possible cause of primary leptomeningeal disease.
Footnotes
Acknowledgments
This material was presented as an abstract at the International Society of Pediatric Neuro-Oncology 2010. Anuranjita Nayak participated in gathering case information during her residency at St Louis University. Dr Nayak is currently at the Pediatric Neurology at Shands Medical Plaza in Gainesville, Florida.
Author Contributions
KMG contributed the majority of the work. BHD provided pathology slides, writing, and review of the manuscript. TJG provided guidance and contributed to writing and review of the manuscript. MM contributed to assembling case information and review of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.